
Abstract
Hydrogels, provided that they integrate strength and toughness at desired high content of water, promise in load-bearing tissues such as articular cartilage, ligaments, tendons. Many developed strategies impart hydrogels with some mechanical properties akin to natural tissues, but compromise water content. Herein, a strategy deprotonation-complexation-reprotonation is proposed to prepare polyvinyl alcohol hydrogels with water content as high as ~80% and favorable mechanical properties, including tensile strength of 7.4 MPa, elongation of around 1350%, and fracture toughness of 12.4 kJ m−2. The key to water holding yet improved mechanical properties lies in controllable nucleation for refinement of crystalline morphology. With nearly constant water content, mechanical properties of as-prepared hydrogels are successfully tailored by tuning crystal nuclei density via deprotonation degree and their distribution uniformity via complexation temperature. This work provides a nucleation concept to design robust hydrogels with desired water content, holding implications for practical application in tissue engineering.
Similar content being viewed by others
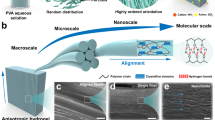
Introduction
Hydrogels are a class of soft and wet materials consisting of a three-dimensional network of hydrophilic polymers swollen with a large amount of water. High equilibrium water content ( > 70%), similar to biological soft tissues1, makes them promising as biocompatible materials for diverse biomedical applications2, such as tissue engineering3, drug delivery4, cancer therapies5, bioadhesives6, soft robots7, and bioelectronics8. Unfortunately, conventional hydrogels with polymer chains crosslinked via covalent bonds into single non-uniform networks are mechanically weak and fragile, hampering their broad bioengineering utility, especially as load-bearing biomaterials. By contrast, crystalline hydrogels have a prominent advantage in strength and toughness because crystallites not only have high cohesive energy to prevent crack from propagation but also act as high-functionality crosslinks for reinforcement by sharing loads among multiple chains (several dozen at least)9, greatly outnumbering the chains interconnected via common covalent crosslinks ( ~ 4)10. In fact, such a crosslink form is widely existed in some biological load-bearing soft tissues that integrate remarkable stiffness, strength and toughness, such as tendon, ligament and articular cartilage11. Recent works based on ice freezing12, solvent exchange13, salting out14,15, air drying16 and wet annealing17 are conducted to prepare crystalline hydrogels with an improved mechanical property. Essentially, these strategies are aimed at forcing polymeric chains to move toward aggregation to promote their crystallization. Substantial increase in crystallinity, especially by salting out, air drying and wet annealing, results in robust hydrogels with ultrahigh strength (σf > 10 MPa) and remarkable fracture toughness (Γ > 10 kJ m−2). Such crystallization processes facilitated by massive chain aggregation, however, are accompanied by a drastic volume shrinkage of polymeric networks, resulting in a reduced water content within the resultant hydrogels, even less than 50% at swelling equilibrium state. Therefore, the substantial dilemma faced at present is how to substantially improve mechanical property of crystalline hydrogels while maintaining a high equilibrium water content, similar to biological soft tissues11.
Such a dilemma arises, to a great extent, from the crystallization processes that focus on promoting crystal growth by compelling polymer chains to move drastically toward aggregation, resulting in an increase in crystallite size and crystallinity, yet a large shrinkage in volume18. In principle, the process of crystallization for a polymeric hydrogel involves two stages, nucleation and crystal growth. It is reasonable to envisage if crystal nuclei are controlled to be dense and uniform in initial nucleation stage, a refined crystalline morphology with decreased crystallite size and shortened intercrystallite distance can be anticipated given that high-density crosslinks of crystal nuclei would impose a limitation on segment movement during crystal growth. Such a refined crystalline morphology will bring about mechanical property enhancement by distributing stress more uniformly. In fact, the strengthening and toughening of polymeric networks by refining crystalline morphology has been proved recently in natural spider silk, one of the strongest and toughest materials known, in which small nanocrystals confined to a few nanometers has higher stiffness, strength and toughness than larger nanocrystals, and control the levels of ultimate mechanical properties of the silk19,20,21. Therefore, the key problem to be out of the above dilemma is how to nucleate densely and uniformly, which are hard to achieve using conventional methods.
