
Abstract
To prevent zinc (Zn) dendrite formation and improve electrochemical stability, it is essential to understand Zn dendrite growth, particularly in terms of morphology and relation with the solid electrolyte interface (SEI) film. In this study, we employ in-situ scanning transmission X-ray microscopy (STXM) and spectro-ptychography to monitor the morphology evolution of Zn dendrites and to identify their chemical composition and distribution on the Zn surface during the stripping/plating progress. Our findings reveal that in 50 mM ZnSO4, the initiation of moss/whisker dendrites is chemically controlled, while their continued growth over extended cycles is kinetically governed. The presence of a dense and stable SEI film is critical for inhibiting the formation and growth of Zn dendrites. By adding 50 mM lithium chloride (LiCl) as an electrolyte additive, we successfully construct a dense and stable SEI film composed of Li2S2O7 and Li2CO3, which significantly improves cycling performance. Moreover, the symmetric cell achieves a prolonged cycle life of up to 3900 h with the incorporation of 5% 12-crown-4 additives. This work offers a strategy for in-situ observation and analysis of Zn dendrite formation mechanisms and provides an effective approach for designing high-performance Zn-ion batteries.
Similar content being viewed by others
Introduction
To date, lithium-ion batteries (LIBs) are widely used due to their high specific capacity, no memory effect, and long cycle life1. However, challenges such as limited lithium mineral resources, high production costs, and safety issues with organic electrolytes (e.g. toxic and flammable) restrict the further applications of LIBs2,3. In this regard, aqueous zinc-based batteries (AZBs) have emerged as promising next-generation energy storage technologies with potential for industrialization. AZBs utilize zinc (Zn) metal as the negative electrode and a neutral or acidic solution as the electrolyte. AZBs offer advantages such as high ionic conductivity, low cost, abundant Zn reserves, environmental friendliness, and enhanced safety4,5,6.
With the rapidly increasing demand for large-scale energy storage solutions, there is an urgent need for high-performance AZBs. Achieving the desired high energy density in AZBs hinges upon designing Zn negative electrodes with high Zn utilization efficiency, particularly at high current densities. However, under such conditions, the heterogeneous and rapid deposition of Zn2+ ions accelerates the formation of Zn dendrites. Moreover, the rough surface of the Zn foil contributes to an uneven electric field on the surface of the Zn negative electrode, resulting in a sharp point effect. This effect leads to preferential deposition of protruding parts of Zn2+ ions, culminating in dendrite formation. These dendrites not only cause capacity degradation but also pose the risk of short circuits. Zn dendrites exhibit various morphologies, including one-dimensional branched cone and two-dimensional hexagonal shapes, and unbranched dendrite morphologies. The morphology difference of the Zn dendrites reflects their ability to penetrate the separator6,7. Therefore, understanding the morphology of Zn deposition is crucial for mitigating Zn dendrites growth. Although mildly acid electrolytes like ZnSO4 are commonly used in AZBs8,9,10,11,12,13,14, the high activity of H+ ions promotes the thermodynamic tendency of the hydrogen evolution reaction (HER)15. The hydrogen (H2) gas generated during HER disrupts the stability of the SEI film and promotes the formation of zinc oxide (ZnO)/zinc sulfate hydroxide hydrate (ZSH), fostering the growth of Zn dendrite16,17. Additionally, the rapid corrosion of the Zn negative electrode exacerbates the dendrite formation. Therefore, understanding the chemical origin of Zn dendrite formation and its relation with SEI film through advanced characterization techniques is paramount in addressing the dendrite-related issues in Zn-based batteries.
From this perspective, numerous characterization techniques have been employed in the research of AZBs. For instance, the in situ X-ray diffraction (XRD) has been utilized to investigate the crystallography of Zn dendrite by observing the phase transformation between Zn and ZnO18. Moreover, the in situ XRD has been employed to demonstrate the phase transformation of Zn (102)/ZnO (002) during charge/discharge cycles, revealing the effectiveness of the anion-exchange ionomer coatings in inhibiting Zn dendrite growth19. Although many previous studies have focused on the relationship between the crystal phases and dendrite morphology using XRD, this technique cannot directly observe the morphology of Zn dendrites and the influence of SEI film composition on dendrite nucleation. Therefore, other spectroscopic techniques such as X-ray photoelectron spectroscopy (XPS), Fourier transform infrared spectroscopy (FTIR), and Raman spectroscopy have been employed to study the mechanisms of SEI film formation and dendrite growth20,21,22,23. Nevertheless, it is important to note that the utilization of XPS, Raman and FTIR primarily provides averaged information from a large sample area, which limits the ability to explore the morphology of Zn dendrites and the SEI film on the Zn surface.
Microscopic techniques have been extensively utilized to monitor the growth direction, nucleation speed, and morphology of Zn dendrites. For example, in situ optical microscope24 and scanning electron microscopy (SEM)25 can directly observe the dendritic morphology on the surface of Zn metal. Liu et al. reported mossy and poorly nucleated Zn dendrite on carbon paper using operando optical microscopy24. Qian et al. used laboratory-scale Sigray PrismaXRM X-ray microscopy to study the visualization of Zn plating and peeling behavior on 3D Cu foam, demonstrating that zinc ions tend to nucleate preferentially on convex substrates with positive local curvature. Additionally, they observed that the spontaneous reduction of Cu substrates can significantly affect the nucleation and growth behavior of Zn deposits26. Moreover, transmission electron microscopy (TEM)27 and atomic force microscopy (AFM) have been utilized to monitor the nucleation of Zn dendrites. For example, Li et al. used in situ AFM to characterize the morphological evolution and mechanisms of Zn deposition. Their AFM results demonstrated that dense, blocky Zn growth was controlled by instantaneous nucleation in concentrated electrolytes (≥0.4 M). Conversely, in electrolytes with a concentration below 0.3 M, mossy Zn formations occurred due to the low nucleation rates28. While these characterization methods are effective in testing protection strategies, the structure of Zn dendrite and the composition of the SEI film may be compromised due to the highly energetic beam spot employed in techniques such as TEM, SEM and XPS. Moreover, these technologies lack the capability to directly detect Zn corrosion and H2 evolution. Instead, they can only infer conclusions regarding hydrogen evolution or Zn corrosion based on their results. Therefore, it is necessary to develop in situ characterization technologies with spectroscopic imaging capabilities, offering high spatial resolution and non-destructive capabilities, to detect the morphology of Zn dendrites and identify the chemical composition of the SEI film.
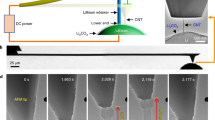
Synchrotron-based scanning transmission X-ray microscopy (STXM) has been proven invaluable for monitoring the dynamic evolution of electrode material structure and identifying chemical component distributions in both bulk and surface during the charge and discharge progress, due to its high spatial resolution, improved elemental selectivity, high chemical sensitivity, and non-destructive nature29,30,31. Therefore, the STXM characterization technique holds great promise for directly exploring the dynamic process of Zn deposition and understanding the dendrite formation, particularly regarding their chemical origins and relationship with the SEI film. Figure 1a illustrates the setup for in situ STXM, utilizing a spatially and temporally coherent soft X-ray beam. In both cases, STXM enables chemical mapping, providing valuable insights into the chemical composition and structure of deposited Zn and the SEI.

a Schematic diagram of in situ STXM setup (OSA: order sorting aperture). b The linear combination fitting result of different thicknesses (red color: 150 nm; green color: 75 nm; blue color: 15 nm, scale bar: 2 µm) and species of the deposited Zn by in situ STXM (Region I: dashed oval; Region II: dashed square). c Zn L-edge X-ray absorption spectroscopy (XAS) spectra in different color regions extracted from (b). d Zn L-edge XAS reference spectra of the standard samples (Zn foil, ZnO, ZnSO4 and Zn(OH)2). e The evolution of the distribution and morphology of the deposited Zn (Region I: dashed oval; Region II: dashed square; Region III: dashed circle, scale bar: 4 µm). f The operando STXM observations of the Zn deposition process (A: red dashed oval; B: orange dashed oval; C: blue dashed square; D: green dashed oval; E: cyan dashed circle, scale bar: 4 µm). The colored regions in (e) and (f) are created using masks derived from the corresponding colored regions in (b).
Results
In situ optical and STXM study of Zn deposition
In Zn-ion batteries, the initial morphology and the stability of the deposited Zn on the Zn electrode have a significant influence on the later Zn2+ ion deposition/stripping (e.g., dendrite formation). Optical images of the Zn foil electrode were taken during the Zn plating/stripping experiments to investigate the growth and dissolution of Zn dendrites. As shown in Supplementary Fig. 1a, during the Zn plating cycle, in the 50 mM ZnSO4 electrolyte, Zn dendrites began to grow on the Zn foil surface. ‘Clusters’ of Zn dendrites initiated/grew at various sites on the surface of the Zn electrode. For example, some tiny and uneven tips can be observed on the Zn surface after 30 min of plating. When the electroplating time reaches 40 minutes, Zn ‘clusters’/dendrites tend to deposit on the convex parts compared to flat areas of the Zn foil. This phenomenon is consistent with the conclusion of Liu et al, who utilized the in situ and operando X-ray microscopy methods to visualize the Zn plating and stripping behaviors26. In addition, according to previous reports32 in neutral solution, the equilibrium potential of H2O/hydrogen (H2, 0 V vs standard hydrogen electrode (SHE)) is higher than that of Zn2+/Zn (−0.76 V vs SHE), and the coexistence of Zn and H2O is thermodynamically unstable, meaning that they will react spontaneously and release H2. During the plating process, there is a competition between Zn deposition and H2 evolution; theoretically, hydrogen evolution takes precedence over Zn deposition. The accumulation of H2 during the HER process, adhering to the surface of the negative electrode, could impede the nucleation of Zn, thereby facilitating the growth of Zn dendrites33. Additionally, as the plating time gradually increased, mossy Zn was formed after 120 min due to the rough and uneven Zn surface. On the other hand, during the subsequent Zn stripping process, as shown in Supplementary Fig. 1b, the Zn dendrites gradually dissolve and diminish in size with the increases in stripping time. Interestingly, comparing the initial and final states of the Zn electrode (i.e., before the plating cycle and the end of the stripping process; the durations of both processes are 120 min), it is obvious that after the completion of the stripping process, there is a small amount of undissolved Zn dendrite could be observed on the Zn electrode surface, which could be attributed to the dead Zn or by-products (e.g. ZnO, ZSH). These insoluble Zn metals serve as the preferential deposition sites of Zn2+ ions in the following plating cycle due to the tip effect, thereby promoting the growth of Zn dendrites. Moreover, as shown in Supplementary Fig. 2, online gas chromatography (GC) analysis indicates that H2 is produced during Zn2+ ion deposition progress, in the 50 mM ZnSO4 electrolyte, indicating that hydrogen evolution may also affect the deposition of Zn2+ ions and thereby promote the growth of Zn dendrites.
The limited resolution of the optical observation cannot reveal the specific composition of deposited Zn in real-time. Therefore, to further monitor the stages, composition, and stability of Zn deposition, the in situ/operando STXM investigations were conducted. Figure 1a and Supplementary Fig. 3 show the schematic diagram and digital photograph of the in situ STXM set-up. The aurum foil (Au) was used as the current collector on the in situ electrochemical Si3N4 window in the set-up, 50 mM ZnSO4 solution was used, and Zn metal was pre-deposited at a high constant voltage of −1.7 V (vs. Ag/AgCl) for 90 s. To determine the spatial distribution of the chemical composition of various deposited Zn species, a linear combination fitting was performed on the acquired STXM image stack as a function of photon energy. Figure 1b shows the chemical imaging of the deposited Zn on the surface of the Au electrode. The red, green, and blue regions represent the deposited Zn species with thicknesses of 150, 75, and 15 nm, respectively, determined from their absorption edge jumps at the Zn L-edge. As shown in Fig. 1b, at the thick region (150 nm), the deposited Zn exhibits moss-like, whisker-like (region I) and flake-like (region II) morphologies. In the thin region (75 nm), it shows granular and moss-like morphologies. In addition, the more evenly distributed signals (blue color) at the right edge of the Au electrode are likely to be from the electrolyte, indicating that most of the signals in the thinnest region (15 nm) come from the ZnSO4 electrolyte, along with a small amount of the deposited Zn (Fig. 1b). The existence of this 15 nm-thick region only on the far right side of the STXM images is due to the spatial distribution of these species. Figure 1c presents the corresponding XANES spectra of Zn L-edges with different species and thicknesses in Fig. 1b, and Fig. 1d shows the Zn L-edge spectra of four standard Zn-containing samples (Zn foil, ZnO, ZnSO4 and Zn(OH)2). Subsequently, the evolution of the morphologies of the deposited Zn was monitored by the in situ STXM electrochemical investigations (Fig. 1e). The pre-edge region (circled in red) in Fig. 1c may be attributed to the transitions of Zn 2p electrons to the Zn 4s and antibonding 3d states34. Supplementary Fig. 4 shows the spatial distribution of ZnO, ZnSO4, Zn foil, and Zn(OH)2. Supplementary Fig. 4a indicates that the composition of mossy Zn is mainly metallic Zn, while the composition of flaky and whisker-like deposited Zn is ZnO, Zn(OH)2, ZnSO4/ZSH (Supplementary Fig. 4b–d). To further interpret the information in Fig. 1e, the results derived from Fig. 1c, d and Supplementary Fig. 4 should be collectively considered. As shown in Fig. 1e, the deposited Zn species on the Au electrode surface with a loose and porous form can be observed, indicating that the deposition of Zn2+ ions is not uniform, which promotes the formation of Zn dendrites. Moreover, at low energy between 1010.0 and 1025.5 eV, the morphology of Zn2+ ion deposition is mainly granular (region III) and mossy. When the photon energy reaches above 1030.3 eV, the morphology of deposited Zn begins to exhibit whisker (region I) and flake (region II) topography. Additionally, the size of whiskers and flakes increases with the increase of the photon energy (from 1033.0 to 1060.0 eV). Interestingly, from 1010.0 to 1021.5 eV, in region III of Fig. 1e, the composition is mainly metallic Zn (Supplementary Fig. 4a). However, from 1023.0 to 1060.0 eV, in region III, the green part gradually disappears and is gradually replaced by the red part, which also means that the composition of the deposited Zn gradually changes from metallic Zn to ZnSO4 or Zn(OH)2 or ZSH (Supplementary Fig. 4c, d). These results indicate the existence of the self-corrosion phenomenon of Zn. As commonly understood, the corrosion process involves the dissolution of Zn metal at the corrosion site, resulting from the loss of electrons. In a neutral solution, H2O gains electrons, generating H2 and OH-35,36,37. Moreover, the formed OH- and H2O react with the electrolyte, producing electrochemically inert corrosion by-products that accumulate on the Zn negative electrode surface. These deposits impede ion transmission, exacerbate electrode polarization, and consequently facilitate the formation of Zn dendrites32,38. Therefore, the self-corrosion of the deposited Zn produces by-products such as ZSH, which in turn promote the formation of Zn dendrites and affect the electrochemical performance of Zn ion batteries. Moreover, the blue part exhibits its maximum brightness at the beginning of the Zn deposition reaction (from 1010.0 to 1011.0 eV). However, as the Zn deposition reaction progresses (from 1021.5 to 1060.0 eV), the blue segment gradually dims without entirely vanishing. This observation suggests that the electrolyte undergoes depletion during Zn deposition, which can be attributed to the formation of ZSH or the occurrence of HER.
To further investigate the effect of HER on the morphology of the deposited Zn, the operando STXM experiment was performed, based on the findings illustrated in Fig. 1e. The experiments were carried out at −1.6 V (vs Ag/AgCl) using the potentiostatic deposition method, as detailed in Supplementary Figs. 5 and 6. Supplementary Fig. 5f shows both the current vs. time curve and STXM map generated during these tests. Additionally, it displays the time corresponding to different photon energies. For this analysis, six X-ray energy points (1015.0, 1023.5, 1025.5, 1029.3, 1032.5, and 1038.3 eV) were chosen to represent the characteristic XANES features of the deposited Zn (Fig. 1c, d). In Fig. 1f at 1015.0 eV (Stage 1), there is a minimal disparity between the map and the results in Fig. 1e, except for a slight change in the current value (Supplementary Fig. 5f) which could be attributed to the migration of Zn2+ ions. To more accurately identify the changes from Fig. 1f-Stage 1, new maps of Stage 1 (from 1023.5 to 1038.3 eV) are obtained by subtracting the maps of Fig. 1f-1015.0 eV (Stage 1) on the absorption scale (i.e., optical density), as depicted in Supplementary Fig. 5a–e. In Supplementary Fig. 5a–e, the bright color represents the Zn species that are formed, while the dark color represents the Zn dendrites that disappear. The positive signal observed in Supplementary Fig. 5a–e indicates the formation of Zn species during the deposition process. These figures demonstrate the moss shapes of the generated Zn species, attributed to the strong electric field at the original moss-shaped Zn, which attracts Zn2+ ions, as reflected in the increased current in Supplementary Fig. 5f. In region B of Supplementary Fig. 5, the gradually brightening color indicates the deposition of Zn2+ ions. This observation supports the notion that Zn2+ ions are preferentially deposited on convex substrates with positive local curvature26. However, region A in Supplementary Fig. 5d appears darker compared to Supplementary Fig. 5a–c, indicating the disappearance of deposited Zn due to corrosion by the ZnSO4 electrolyte. The corrosion current results from the electrochemical corrosion process of the deposited Zn by the ZnSO4 electrolyte, leading to a decrease in deposition current. Therefore, in the i-t curve (Supplementary Fig. 5f), the current gradually returns to zero between 250 and 300 s. In the range of 300–382 s (stage 1), the current fluctuates around zero mA (Supplementary Fig. 5f), which indicates that only a small amount of Zn2+ ions are deposited during the plating process. This phenomenon is due to the presence of a ZnO/ZSH passivation film on the outer surface of the deposited Zn layer, hindering the rapid capture of electrons by Zn2+ ions to generate Zn metal. This result suggests that the later deposition of Zn2+ ions is controlled by ion transport kinetics. In the initial state of Stage 2 (Fig. 1f), the current experiences a sharp increase followed by a gradual decrease. This behavior could be attributed to the uneven deposition of Zn2+ ions, the generation of by-products, and the significant fluctuations in incident energy during the X-ray measurement process. The map of Fig. 1f at 1015.0 eV monitored in Stage 2 is similar to that in State 1 (Figs. 1f, 1015.0 and 1038.3 eV). However, as the deposition reaction progresses, the i-t curve (Supplementary Fig. 5f) in Stage 2 exhibits a very poor signal-to-noise ratio, indicating the potential generation of H2 gas or an increased generation of corrosion by-products. The generated H2 gas can disrupt the electrode structure, leading to the dissolution of Zn dendrites. Therefore, compared to the map in Fig. 1f at 1015.0 eV (Stage 2), the morphologies of the maps from 1023.5 to 1038.3 eV (Fig. 1f, Stage 2) gradually change: first, the whisker-like dendrites (region B in Fig. 1f) begin to disappear, followed by the disappearance of the sheet-like (region A and D in Fig. 1f) dendrites, and eventually, the moss (region C in Fig. 1f) and granular (region E in Fig. 1f) dendrites start to vanish. This observation indicates that H2 production initiates at 1023.5 eV in Fig. 1f (Stage 2), albeit at a minimal level initially, primarily affecting the most unstable whisker-like dendrites. With the gradual increase of H2 production, flaky (region A and D in Fig. 1f) and mossy (region C in Fig. 1f) dendrites progressively disappear as well. In Supplementary Fig. 6a-c, brighter colors indicate the formation of more products, while darker colors (Supplementary Fig. 6d–f) indicate the disappearance of dendrites. Supplementary Fig. 6a-c reveal an increase in topography during Stage 2 compared to Stage 1. Supplementary Fig. 6d–f shows a reduction in the topography of Stage 2 compared to Stage 1. As shown in Supplementary Fig. 6a, d, in stage 2, despite the formation of only a small amount of deposited Zn, a significant number of whisker-like dendrites disappear (region B in Fig. 1f). In Supplementary Fig. 6e, f, the disappearance of the sheet-like dendrites commences (region A and D in Fig. 1f), while the whisker-like dendrites (region B in Fig. 1f) disappear at a slower rate due to their substantial loss during the initial stages. Supplementary Fig. 6b, c indicate the continued appearance of a small amount of products, which can be attributed to the by-products (ZnO or ZSH) produced by HER.