Herein, we propose a deprotonation-complexation-reprotonation strategy to manipulate nucleation-dominant crystallization for refining crystalline morphology of polymeric network (Fig. 1), thereby obtaining water-laden, yet strong and tough crystalline hydrogels. The key is the ability to control the density and uniformity of crystal nuclei by the deprotonation and complexation before reprotonation-induced crystal growth. Concretely, polymers such as poly(vinyl alcohol) (PVA) dissolved in aprotic solvent is deprotonated from its hydroxyl groups to form a polyanion solution in which the polymer chains would become more extended in conformation by electrostatic repulsion (Figs. 1a, b). Complexing the polyanion with metal cation makes adjacent chain segments closer and provides numerous sites for uniform nucleation (Fig. 1c). Re-protonating the polyanion by a protic solvent drives a limited chain movement for moderate growth of crystal nuclei (Fig. 1d), leading to a water-laden ( ~ 80%), yet strong (σf = 7.5 MPa) and tough (Γ = 12.4 kJ m−2) crystalline hydrogel with increased crystallinity, decreased crystallite size, and shortened intercrystallite distance, in stark contrast with conventional strategies such as salting out, air drying and wet annealing that tend to generate strong and tough, but low-water-content hydrogels with a coarsened crystalline morphology (Fig. 1e). We analyze the process of crystallization, highlight the control of density and uniformity of crystal nuclei, discuss the correlation between crystalline morphology refinement and mechanical property enhancement, and clarify the strengthening and toughening mechanisms of the crystalline hydrogels.

The strategy involves deprotonation of PVA for chain extension (a, b), complexation for dense and uniform nucleation(c), and reprotonation for moderate crystal growth (d), leading to a refined crystalline morphology. In contrast, conventional strategy such as salting out, air drying and wet annealing tends to form a coarsened crystalline morphology with drastic shrinkage in volume by massive chain aggregation (a, e).
Results
Crystallization process analysis
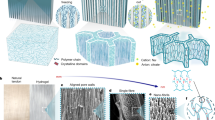
The experimental process involves three successive steps for crystallizing PVA, as shown in Fig. 2a. i) Adding potassium t-butoxide (C4H9OK) into a PVA solution with a mass concentration of 20% in dimethyl sulfoxide (DMSO) to partially deprotonate hydroxyl groups at 90 °C, forming a dark red PVA-O- solution (Supplementary Figs. 1a, b); ii) Cooling the solution at ambient temperature to complex the oxyanion with potassium cation for inducing nucleation, forming a PVA-O-K+ organogel (Supplementary Fig. 1c); iii) Immersing the organogel into water to reprotonate the oxyanion for crystal growth, leading to a crystalline PVA-OH hydrogel (Supplementary Fig. 1d). In order to analyze the crystallization process step-by-step, we fix the cooling temperature and C4H9OK amount to be 25 °C and 4 mol% in relative to the OH functionality of PVA.

C4H9OK amount and cooling temperature are fixed to be 4 mol% and 25 °C to analyze the whole crystallization process. a Schematic of crystallization process. b FTIR spectra and (c) 13C-NMR spectra for PVA and PVA-O- solutions. d Radius of gyration for PVA and PVA-O- chains by molecular dynamics simulation, the inserted are corresponding molecular conformational diagrams. e FTIR spectra for PVA-O- solution and PVA-O-K+ organogel. f K 2p XPS spectrum of PVA-O-K+ organogel. g Dependence of storage modulus (G′) and loss modulus (G″) on temperature for PVA-O-K+ organogel. h FTIR spectra, (i) 1D SAXS, and (j) WAXD profiles for PVA-O-K+ organogel and PVA-OH hydrogel.
Partial deprotonation of hydroxyl groups on PVA by C4H9OK in the first step is proved by FTIR spectra, in which the O-H stretching vibration band ranging from 3700 to 3000 cm−1 shows a decrease in area (Fig. 2b). Their bending vibration band around 1419 cm−1 also fades away from observation in Raman spectra (Supplementary Fig. 2)22. 13C-NMR spectra reveal two peaks at 31.7 ppm and 25.6 ppm (Fig. 2c). The former is assigned to the resonance of methyl in C4H9OK, while the latter arises from the methyne connected to oxyanion in PVA-O- chains23. The partial deprotonation reduces intrachain hydrogen bond because of oxyanion-caused electrostatic repulsion between adjacent segments. Deconvoluting O-H stretching vibration peak reveals two main peaks at around 3400 cm−1 and 3245 cm−1 (Supplementary Fig. 3), corresponding to the hydrogen bond between residual O-H groups and DMSO, as well as between residual O-H groups on PVA-O- chains24. The area ratio of peak (3400 cm−1) to peak (3245 cm−1) is increased in comparison to initial PVA solution, indicating that the deprotonation facilitates residual hydroxyl groups on PVA-O- chains to be solvated by DMSO. The deprotonation makes the polymer chains to be more extended, which is further revealed by molecular dynamics simulation. The average gyration radius of deprotonted PVA-O- chains is larger than that of non-deprotonated PVA chains (Fig. 2d, Supplementary Data 1). Molecular conformational diagrams inserted intuitively reflect the extension of deprotonated PVA-O- chain.
We study the conditions of base and solvent that form PVA-O- polyanion. The deprotonation process is an acid-base reaction, and would proceed provided that the pKa for conjugated acid of any base in DMSO is greater than that for PVA in principle25. It is found that, besides C4H9OK (tert-butanol, pKa = 32), other bases including NaOH (water, pKa = 31.4), sodium ethoxide (ethanol, pKa = 29.8), sodium methoxide (methanol, pKa = 29) and sodium trifluoroethanol (trifluoroethanol, pKa = 23.5)26 are available for the deprotonation (Supplementary Fig. 4). Different bases have different effects on deprotonation degree, which would be raised with an increase in pKa for conjugated acid of base. Aside from DMSO, other aprotic solvents that have the ability to dissolve PVA, such as N-methylpyrrolidone and N,N-dimethylacetamide, can also be used as reaction medium (Supplementary Fig. 5). The aprotic solvents do not participate in the deprotonation reaction, which is proved by the fact that adding C4H9OK into DMSO in the absence of PVA only generates dissolution with no chemical changes (Supplementary Fig. 6). By contrast, protic solvents such as water and ethylene glycol are unavailable in spite of their ability to dissolve PVA, because they shift the reaction equilibrium toward protonation.
The complexation-induced nucleation in the second step through cooling PVA-O- solution is proved from FTIR spectra (Fig. 2e), in which two peaks at 1103 cm−1 and 1143 cm−1 emerge in the PVA-O-K+ organogel. The peak at 1103 cm−1 is an amorphous complexation peak27 and assigned to the stretching vibration of C-O-K+ where the oxyanion coordinates with potassium ion by offering lone pair electrons28. The coordination increases the electron cloud density of potassium ion and thus lower the electron binding energy of K 2p core level29, as shown in Fig. 2f. By contrast, the oxyanion has a reduced electron cloud density and generates an increased electron binding energy of O 1s core level30 (Supplementary Fig. 7). In addition, the peak at 1143 cm−1 is assigned to C-C symmetric stretching vibrations of the carbon framework of the polymer chain in the crystalline region31 or C-O stretching vibrations in C-OH groups of the crystalline region28. And the intensity of this peak is highly sensitive to crystalline portion of polymer chain28,32. Therefore, the peak at 1143 cm−1 is considered as the fingerprint of crystallinity of PVA in many literatures27,32,33,34. The faint crystalline peak emerged in our PVA-O-K+ organogel implies that the complexation induces a weak crystallization, which is related to the formation of crystal nuclei. We infer that non-deprotonated PVA segments adjacent to complexation sites are close to each other, and regularly stack together by hydrogen bonding, thus generating the crystal nuclei (Fig. 2a, purple region). SAXS curve shows a weak scattering (Fig. 2i, purple curve), due to very small size of the crystal nuclei35. WAXD measurement reveals an almost negligible crystalline peak at 19.4° and 40.5° (Fig. 2j, purple curve), implying that the crystal nuclei contribute a faint crystallization. The formed crystal nuclei are sensitive to temperature and can be eliminated at elevated temperature and re-formed once cooling down again (Supplementary Fig. 8), leading to a reversible transition between PVA-O-K+ organogel and PVA-O- solution (Fig. 2g). As temperature rises in a real-time programmed heating process of 3 °C/min, the loss modulus starts to be higher than the storage modulus at 82 °C, corresponding to a transition from organogel to solution. The storage and loss moduli reverse in a real-time programmed cooling process with the same temperature change rate, and PVA-O- solution start to restore to the organogel at a temperature of 40 °C. Besides temperature, metal cation species also have an effect on the nucleation. It is found that that potassium ion has the ability to induce nucleation comparable to sodium ion, but obviously higher than lithium ion (Supplementary Fig. 9).