The above studies present the visualization of the morphology (granular, mossy, flake, and whisker-like) of the deposited Zn and its corrosion using the in situ STXM technique. Notably, it reveals that the granular deposited Zn is unstable and prone to rapid dissolution. Interestingly, previous literature primarily inferred the existence of corrosion phenomena based on the intrinsic property (high H+ activity) of ZnSO4 solution15,16,39,40, with few reports employing advanced characterization techniques to prove this41,42. In this work, we directly observe dynamic corrosion phenomena and HER using the in situ STXM technique. Furthermore, to mitigate Zn negative electrode corrosion and enhance Zn utilization, it is necessary to create a protective layer on the Zn negative electrode surface.
STXM spectro-ptychography and in situ synchrotron-based X-ray diffraction of Zn dendrites
To further investigate the relationship between Zn dendrite morphology and chemical composition, ex situ spectro-ptychography STXM measurements were carried out following the cyclic voltammetry (CV) test (Supplementary Fig. 7a). In the initial cycle, the peaks at −0.75 and 0.25 V correspond to the oxidation and reduction peaks of Zn2+ ions, respectively. However, after 7 cycles, the curve is notably different from the first cycle due to the dendrite formation and the resultant dead Zn, indicating significant irreversibility. The STXM image corresponding to the CV curve in the 7th cycle is shown in Supplementary Fig. 7b. The conventional STXM image depicts increased Zn deposition at the edge of the Au electrode in Region 1 and dead Zn fall-off in Region 2 (Supplementary Fig. 7b). To better distinguish the morphology and chemical composition of Region 1 and Region 2, a series of high-resolution multi-energy spectro-ptychographic images and XAS spectra were obtained, as displayed in Fig. 2a–j.

a, b Ptychography averaged amplitude (optical density) and phase images from O K-edge and Zn L-edge in region 1 after 7 plating/stripping cycles, respectively. c Chemical mapping of Zn dendrites by ptychography amplitude images in region 1. d, e Ptychography averaged amplitude (optical density) and phase images from O K-edge and Zn L-edge in region 2 after 7 plating/stripping cycles, respectively. f Chemical mapping of Zn dendrites by ptychography amplitude images in region 2 (a–f: scale bar: 1 µm). g–j Amplitude (absorption) and phase spectra extracted from “Thoracic” and “Cluster”-like dendrites in regions 1 and 2.
The amplitude (absorption) images and phase images of Region 1 and Region 2 are displayed in Fig. 2a, d and Fig. 2b, e, respectively. In principle, the absorption image can highlight the denser areas and specific target element/chemical species within the sample, whereas the phase contrast image can resolve tiny structures and other elements beyond the measured elemental edge with high precision. Moreover, the absorption images, which provide details based on the attenuation of the X-ray beam, are crucial for identifying the presence and concentration of materials. The phase images, on the other hand, provide valuable insights through the detection of the phase shift experienced by the beam, revealing structural and compositional details that may not be readily apparent in absorption images. Combining the amplitude (Fig. 2a, d) and phase (Fig. 2b, e) images provides information on morphology with higher spatial resolution43. As shown in Fig. 2a, b, the Zn dendrites at the edge of the Au electrode exhibit “thoracic” (green) and “cluster”-like (red) morphologies. Figure 2d, e reveals that the exfoliated dead Zn has a regular hexagonal and dendritic morphology, possibly resulting from the production of H2. To analyze the chemical phase distribution in different regions, principal component analysis (PCA) was used, and the results are shown in Fig. 2c, f. The various colorful clusters in these figures correspond to distinct chemical phases. Figure 2g, i and Fig. 2h, j present the average O K-edge and Zn L-edge spectra extracted from the various corresponding clusters in Fig. 2c, f, respectively. The colors in these spectra align with those of clusters in Fig. 2c, f. The solid lines represent the absorption spectra and the dotted lines represent the phase spectra. In region 1, as evidenced by Fig. 2g, h (solid line) and Fig. 1d, the components of “thoracic” and “cluster”-like Zn dendrites are mainly ZnO and Zn(OH)2. Moreover, in Fig. 2h (red and green spectra, solid line), the peak at around 1023 eV indicates the presence of metallic Zn. In region 2, Fig. 2j (red solid line), the peak at around 1023 eV demonstrates the presence of metallic Zn, while in the green solid line, a peak at around 1023.6 eV suggests the presence of Zn(OH)2. Moreover, as shown in Fig. 2i, the peak at 537.1 eV is attributed to water43, and this peak is formed due to the O-H bond in ZSH. Therefore, the components of planar dead Zn encompass ZnO, ZSH, and metallic Zn (red region). Furthermore, compared to the absorption spectra (solid line), the phase spectra (dotted line) at the O K-edge shifted to lower energies by approximately 0.9 eV (peak A) and 1.5 eV (peak B) in Region 1, and 1.8 eV (peak D) in Region 2. The Zn L-edge phase spectra of peak C in Region 1 and peak E in Region 2 also shifted to lower energies by roughly 1.6 eV and 2.1 eV, respectively, compared to their corresponding absorption peaks.
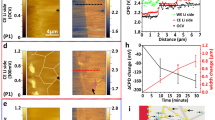
Based on the above discussions, in 50 mM ZnSO4 electrolyte, the occurrence of HER and self-corrosion gives rise to Zn dendrites with moss, plate and whisker-like morphologies, thereby forming an SEI film containing ZnO and ZSH. This SEI film with poor ionic conductivity further facilitates Zn dendrite formation and manifests inadequate electrochemical stability (Fig. 1e, f). The planar dead Zn and regular hexagonal Zn dendrites are prone to detach from the electrode surface due to their weak adhesion and relatively inert electrochemical reactivity, coupled with H2 production (Supplementary Fig. 7b and Fig. 2f). Consequently, the Zn negative electrode exhibits poor cycle stability in 50 mM ZnSO4 electrolyte due to the presence of ZnO and ZSH (Fig. 3a). As illustrated in Fig. 3a, the cycling stability of the Zn negative electrode in the 50 mM ZnSO4 electrolyte was assessed at a current density of 1.0 mA cm−2/1.0 mAh cm−2. The Zn | |Zn symmetric cell with the 50 mM ZnSO4 electrolyte exhibited poor cycling stability (46 h, overpotential: approximately 135 mV) due to the unstable and porous SEI film. Notably, Supplementary Fig. 8a shows a top-view SEM image of Zn deposition (plating progress) on Cu foil in the 50 mM ZnSO4 electrolyte (plating time: 3 h). It clearly indicates the growth of regular hexagonal flakes, leading to a loose and porous surface, which is consistent with the observations in Fig. 1e. Therefore, in the 50 mM ZnSO4 electrolyte system, this porous structure cannot effectively prevent the electrolyte contact with fresh Zn, which accelerates the Zn surface corrosion and leads to the formation of by-products ZSH and ZnO. Supplementary Fig. 8b shows a top-view SEM image of the Zn negative electrode in the 50 mM ZnSO4 electrolyte after 20 plating/stripping cycles. It demonstrates that the dead Zn, massive mossy Zn, dendritic Zn and cracks emerge on the Zn surface, leading to the formation of Zn dendrites. Moreover, to ascertain the composition of the Zn negative electrode, ex situ synchrotron-based X-ray diffraction (SXRD) was conducted, as displayed in Fig. 3b, c and Supplementary Fig. 9. Figure 3b shows the collection area of the diffraction pattern, and Fig. 3c shows the most representative 48 SXRD spectra. As presented in Fig. 3c, the peaks at ~4.7°, ~5.1°, ~5.6°, ~6.9°, ~8.7°, ~8.8°, ~9.4°, ~9.9°, ~10.1° and ~10.3° are attributed to Zn metal. The peaks at ~7.16° and ~10.7° may be attributed to 6Zn(OH)2·ZnSO4·4H2O and ZnO, respectively (Supplementary Fig. 9). These findings suggest that the by-products may be attributed to ZnO and 6Zn(OH)2·ZnSO4·4H2O. As shown in Fig. 3b (orange box) and Supplementary Fig. 9a, the presence of 6Zn(OH)2·ZnSO4·4H2O is unevenly dispersed across the grids, indicating the random occurrence of HER on the Zn surface. Moreover, the peak intensity and full width at half maximum (FWHM) of 6Zn(OH)2·ZnSO4·4H2O (Supplementary Fig. 9a) and ZnO (Supplementary Fig. 9b) exhibit constant variation, leading to diverse crystal forms and grain sizes of 6Zn(OH)2·ZnSO4·4H2O and ZnO. These results indicate that the distribution of 6Zn(OH)2·ZnSO4·4H2O and ZnO on the Zn surface is not uniform, which hinders the deposition of Zn ions and promotes the growth of Zn dendrites. Therefore, based on the STXM results, to mitigate self-corrosion, inhibit the formation of zinc dendrites and hydrogen evolution reactions, and reduce by-product generation, it is imperative to explore new and efficient interface protection strategies. The electrolyte additive approach emerges as a simple, rapid, and cost-effective method for constructing a dense and stable protective film on the Zn negative electrode surface.

a The long cycle performance of Zn||Zn symmetric cell in the 50 mM ZnSO4 electrolyte at 1.0 mA cm−2/1.0 mAh cm−2 (Zn utilization: 2.4%, Blank: 50 mM ZnSO4). b A schematic illustration of X-ray diffraction mapping employing a 21 × 21 grid, with each grid segment being 1 × 1 μm2 and featuring no overlap. c The ex situ synchrotron-based X-ray diffraction (SXRD) spectra of Zn negative electrode in the 50 mMZnSO4 electrolyte after 20 cycles at a current density of 1 mA cm−2/1mAh cm−2. d The 1st cycle curve of Zn||Zn symmetric with the 50 mM ZnSO4 electrolyte containing 50 mM LiCl additive at a current density of 0.5 mA cm−2/1.5 mAh cm−2. e and f The in situ SXRD spectra of Zn||Zn symmetric cell for Zn Stripping state Zn Plating state, respectively, with the 50 mM ZnSO4 electrolyte containing 50 mM LiCl additive at a current density of 0.5 mA cm−2/1.5 mAh cm−2. These spectra are labeled in (d).
To validate the conclusions drawn from STXM characterizations, lithium chloride (LiCl) material was employed as an electrolyte additive to inhibit Zn dendrite growth due to its low cost and high solubility32,44. Supplementary Fig. 10 illustrates the cycling stability of Zn negative electrode in 50 mM ZnSO4 electrolyte with 50 mM LiCl additive at a current density of 1.0 mA cm−2/1.0 mAh cm−2. The inclusion of LiCl additive in the cell demonstrated higher cycling stability (overpotential: approximately 118 mV), lasting up to 138 h compared to the Zn negative electrode in the electrolyte of 50 mM ZnSO4 (Fig. 3a). This enhancement can be attributed to the formation of a stable and dense SEI film, consisting of Li2CO3 and Li2S2O7, on the Zn surface when the 50 mM LiCl additive was added into the 50 mM ZnSO4 electrolyte. This film effectively hinders direct contact between water and Zn, thus preventing the generation of by-products (ZSH, ZnO).
To further confirm the protective effect of the LiCl additive on the Zn negative electrode, in situ SXRD, ex situ XRD, XPS and SEM characterizations were employed, and the results are displayed in Fig. 3d–f, Supplementary Figs. 11–17. As presented in Fig. 3d–f, during the initial stripping/plating progress, in situ SXRD was used to characterize the changes in the composition of the Zn electrode surface in the 50 mM ZnSO4 electrolyte containing 50 mM LiCl additive at 0.5 mA cm−2/1.5 mAh cm−2. Figure 3d illustrates the first cycle curve at 0.5 mA cm−2/1.5 mAh cm−2, while the SXRD spectra at different times are shown in Fig. 3e, f. After introducing the LiCl additive into the 50 mM ZnSO4 electrolyte, signals corresponding to the metallic Zn signal were observed at ~4.7°, ~5.1°, ~5.6°, ~6.9°, ~8.7°, ~8.8°, ~9.4°, ~9.9°, ~10.1° and ~10.3°. Moreover, the peaks at ~6.49°, ~9.18° and ~10.78° are also obviously detected, which may be attributed to Li2S2O7 (Fig. 3e, f). This indicates that the Li+ ion participated in the SEI film-forming reaction, generating Li2S2O7 during the first cycle. Additionally, the shoulder peak at~10.71° is attributed to ZnO (Supplementary Fig. 11a, b), which differs from the spike peak of ZnO in Supplementary Fig. 9b. Furthermore, as delivered in Supplementary Fig. 12a, during the Zn stripping process, a weak peak of 6Zn(OH)2·ZnSO4·4H2O inevitably emerged. However, during Zn plating, this peak disappeared (Supplementary Fig. 12b). Moreover, the presence of 50 mM LiCl additive in the 50 mM ZnSO4 electrolyte (Supplementary Fig. 13) effectively suppresses HER, which is 0.22 times that observed in the 50 mM ZnSO4 electrolyte (Supplementary Fig. 2). These findings indicate that the introduction of LiCl additive effectively suppresses the generation of ZnO and ZSH by-products and improves the cycle life of Zn metal. As shown in Supplementary Fig. 14a, after introducing the LiCl additive into the 50 mM ZnSO4 electrolyte, a dense and smooth Zn deposition was detected due to the formation of a stable SEI film. Over time, this robust SEI film inhibits the formation of by-products (ZSH, ZnO) and Zn dendrites during extended cycling. Supplementary Fig. 14b presents a top-view SEM image of the Zn negative electrode after 20 cycles at 1.0 mA cm−2/1.0 mAh cm−2, revealing a smooth surface. Based on these observations, it can be inferred that the growth of Zn dendrite is effectively suppressed by using LiCl as an additive to form a stable and dense SEI film, thus enhancing the cycling stability of the Zn negative electrode (Supplementary Fig. 10). To study the inhibitory effect of the SEI film after adding the LiCl additive, XRD (λ = 0.1541 nm) characterization was conducted (Supplementary Fig. 14c). When the 50 mM LiCl was added to the 50 mM ZnSO4 electrolyte, most peaks of ZnO disappeared and the intensity of the peak at 11.2° weakened, confirming the inhibition of ZSH and ZnO growth via a stable and dense SEI film. This result is consistent with Supplementary Fig. 12. To further determine the composition of the SEI film, XPS measurements were carried out. Supplementary Fig. 14d presents the O 1 s spectrum of the Zn surface after 10 cycles at a current density of 1.0 mA cm−2/1.0 mAh cm−2. The O 1 s spectrum was deconvoluted into four peaks at 531.3 eV, 532.1 eV, 533.5 eV and 534.5 eV, corresponding to Li2CO3, Li2ZnCl4·9H2O, H2O and surface-adsorbed oxygen, respectively45,46,47,48. Moreover, Cl 2p and Zn 2p3/2 XPS spectra were also examined (Supplementary Fig. 15). When LiCl is introduced into the 50 mM ZnSO4 electrolyte, the Zn-Cl bond and Li2ZnCl4·9H2O were detected (Supplementary Fig. 15a, b)48,49,50. This SEI film (Li2S2O7, Li2CO3, Zn-Cl and Li2ZnCl4·9H2O) not only inhibits the occurrence of HER but also promotes the transmission of Zn2+ ions, resulting in a uniform and smooth deposition surface, thereby preventing Zn dendrite growth. Moreover, to further validate the in situ formation of dense SEI films by the LiCl additive, and confirm their chemical compositions as Li2S2O7 and Li2CO3, the XRD, SEM, energy-dispersive X-ray spectroscopy (EDX), high-resolution transmission electron microscopy (HRTEM), and TEM-EDX analyses were conducted and presented in Supplementary Figs. 16, 17. Prior to the measurements, the Zn electrode was rinsed with deionized water to remove any additional electrolytes adsorbed on its surface. As shown in Supplementary Fig. 16a, Li2S2O7 and Li2S2O6 2H2O were detected following immersion of the Zn electrode in the 50 mM ZnSO4 electrolyte containing 50 mM LiCl additive for serval days. Subsequently, SEM-EDX analysis was carried out to study the element distribution on the Zn surface at a constant potential of −0.8 V for 3000 s. In the electrolyte of 50 mM ZnSO4, the deposited Zn appeared loose, porous and mostly mossy (Supplementary Fig. 16b). Conversely, in the 50 mM ZnSO4 electrolyte with 50 mM LiCl additive, a densely deposited layer is observed on the Zn surface (Supplementary Fig. 16c). Moreover, SEM-EDX results (Supplementary Fig. 16b–d) reveal the presence of carbon (C), sulfur (S), and chlorine (Cl) elements in the 50 mM ZnSO4 electrolyte containing 50 mM LiCl additive, indicating the formation of an SEI film containing these elements on the Zn surface. In addition, the S content in the electrolyte containing 50 mM LiCl additive is lower than that in the untreated electrolyte of 50 mM ZnSO4 (Supplementary Fig. 16d), suggesting that the dense SEI film inhibited the production of ZSH by-products. Supplementary Fig. 17a illustrates the elemental distribution of the plated Zn electrode 50 mM ZnSO4 electrolyte with 50 mM LiCl additive, with clear mappings of C, O, S, Zn, and Cl. Notably, the outer edge of the Zn electrode is covered with a SEI film containing C, O, S, and Cl elements. As shown in Supplementary Fig. 17b, the TEM image of the Zn electrode reveals a uniform and dense SEI film with a thickness ranging from 8.09 to 9.75 nm. This uniform and dense SEI film can effectively suppress the production of by-products and the growth of Zn dendrites. As displayed in Supplementary Fig. 17c, the interplanar spacing of the deposited metallic Zn is 0.247 nm, which corresponds to the (002) crystal plane of Zn metal (highlighted in the green square). An additional product with a lattice space of 0.262 nm is identified at the outer edge of the Zn electrode (highlighted in red square), corresponding to the (−112) crystal plane of Li2CO3. Furthermore, the presence of Li2S2O7 is also observed (highlighted in the cyan square) with a lattice space of 0.2605 nm. Moreover, since the reaction product containing Cl element may be amorphous, its crystal structure is not discernible in HRTEM analysis. By combining the findings from Supplementary Figs. 15, 16 and 17, as well as Fig. 3e, f, it becomes evident that, after adding LiCl additive to 50 mM ZnSO4 electrolyte, the main components of the SEI film formed on the Zn surface comprise Li2S2O7, Li2CO3, and Li2ZnCl4·9H2O. The Tafel plot was employed to assess the charge transfer kinetics at the electrolyte/Zn negative electrode interface (Supplementary Fig. 18). Compared to the 50 mM ZnSO4 electrolyte, the LiCl-containing electrolyte exhibited a higher i0 value, indicating enhanced charge transfer kinetics (i.e., increased transmission of Zn2+ ions), facilitating uniform Zn deposition and improving the electrochemical performance51,52. Therefore, based on the above discussions, the in situ STXM characterization technique has demonstrated significant utility in investigating Zn dendrite growth and suggesting optimization strategies for Zn batteries.