Soaking the PVA-O-K+ organogel in water for crystal growth in the third step gradually changes the gel’s color from dark red to colorless in appearance. We observe that the soaking does not generate shrinkage in the gel’s volume with the increasing exchange of DMSO with water because the polymeric chains has been crosslinked into a robust network by crystal nuclei. Essentially, the solvent exchange process involves in a reverse reaction of deprotonation, that is, PVA-O-K+ as base accepts protons from water. The recovery of protons causes the segments around crystal nuclei to form hydrogen bonds, leading to crystal growth. FTIR spectra reveal three changes (Fig. 2h): i) The amorphous peak assigned to the stretching vibration of C-O is red-shifted from 1103 cm−1 to 1092 cm−1, proving proton recovery from C-O-K+ to C-OH; ii) The O-H stretching vibration peak is red-shifted from 3420 cm−1 to 3280 cm−1, indicating that the restored hydroxyl groups form hydrogen bonds; iii) The crystalline peak assigned to the stretching vibration of C-C at 1143 cm−1 is obviously intensified, proving crystal growth. WAXD curves show two prominent crystalline peaks at 19.4° and 40.5° (Fig. 2j, red curves), corresponding to (101) and (102) planes of the grown crystallites36. Thanks to the grown crystallites, the crystalline peaks become sharp, compared with PVA-O-K+ organogel. This is consistent with the occurrence of scattering in 1D and 2D SAXS (Fig. 2i, Supplementary Fig. 10).
Deprotonation-degree-controlled nucleation density
The above crystallization process analysis reveals that the complexation of PVA-O- with potassium ions induces surrounding segments to arrange regularly and acts as sites for nucleation. With this in mind, we make an attempt to improve the degree of deprotonation on PVA to form more complexation sites for increasing the density of crystal nuclei and thereby refine crystalline morphology (Figs. 3a1, a2). The deprotonation degree is improved by increasing the amount of C4H9OK (Cbase = 0, 1, 2, 4, 6 mol%), while the cooling temperature is fixed (T = 25 °C). As expected, the O-H stretching vibration peak for PVA-O- solutions exhibits a decrement in intensity with the increase of Cbase (Supplementary Fig. 11). For PVA-O-K+ organogels cooled from these PVA-O- solutions, both the C-O-K+ stretching vibration peak related to the complexation and the C-C stretching vibration peak related to crystal nuclei show an increase in intensity (Fig. 3b), suggesting that more complexation sites are formed and causes denser crystal nuclei.

An increasing amount of C4H9OK (Cbase = 0, 1, 2, 4, 6 mol%) is applied to increase the number of crystal nuclei for refining crystalline morphology, while the cooling temperature is fixed to be 25 °C. a Schematic of crystalline morphology refinement. b FTIR spectra of PVA-O-K+ organogels. c Plots of Iq2 versus q obtained from SAXS measurement, d the corresponding average crystallite size (Rg) and intercrystallite distance (Rd). e WAXD profiles. f Representative SEM images. Scale bars: 5 μm. g Crystallinity and water content. h Representative tensile curves, and i, j the extracted mechanical parameters, including elastic modulus (E), ultimate strength (σf) and fracture toughness (Γ). Data are presented as mean values ± SD, n = 3.