The performance of ZIBs with 12-crown-4 additive
Moreover, as depicted in Fig. 1e, f and Supplementary Fig. 8a, the presence of a loose and porous Zn2+ ion deposition layer exacerbates Zn dendrite growth during the repeated plating/stripping process. Therefore, it is necessary to introduce a Zn2+ ion deposition inducer into the electrolyte, which can inhibit the formation of Zn dendrites and promote the commercial application of ZIBs. In this study, the 12-crown-4 (12-C-4), 15-crown-5 (15-C-5) and 18-crown-6 (18-C-6) are used as Zn2+ ion deposition inducers to suppress Zn dendrite growth. To validate the absorption effect of various additives on the Zn surface, the differential capacitance was measured in different electrolyte systems (Supplementary Fig. 19). The potential of zero charge (PZC) means that the capacitance on a surface is minimal53. A distinct absorption behavior is observed due to different PZC reflections53. As illustrated in Supplementary Fig. 19a, the introduction of 12-C-4 and 15-C-5 additives into the 2 M ZnSO4 electrolyte alters the PZC, indicating that additives are absorbed on the Zn surface. Although the PZC of 18-C-6 is similar to that of untreated electrolyte of 2 M ZnSO4, the capacitance is lower51, indicating that the 18-C-6 additive is also absorbed on the Zn surface. Moreover, the adsorption behavior of the electrolyte with various additives at the inner Helmholtz plane (IHP) was analyzed by measuring the differential capacitance curves, as shown in Supplementary Fig. 19b. The capacitance is obtained by Eqs. (1) and (2)54:
where C, d, ε and S represent the capacitance, the thickness of adsorbed species on the Zn surface, dielectric constant and the area of the electrode, respectively. The capacitance is inversely proportional to the thickness, d. Notably, with 2 M ZnSO4 electrolyte containing 5% 12-C-4 additive, the value of C is lower than that of the 2 M ZnSO4 electrolyte and electrolytes with 5% 15-C-5 and 5% 18-C-6 additives. This observation suggests the presence of a loose adsorption layer in the IHP, which may facilitate the transport of Zn2+ ions and inhibit the formation of Zn dendrites. Moreover, Supplementary Fig. 20 shows the results of electrochemical impedance spectroscopy (EIS) conducted in electrolytes with various additives at a current density of 2 mA cm−2 with a capacity of 2 mAh cm−2. The 2 M ZnSO4 electrolyte containing 5% 12-C-4 additive exhibits the lowest resistance compared to those with 5% 15-C-5 and 5% 18-C-6 additives. The low resistance indicates that the electrolyte containing 5% 12-C-4 additive enhances the transport of Zn2+ ions. Hence, combining the data from both the differential capacitance and EIS analyses, it is apparent that the introduction of 15-C-5 and 18-C-6 additives not only changes the structure of the IHP layer but also increases the resistance at the electrode/electrolyte interface, consequently promoting the formation of Zn dendrites. Therefore, as demonstrated in Supplementary Fig. 21, the 2 M ZnSO4 electrolyte containing the 5% 12-C-4 additive exhibits the best performance (230 h, overpotential: approximately 277.1 mV) compared to the electrolyte with 5% 15-C-5 (166 h, overpotential: approximately 299.4 mV) and 5% 18-C-6 (170 h, overpotential: approximately 304.4 mV). This is because 5% 15-C-5 and 5% 18-C-6 additives contain abundant ether-based functional groups, consequently promoting Zn dendrite formation Zn negative electrode surface. Furthermore, the long cycle stability of the Zn | |Zn symmetric cell without or with 5% 12-C-4 additive was evaluated. As shown in Supplementary Fig. 22, when operating at a current density of 4.0 mA cm−2 with a capacity of 10.0 mAh cm−2, the 2 M ZnSO4 electrolyte containing 5% 12-C-4 additives demonstrates improved cycle stability for up to 700 h (equivalent to 140 cycles, overpotential: approximately 306.2 mV). This performance is 10.8 times superior to that observed with untreated electrolyte of 2 M ZnSO4, which only lasts for 65 h (or 13 cycles). These findings indicate that the 5% 12-C-4 additive effectively inhibits Zn dendrite growth, particularly at high current density conditions and with a high capacity. Moreover, based on the discussions above (Fig. 1b–e and Fig. 3b, c), it becomes evident that the by-products of Zn corrosion accelerate Zn dendrite growth. The lower corrosion current density (icorr) and higher inhibition efficiency (η%) represent the reduced Zn corrosion rate55. As exhibited in Supplementary Fig. 23a, among all the additives investigated in this study, the 2 M ZnSO4 electrolyte containing the 5% 12-C-4 additive exhibits the best performance. Furthermore, the difference of η% also reflects the compactness and the stability of the 5% 12-C-4 derived SEI film. The η% value is calculated using Eq. 3, where i0corr and icorr are the corrosion current densities of Zn without and with 5% 12-C-4 additive in 2 M ZnSO4 electrolyte, respectively55,56. Thus, for the 2 M ZnSO4 electrolyte with 5% 15-C-5 additive, the η% is 46.8%, for 5% 18-C-6 additive, it is 43.5%, while for the 2 M ZnSO4 electrolyte containing 5% 12-C-4 additive, it reaches 74.2%, indicating that the 5% 12-C-4 derived SEI film is more compact and stable than those derived from the other two additives (Supplementary Fig. 23b). Therefore, the 2 M ZnSO4 electrolyte with 5% 12-C-4 additive demonstrates the best cycle stability for the Zn negative electrode (Supplementary Fig. 21, red line). To validate the effect of the 5% 12-C-4 additive on Zn2+ ion deposition, SEM investigations were conducted (Supplementary Fig. 24). Supplementary Fig. 24a demonstrates that, in 2 M ZnSO4 electrolyte, the deposition of Zn2+ ions is inhomogeneous and characterized by large-grains, leading to Zn dendrite growth. By contrast, in the 2 M ZnSO4 electrolyte with 5% 12-C-4 additive, the Zn negative electrode surface shows a dense and smooth deposition layer, which indicates improved Zn2+ ion deposition inducibility (Supplementary Fig. 24b). This improved inductivity can be attributed to the change of the electrolyte’s wettability (the contact angle) on the Zn surface by the 12-C-4 additive. As shown in Supplementary Fig. 25, upon introducing the 5% 12-C-4 additive into the 2 M ZnSO4 electrolyte, the contact angle decreased from 66° to 53°. The lower contact angle may enhance the wettability between the Zn negative electrode and the electrolyte, thereby accelerating the Zn2+ ion transfer number and promoting uniform Zn2+ ion nucleation. The Zn2+ ion transfer number (tZn2+) was determined through direct current polarization and EIS measurements, which are displayed in Supplementary Fig. 26. The tZn2+ for the 5% 12-C-4 additive is 0.7 (as shown in Supplementary Fig. 26b), which is higher than that of the 2 M ZnSO4 electrolyte (0.2, as depicted in Supplementary Fig. 26a). A high tZn2+ promotes Zn2+ transmission within the SEI film and suppresses Zn dendrites growth51. Therefore, at a current density of 0.5 mA cm−2 with a capacity of 1.5 mAh cm−2, the 2 M ZnSO4 electrolyte with 5% 12-C-4 additives maintains exceptional cycle stability up to 3900 h (650 cycles, overpotential: approximately 235.2 mV), which is 10.5 times better than that with untreated electrolyte of 2 M ZnSO4 (Supplementary Fig. 27). After optimizing the composition of the electrolyte additive, the influence of the 12-C-4 additive concentration on the cycling stability of Zn | |Zn symmetric cells was carried out and is displayed in Supplementary Fig. 28. As displayed in Supplementary Fig. 28, although the overpotentials of 0.5% 12-C-4 (overpotential: approximately 202.3 mV) and 2% 12-C-4 (overpotential: approximately 182.2 mV) are smaller than that of 5% 12-C-4, their cycling stabilities are lower than that of 5% 12-C-4. Because at excessively low additive concentrations, 12-C-4 cannot induce the uniform deposition of Zn2+ ions, resulting in Zn dendrite growth. Therefore, the 2 M ZnSO4 electrolyte containing 5% 12-C-4 additive presents the best cycle stability.
After pairing Zn negative electrode with MnO2, using 2 M ZnSO4 electrolyte with 5% 12-C-4 additive, it maintains a high specific capacity of 136.86 mAh g−1 (3.4 times that when using untreated electrolyte of 2 M ZnSO4, 40.32 mAh g−1) at a specific current of 500 mA g−1 after 50 cycles (Supplementary Fig. 29a). Moreover, in the 2 M ZnSO4 electrolyte, the columbic efficiency (CE) is very low (87.87%) after 50 cycles due to the presence of self-discharge in ZIBs (Supplementary Fig. 29b)57. The HER, Zn corrosion, Zn dendrite growth and shape change of the Zn negative electrode during the plating/stripping process may promote self-discharge57,58. As illustrated in Supplementary Fig. 23a, the untreated electrolyte of 2 M ZnSO4 exhibits a higher icorrr (0.62) and lower corrosion potential (cV) (-0.027 V) compared to the 2 M ZnSO4 electrolyte containing 5% 12-C-4 additive (icorr: 0.16, cV: −0.015 V). Moreover, the cV shifts towards a more positive value may indicate that the additive-derived SEI film can better inhibit Zn dissolution55. Therefore, the untreated electrolyte of 2 M ZnSO4 accelerates Zn corrosion, which is consistent with the results in Fig. 1. This outcome demonstrates that the 5% 12-C-4 additive is adsorbed on the Zn surface to form a stable protective film, thereby mitigating the self-discharge of ZIBs. Therefore, the introduction of the 5% 12-C-4 additive improves the electrochemical performance of Zn||MnO2 full-cell.
Based on the aforementioned findings, the introduction of electrolyte additives (LiCl, 12-C-4) into the untreated electrolyte effectively inhibits the growth of Zn dendrites by suppressing HER and self-corrosion. Phase field simulation was used to further validate the necessity of introducing additives (Fig. 4). To statistically characterize the dendritic pattern, we simulated the dendrite growth with or without the presence of ZnO. In the simulation, the left side of the area represents a thin layer of initial Zn crystal, while the right side represents the formation of dendrite. In the presence of ZnO by-products, at t = 0 s, the solid-liquid interface of the Zn negative electrode appears smooth without any unit cell bulges (Fig. 4a, a’). However, by t = 0.1 s, the existence of ZnO by-products on the Zn surface disrupts the kinetic balance at the interface of Zn/electrolyte interface, resulting in numerous protruding unit cells on the Zn surface (Fig. 4b). In comparison, the absence of ZnO by-products results in minimal unit cell protrusions (Fig. 4b’). Between 0.4 < t ≤ 0.9 s, the Zn dendrites on the Zn negative electrode surface experience rapid growth, accompanied by the tip-splitting phenomenon along the Zn dendrites (Fig. 4c–e). Tip splitting occurs due to the competitive growth among Zn dendrites, where the electric field gradients at the tips of the most favorable Zn dendrites facilitate easier capture of Zn2+ ions from the electrolyte, leading to the formation of new dendrites. At t > 0.9 s, the electric field gradient difference within the simulated region basically diminishes, halting further growth of Zn dendrites (Fig. 4f). Conversely, in the absence of ZnO by-products, the shape and number of Zn dendrites remain unchanged over time (Fig. 4c’–f’). This stability arises from better interfacial kinetic properties of the Zn negative electrode surface, which reduces the driving force for Zn dendrite growth, inhibiting their further formation at the Zn/electrolyte interface.

a–f Phase field simulation of Zn dendrite growth with ZnO by-product. a’–f’ Phase field simulation of Zn dendrite growth without ZnO by-product.
Discussion
In this study, we present the employment of the synchrotron-based in situ STXM technique to investigate the relationship between Zn dendrite morphology and the SEI film. In the electrolyte of 50 mM ZnSO4, we not only visualized the evolution of Zn dendrites, but also revealed that the formation of a dense and stable SEI film is crucial for mitigating Zn dendrite growth. During the initial stages of dendrite formation, the hydrogen evolution reaction accelerates Zn dissolution and promotes the formation of Zn dendrites with whisker and moss morphologies on the Zn surface. Moreover, the in situ STXM and ptychography-based XAS characterizations revealed that the main components of these morphologies are ZSH and ZnO (a component of the SEI film). As Zn deposition progresses, the existence of the loose and porous SEI film not only hinders the transmission of Zn2+ ions, favoring the deposition of Zn2+ ions at the outer edge of the electrode, but also increases the contact area with water, leading to enhanced H2 gas generation. This combination of poor ion transmission kinetics and the H2 gas evolution promotes the exfoliation of the partially deposited metallic Zn from the electrode, resulting in dead Zn with a planar hexagonal morphology. To address these challenges, we introduced LiCl as an electrolyte additive to effectively construct a dense and stable SEI film on the Zn surface. After the addition of the 50 mM LiCl additive into the electrolyte of 50 mM ZnSO4, the Zn negative electrode exhibited the ability to suppress HER and maintain stable cycling for 138 h at a current density of 1 mA cm−2/1 mAh cm−2, compared to 46 h for the cell without the additive. Furthermore, the in situ synchrotron-based XRD analysis revealed that the LiCl additive facilitated the formation of a LiS2S2O7-based SEI film, which effectively mitigated the generation of ZnO and ZSH by-products during the stripping/plating progress, thereby improving the electrochemical performance of ZIBs. Additionally, with the introduction of the 5% 12-C-4 additive into the 2 M ZnSO4 electrolyte, the Zn negative electrode demonstrated remarkable stability, cycling stably for 3900 h at a current density of 0.5 mA cm−2/1.5 mAh cm−2, in comparison to 370 h for the cell without the additive.
Methods
Materials
Manganese oxide (reagent grade, ≥90%), lithium chloride (≥99%), Separator (Whatman glass microfiber filters, grade GF/A, diameter = 16 mm), zinc sulfate 7H2O (ACS reagent, 99%) and 12-crown-4 (98%) were purchased from Sigma-Aldrich. Zn foil (70 µm thickness) was purchased from Alibaba.
Characterization
STXM
The CLS SM (10ID-1) beamline uses an undulator to emit a continuous spectrum of X-ray beams, which subsequently pass through the monochromator, generating the X-ray beam with the desired energy. Then, the beam is directed to the Fresnel zone plate (FZP) and the order sorting aperture (OSA) to form a 30-50 nm spot on the sample in the STXM chamber. The transmitted X-ray intensity is recorded by a phosphor scintillator coupled photomultiplier tube (PMT) for the conventional STXM measurement. Chemical mapping by STXM provides insights into the chemical composition and structures of the deposited Zn and SEI. All STXM data analysis, including alignment of stack images, optical density (O.D.) conversion and principal component analysis-cluster analysis (PCA-CA), were performed using the aXis2000 software package (Adam Hitchcock’s group, McMaster University, Canada).
Spectro-ptychography measurement
The Zn dendrite samples used for STXM-ptychography were the same silicon nitride window samples as those used for the conventional in situ STXM measurements. The ptychography measurement in defocus mode under a high vacuum of 10-5 mbar utilized a 40 nm outer-zone-width ZP to focus the monochromatic soft X-ray beam to the focal plane of the sample, producing a 2.5 µm beam spot on the sample. The sample underwent raster scanning in 8 × 8 pixels with 0.5 µm increments, ensuring sufficient overlap of the scanned area. To block all but the first-order beam from the zone plate, a 50 µm order-sorting aperture (OSA) was used. The diffraction images were captured by a CCD (AndorTM DX434-BN), with a resolution of 1024 × 1024 pixels, which was cooled to −45 °C and positioned 60 mm downstream from the sample. Moreover, the spectro-ptychography technique was used to conduct energy stacks at the Zn L-edge and O K-edge. The energy was scanned from 525-560 eV at the O K-edge and 1010-1060 eV at the Zn L-edge, all with a varied energy step size of 0.2-0.5 eV at the XANES features and coarser in the pre- and post-edges, and using 1 s exposure time. The ptychography results were obtained through batch reconstruction using the PyPIE software (SM beamline team, Canadian Light Source, Canada).
SEM characterizations
The morphology of Zn foil and the deposition process of Zn2+ ions were characterized by SEM (Bruker, Quanta 450 ESEM, FEI).
XPS and XRD analysis
XPS (VG ESCALAB 220i-XL) experiments were conducted using the monochromatic 1486.6 eV Al Kα radiation. The peak energies were calibrated by assigning the graphite C 1s peak to 284.6 eV. The spectra were fitted with mixed Gaussian-Lorentzian component profiles after a Shirley background subtraction. The surface composition was verified by X-ray diffraction (XRD) (40 kV, 25 mA, Cu Kα radiation, λ = 1.5418 Å).