The PVA-OH hydrogels obtained by soaking these PVA-O-K+ organogels in water were characterized by SAXS to analyze the change of their crystallite morphology. Their scattering peaks corrected through multiplying the scattering intensity I(q) with the squared scattering vector q2 shows a gradual intensification (Fig. 3c), implying an increased number of crystallites. Moreover, the peak position shifts toward higher qpeak, indicating a decrement in intercrystallite distance (Rd). The exact values calculated from equation Rd = 2π/qpeak is decreased significantly from 25.2 ± 0.5 nm to 18.9 ± 0.4 nm (Fig. 3d, blue spheres). Meanwhile, the crystallite size (Rg) obtained by applying the Guinier equation is decreased by about 1 nm, from 4.2 ± 0.5 to 3.2 ± 0.5 nm (Supplementary Fig. 12 and Fig. 3d, black spheres). Although the crystallite size is decreased, the crystallinity is improved substantially. WAXD curves show that the crystalline peaks at 19.4° and 40.5° are sharpened with the increase of Cbase from 0 mol% to 6 mol% (Fig. 3e). DSC analysis reveals that the crystallinity is improved by about 10 times, from 0.7% to 7.3% (Fig. 3g and Supplementary Fig. 13). The improved crystallinity, increased crystallite number and decreased crystallite size suggest that the whole crystallization process is indeed dominated by nucleation. Thanks to the nucleation-dominant crystallization that does not bring about shrinkage in the volume of PVA network, the hydrogels retain a high content of water about 80% (Fig. 3g, black spheres), similar to the liquid content in initial PVA solution. The final water content of PVA-OH hydrogels is almost controlled by initial solvent content, which is difficult to achieve for PVA hydrogels by salting-out, annealing, nonsolvent-quenching or confined drying (Supplementary Table 1).
The refinement of crystalline morphology supports a gradual homogenization of porous morphology within PVA-OH hydrogels. As shown in Fig. 3f, the size of pores presents an eye-catching decrease as Cbase increases. Statistical results show that average diameter of the pores is reduced sharply from 3.5 ± 0.8 μm to 1.1 ± 0.3 μm as Cbase increases from 0 to 4 mol%, and then levels off with further increase of Cbase (Supplementary Fig. 14). The homogenization of porous morphology at the microscale is beneficial to avoiding stress concentration when the hydrogels are loaded. It is observed that their ultimate elongation is increased continuously from 4.4 to 11.8, as shown in Fig. 3h and Supplementary Fig. 15. The refinement of crystalline morphology at the nanoscale along with the homogenization of porous morphology at the microscale leads to simultaneous improvement of multiple conflicting mechanical parameters, including elastic modulus, fracture strength and toughness. These mechanical parameters are improved to 1.6 ± 0.1 MPa, 4.5 ± 0.1 MPa, and 9.9 ± 0.6 kJ m−2, which are about 16, 10 and 26 times higher than those of the PVA-OH hydrogel without the use of base (Cbase = 0 mol%), i.e., solvent-exchanged PVA hydrogel by displacement of DMSO with water (Fig. 3i, j). The substantial changes highlight the important role of dense-nucleation-induced refinement of crystalline morphology in improving the hydrogel’s mechanical properties.