H2 test
The H2 yield analysis was performed using a three-electrode system employing the potentiostatic method on a CHI-760D electrochemical workstation. Both the working electrode and counter electrode are Zn plates. The reference electrode was an Ag/AgCl electrode with a saturated KCl filling solution. 50 mL ZnSO4 solution (with and without the LiCl additive) was applied as the electrolyte. N2 gas (Praxair, 99.999%) was bubbled through the electrolyte with a flow rate of 20 standard cubic centimeters per minute (SCCM) for at least 30 min. Then, during the electrochemical chronoamperometry tests, the gas outlet was directed into a gas-sampling loop of the gas chromatography (GC; 9790 II, Fuli) for the online GC quantification of H2. The GC was equipped with a packed HaySep A column and a packed MolSieve 5A column with argon (Praxair, 99.999%) as the carrier gas. A thermal conductivity detector (TCD) was used for the qualification and quantification of H2.
Electrochemical test
All experiments were conducted at room temperature. CV test was conducted using a three-electrode electrochemical flow cell in 50 mM ZnSO4 solution (3 µL). The Ag/AgCl and Au (200 µm × 30 µm) were used as reference and counter electrodes, respectively. The CV test was conducted by using a BioLogic SP-200 potentiostat with a scan rate of 20 mV s−1, the open-circuit voltage was scanned backward to −1 V, then forward swept to 1.0 V. All the assembled batteries (coin cells) were operated in ZnSO4-based electrolyte (100 µL). The concentrations of LiCl and 12-crown-4 additives are 50 mM and 5% (volume ratio), respectively. The pH values of 50 mM ZnSO4, 50 mM ZnSO4 + 50 Mm LiCl, 2 M ZnSO4 and 2 M ZnSO4 + 5% 12-C-4 are 4.46, 4.52, 3.47 and 2.36, respectively. The pH tested temperature is 22.4 °C. The ionic conductivity of 50 mM ZnSO4, 50 mM ZnSO4 + 50 Mm LiCl, 2 M ZnSO4 and 2 M ZnSO4 + 5% 12-C-4 are 8.36, 13.57, 54.2 and 42.7 mS/cm, respectively. The ionic conductivity tested temperature is 22.3 °C. The pH was tested by FiveEasy meters (Mettler Toledo). The ionic conductivity experiments were measured by the OAKTON CON 11 conductivity meter. To evaluate the cycling performance of Zn||Zn symmetric coin cells in different electrolyte systems (e.g., 50 mM ZnSO4, 50 mM ZnSO4 with 50 mM LiCl, 2 M ZnSO4, 2 M ZnSO4 with 5% 12-C-4, 15-C-5, and 18-C-6 additives), cyclic plating/stripping and galvanostatic testing were conducted at current densities of 0.5, 1.0, 2.0 and 4.0 mA cm−2, respectively. The cycle performance was tested by a Neware battery tester. Both the working electrode and counter electrode were Zn foil (1.13 cm2). To study the electrochemical performance of the Zn||MnO2 full-cell in 2 M ZnSO4 with or without 5% 12-C-4 additive, MnO2 and Zn foil were used as positive and negative electrode, respectively. To prepare the MnO2 positive electrode, MnO2 powder (purchased from Sigma) was mixed with carbon black and polyvinylidene fluoride with a mass ratio of 7:2:1 using N-methyl-2-pyrrolidone as the solvent to form a homogenous slurry. The resulting slurries were uniformly coated onto carbon cloth, followed by drying the foils at 60 ̊C under vacuum for 12 h. The carbon cloth was then cut into discs with a diameter of 12 mm. The charge and discharge cut-off potentials were measured at 1.85 and 1.0 V (vs. Zn2+/Zn), respectively. The mass loading of MnO2 is 1–2.5 mg cm−2 (N/P = 29.2, Zn utilization is 1.1%). The capacitance-potential curves of the cell were collected based on the following Eq. 4, where C is the capacitance, f is the frequency (1 kHz), Zim is the imaginary component of the impedance59. The potential of the polarization was scanned positively from −0.5 to +0.5 V with a sweep rate of 10.0 mV s−1 using a CHI-760D electrochemical workstation. The Zn2+ ion transfer number using the Zn | |Zn symmetric cell can be calculated according to the following Eq. 5, where ΔV is the constant voltage (10 mV), ibp and iap are the values of the before polarization and after polarization currents; and Rbp and Rap are the interfacial resistances of before polarization and after polarization, respectively51.
In situ synchrotron XRD annealing was used to investigate changes in the surface composition of Zn metal during the stripping/plating process. The study was carried out at the Canadian Light Source (CLS, Saskatoon Canada) at their Brockhouse high-energy wiggler beamline. The beam energy used was 65.07 keV (λ = 0.0238 nm) and diffraction patterns were acquired using a Perkin Elmer XRD 1621 area detector with a collection time of 15 s for each measurement (20 × 20 μm2 in area). All data collected from the experiment were analyzed using the GSAS-II software60.
Data availability
The data that support the findings of this study are available within the text, including the Methods, and Supplementary information and the source data are deposited in figshare database (https://doi.org/10.6084/m9.figshare.26885332). Raw datasets related to the current work are available from the corresponding author on request. Source data are provided with this paper.
References
Xing, J., Bliznakov, S., Bonville, L., Oljaca, M. & Maric, R. A review of nonaqueous electrolytes, binders, and separators for lithium-ion batteries. Electrochem. Energy Rev. 5, 14 (2022).
Zhou, T. et al. Cathode materials for aqueous zinc-ion batteries: a mini review. J. Colloid Interface Sci. 605, 828–850 (2022).
Selvakumaran, D., Pan, A., Liang, S. & Cao, G. A review on recent developments and challenges of cathode materials for rechargeable aqueous Zn-ion batteries. J. Mater. Chem. A 7, 18209–18236 (2019).
Sun, T. et al. Poly(2,5‐dihydroxy‐1,4‐benzoquinonyl sulfide) as an efficient cathode for high-performance aqueous zinc-organic batteries. Adv. Funct. Mater. 31, 2010049 (2021).
Guo, S. et al. Fundamentals and perspectives of electrolyte additives for aqueous zinc-ion batteries. Energy Storage Mater. 34, 545–562 (2021).
Yang, Q. et al. Dendrites in Zn-based batteries. Adv. Mater. 32, 2001854 (2020).
Zhang, Z. et al. Recent progress, mechanisms, and perspectives for crystal and interface chemistry applying to the Zn metal anodes in aqueous zinc‐ion batteries. SusMat 2, 114–141 (2022).
Zhao, K. et al. Stabilizing zinc electrodes with a vanillin additive in mild aqueous electrolytes. ACS Appl. Mater. Interfaces 13, 47650–47658 (2021).
Xu, Y. et al. A rechargeable aqueous zinc/sodium manganese oxides battery with robust performance enabled by Na2SO4 electrolyte additive. Energy Storage Mater. 38, 299–308 (2021).
Xiong, W. et al. Electrolyte engineering for a highly stable, rechargeable hybrid aqueous battery. J. Energy Storage 26, 100920 (2019).
Olbasa, B. W. et al. High-rate and long-cycle stability with a dendrite-free zinc anode in an aqueous zn-ion battery using concentrated electrolytes. ACS Appl. Energy Mater. 3, 4499–4508 (2020).
Li, Z. et al. Mechanistic insight into the electrochemical performance of Zn/VO2 batteries with an aqueous ZnSO4 electrolyte. Adv. Energy Mater. 9, 1900237 (2019).
Duan, J. et al. Hybrid electrolyte-mediated nano-scaled γ-Fe2O3 cathode for emerging aqueous zinc battery. Electrochim. Acta 390, 138883 (2021).
Dong, Y. et al. Long-life zinc/vanadium pentoxide battery enabled by a concentrated aqueous ZnSO4 electrolyte with proton and zinc ion co-intercalation. ACS Appl. Energy Mater. 3, 11183–11192 (2020).
Han, C., Li, W., Liu, H. K., Dou, S. & Wang, J. Principals and strategies for constructing a highly reversible zinc metal anode in aqueous batteries. Nano Energy 74, 104880 (2020).
Du, W. et al. Challenges in the material and structural design of zinc anode towards high-performance aqueous zinc-ion batteries. Energy Environ. Sci. 13, 3330–3360 (2020).
Chen, X., Li, W., Reed, D., Li, X. & Liu, X. On energy storage chemistry of aqueous Zn-ion batteries: from cathode to anode. Electrochem. Energy Rev. 6, 33 (2023).
Nakata, A. et al. Transformation of leaf-like zinc dendrite in oxidation and reduction cycle. Electrochim. Acta 166, 82–87 (2015).
Stock, D. et al. Towards zinc-oxygen batteries with enhanced cycling stability: the benefit of anion-exchange ionomer for zinc sponge anodes. J. Power Sources 395, 195–204 (2018).
Wang, F. et al. Highly reversible zinc metal anode for aqueous batteries. Nat. Mater. 17, 543–549 (2018).
Xie, F. et al. Mechanism for zincophilic sites on zinc‐metal anode hosts in aqueous batteries. Adv. Energy Mater. 11, 2003419 (2021).
Liu, Z. et al. Tuning the electronic environment of zinc ions with a ligand for dendrite-free zinc deposition in an ionic liquid. Phys. Chem. Chem. Phys. 19, 25989–25995 (2017).
Li, B. et al. A review of solid electrolyte interphase (SEI) and dendrite formation in lithium batteries. Electrochem. Electrochem. Energy Rev. 6, 7 (2023).
Zhang, Y., Howe, J. D., Ben-Yoseph, S., Wu, Y. & Liu, N. Unveiling the origin of alloy-seeded and nondendritic growth of Zn for rechargeable aqueous Zn batteries. ACS Energy Lett. 6, 404–412 (2021).
Biton, M., Tariq, F., Yufit, V., Chen, Z. & Brandon, N. Integrating multi-length scale high resolution 3D imaging and modelling in the characterisation and identification of mechanical failure sites in electrochemical dendrites. Acta Mater. 141, 39–46 (2017).
Qian, G. et al. Structural, dynamic, and chemical complexities in zinc anode of an operating aqueous Zn‐ion battery. Adv. Energy Mater. 12, 2200255 (2022).
Sasaki, Y. et al. In situ electron microscopy analysis of electrochemical Zn deposition onto an electrode. J. Power Sources 481, 228831 (2021).
Wang, S. et al. A highly reversible zinc deposition for flow batteries regulated by critical concentration induced nucleation. Energy Environ. Sci. 14, 4077–4084 (2021).
Zhou, J. et al. Imaging state of charge and its correlation to interaction variation in an LiMn0.75Fe0.25PO4 nanorods-graphene hybrid. Chem. Commun. 49, 1765–1767 (2013).
Chueh, W. C. et al. Intercalation pathway in many-particle LiFePO4 electrode revealed by nanoscale state-of-charge mapping. Nano Lett. 13, 866–872 (2013).
Tan, X. et al. X-ray spectromicroscopy investigation of heterogeneous sodiation in hard carbon nanosheets with vertically oriented (002) planes. Small 17, 2102109 (2021).
Guo, X. et al. Alleviation of dendrite formation on zinc anodes via electrolyte additives. ACS Energy Lett. 6, 395–403 (2021).
Yang, J. et al. Zinc anode for mild aqueous zinc-ion batteries: challenges, strategies, and perspectives. Nanomicro Lett. 14, 42 (2022).
Sharma, A. et al. Effect of Cu insertion on structural, local electronic/atomic structure and photocatalyst properties of TiO2, ZnO and Ni(OH)2 nanostructures: XANES-EXAFS study. Mater. Chem. Phys. 191, 129–144 (2017).
Bayaguud, A., Fu, Y. & Zhu, C. Interfacial parasitic reactions of zinc anodes in zinc ion batteries: Underestimated corrosion and hydrogen evolution reactions and their suppression strategies. J. Energy Chem. 64, 246–262 (2022).
Zhao, R. et al. Redirected Zn electrodeposition by an anti-corrosion elastic constraint for highly reversible Zn anodes. Adv. Funct. Mater. 31, 2001867 (2021).
Tian, H. et al. Steel anti-corrosion strategy enables long-cycle Zn anode. Adv. Energy Mater. 13, 2202603 (2022).
Zhang, H., Wu, Y., Yu, J., Jiang, T. & Wu, M. Shooting three birds with one stone: the regulation of Zn2+, H2O, and oh- kinetics for stable Zn‐metal anodes with a multifunctional sieve. Adv. Funct. Mater. 34, 2301912 (2023).
Shin, J., Lee, J., Park, Y. & Choi, J. W. Aqueous zinc ion batteries: focus on zinc metal anodes. Chem. Sci. 11, 2028–2044 (2020).
Kwon, K. Y., Jo, T. H., Kim, J. S., Hasan, F. & Yoo, H. D. A chronocoulometric method to measure the corrosion rate on zinc metal electrodes. ACS Appl. Mater. Interfaces 12, 42612–42621 (2020).
Lin, M.-H., Huang, C.-J., Cheng, P.-H., Cheng, J.-H. & Wang, C.-C. Revealing the effect of polyethylenimine on zinc metal anodes in alkaline electrolyte solution for zinc-air batteries: mechanism studies of dendrite suppression and corrosion inhibition. J. Mater. Chem. A 8, 20637–20649 (2020).
Zhang, Y. et al. An effective and accessible cell configuration for testing rechargeable zinc-based alkaline batteries. J. Power Sources 491, 229547 (2021).
Prendergast, D. & Galli, G. X-ray absorption spectra of water from first principles calculations. Phys. Rev. Lett. 96, 215502 (2006).
Zhang, C. et al. The electrolyte comprising more robust water and superhalides transforms Zn‐metal anode reversibly and dendrite‐free. Carbon Energy 3, 339–348 (2020).
Zhang, J., Xu, H., Guo, J., Chen, T. & Liu, H. Superhydrophobic polypyrrole-coated cigarette filters for effective oil/water separation. Appl. Sci. 10, 1985 (2020).
Manickam, M., Singh, P., Issa, T. B., Thurgate, S. & De Marco, R. Lithium insertion into manganese dioxide electrode in MnO2/Zn aqueous battery. J. Power Sources 130, 254–259 (2004).
Nefedov, V., Gati, D., Dzhurinskii, B., Sergushin, N. & Salyn, Y. V. X-ray electron study of oxides of elements. Zh. Neorg. Khim. 20, 2307–2314 (1975).
Yang, C. et al. All-temperature zinc batteries with high-entropy aqueous electrolyte. Nat. Sustain. 6, 325–335 (2023).
Dake, L. S., Baer, D. R. & Zachara, J. M. Auger parameter measurements of zinc compounds relevant to zinc transport in the environment. Surf. Interface Anal. 14, 71–75 (1989).
Kanamura, K., Tamura, H., Shiraishi, S. & Takehara, Z.-i XPS analysis for the lithium surface immersed in tetrahydrofuran containing various salts. Denki Kagaku oyobi Kogyo Butsuri Kagaku 61, 1377–1382 (1993).
Dai, H. et al. Cobalt-phthalocyanine-derived molecular isolation layer for highly stable lithium anode. Angew. Chem. Int. Ed. Engl. 60, 19852–19859 (2021).
Meng, J., Chu, F., Hu, J. & Li, C. Liquid polydimethylsiloxane grafting to enable dendrite-free Li plating for highly reversible Li-metal batteries. Adv. Funct. Mater. 29, 1902220 (2019).
Yan, C. et al. Regulating the inner helmholtz plane for stable solid electrolyte interphase on lithium metal anodes. J. Am. Chem. Soc. 141, 9422–9429 (2019).
Li, R. et al. Hexaoxacyclooctadecane induced interfacial engineering to achieve dendrite-free Zn ion batteries. Energy Storage Mater. 46, 605–612 (2022).
Chen, W. et al. Protection of copper corrosion in 0.5 M NaCl solution by modification of 5-mercapto-3-phenyl-1,3,4-thiadiazole−2-thione potassium self-assembled monolayer. Corros. Sci. 61, 53–62 (2012).
Li, C., Li, L. & Wang, C. Study of the inhibitive effect of mixed self-assembled monolayers on copper with SECM. Electrochim. Acta 115, 531–536 (2014).
Chao, D. et al. Roadmap for advanced aqueous batteries: from design of materials to applications. Sci. Adv. 6, eaba4098 (2020).
Jo, Y., Santhoshkumar, P., Prasanna, K., Vediappan, K. & Lee, C. Improving self-discharge and anti-corrosion performance of Zn-air batteries using conductive polymer-coated Zn active materials. J. Ind. Eng. Chem. 76, 396–402 (2019).
Dai, H., Xi, K., Liu, X., Lai, C. & Zhang, S. Cationic surfactant-based electrolyte additives for uniform lithium deposition via lithiophobic repulsion mechanisms. J. Am. Chem. Soc. 140, 17515–17521 (2018).
Toby, B. H. & Von Dreele, R. B. GSAS-II: the genesis of a modern open-source all purpose crystallography software package. J. Appl. Crystallogr. 46, 544–549 (2013).
Acknowledgements
This work is financially supported by the Natural Sciences and Engineering Research Council of Canada (NSERC), Fonds de Recherche du Québec-Nature et Technologies (FRQNT), Canada Foundation for Innovation (CFI), Centre Québécois sur les Materiaux Fonctionnels (CQMF), Institut National de la Recherche Scientifique (INRS), and École de Technologie Supérieure (ÉTS). G. Zhang thanks for the support from the Marcelle-Gauvreau Engineering Research Chair program. H. Dai thanks for the scholarship from the FRQNT and the support from the CLSI Student Travel Support Program. The synchrotron research described in this paper was performed at the Canadian Light Source, which is supported by CFI, NSERC, the University of Saskatchewan, the Government of Saskatchewan, Western Economic Diversification Canada, the National Research Council Canada, and the Canadian Institutes of Health Research.
Author information
Authors and Affiliations
Contributions
G.Z., J.Z., J.W., and S.S. conceived and supervised the project. H.D. and T.S. contributed equally and drafted the manuscript. Z.C., J.Z., J.W., and G.Z. assisted with materials characterizations and result discussions. S.S. led the project. All authors discussed the results and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interest.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Dai, H., Sun, T., Zhou, J. et al. Unraveling chemical origins of dendrite formation in zinc-ion batteries via in situ/operando X-ray spectroscopy and imaging. Nat Commun 15, 8577 (2024). https://doi.org/10.1038/s41467-024-52651-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-52651-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