Complexation-temperature-controlled nucleation uniformity
The crystallization process analysis reveals that the crystal nuclei are formed by cooling PVA-O- solution and would disappear once temperature rise to 90 °C (Fig. 2g and Supplementary Fig. 8), implying that the nuclei are sensitive to complexation temperature. With this in mind, we attempt to lower the complexation temperature to improve the uniformity of crystal nuclei for refining crystalline morphology and thereby improving the mechanical property of PVA-OH hydrogels (Figs. 4a1, a2). The complexation temperature is lowered from 75 °C to 4 °C, while the amount of C4H9OK is fixed to be 6 mol%. PVA-O-K+ organogels exhibit an increasing C-O-K+ stretching vibration peak at 1103 cm−1 and an increasing C-C stretching vibration peak at 1143 cm−1 (Fig. 4b), proving that the formed crystal nuclei are indeed more stable at low complexation temperature. At a high complexation temperature, molecular thermal motion is intensive and tends to destruct the formed crystal nuclei. PVA-OH hydrogels obtained by soaking these PVA-O-K+ organogels in water have almost the same crystallite size ( ~ 3.2 nm), regardless of different complexation temperature (Fig. 4c, black spheres, Supplementary Fig. 16). This should be related to the same amount of C4H9OK (Cbase = 6 mol%), which determines the number of crystal nuclei in PVA-O-K+ organogels and further controls the ability of segment movement during soaking these organogels in water for crystal growth. We find the complexation temperature influences the distance between crystallites and their distribution. The average intercrystallite distance is shortened from 20.2 nm to 15.3 nm when complexation temperature is lowered from 75 °C to 4 °C (Fig. 4c, blue spheres, Supplementary Fig. 17). The crystallite distribution becomes more uniform, which is reflected from a gradual expansion of high-intensity scattering in 2D SAXS patterns (Supplementary Fig. 18). The corresponding 1D SAXS curves show an intensified scattering, implying that the crystallite distribution is more regular (Supplementary Fig. 19)37. The pair-distribution function P(r) that resolves interparticle distances within each scattering unit provides an intuitive representation about the real space of crystallites in these hydrogels, as shown in Fig. 4d. The profiles show a main peak at 6.3 nm and a shoulder peak around 35 nm. When the complexation temperature decreases, the shoulder peak obviously become weak in relative to the main peak, suggesting that low complexation temperature favors more uniform nucleation. At high complexation temperature, intensive molecular thermal motion tends to destruct crystal nuclei and decreases the uniformity of nuclei distribution, thereby leading to nonuniform distribution of crystallites. Moreover, uniform and stable nucleation at a lowered complexation temperature brings a sharpening in the crystalline peaks of PVA-OH hydrogels (Fig. 4e) and a rapid increase in their crystallinity from 2.2% to 8.6% (Fig. 4g, Supplementary Fig. 20). Although the crystallinity is increased substantially, the equilibrium water content of these hydrogels is retained to be about 80% with little change (Fig. 4g, orange spheres).

A lowered cooling temperature (T = 75, 50, 25, 4 °C) is applied to homogenize the distribution of crystal nuclei for refining crystalline morphology, while the amount of C4H9OK is fixed to be 6 mol%. a Schematic of crystalline morphology refinement. b FTIR spectra of PVA-O-K+ organogels. c Average crystallite size (Rg) and intercrystallite distance (Rd). d The pair-distribution function P(r). (e) WAXD profiles. f Representative SEM images. Scale bars: 5 μm. g Crystallinity and water content. h Representative tensile curves. i Elastic modulus (E) and fracture strength (σf). j Fracture toughness (Γ). Data are presented as mean values ± SD, n = 3.
Besides the refinement of crystalline morphology at nanoscale, uniform nucleation at low complexation temperature homogenizes the porous structure of PVA-OH hydrogels at microscale. SEM images show that large-size pores are gradually eliminated as the complexation temperature decreases from 75 °C to 4 °C (Fig. 4f). Statistic results shows that the average size of pores is reduced from 1.2 μm to 0.8 μm and their distribution range is narrowed obviously (Supplementary Fig. 21). It is associated with the fact that the uniformly distributed and stable crystal nuclei freeze local segments and prevent PVA chains from long-distance movement in subsequent crystal growth process, thus homogenizing the porous structure. At macroscopic level, the surface of PVA-OH hydrogels visibly become smooth (Supplementary Fig. 22). The structural homogenization at multiple scales induced by uniform nucleation at low complexation temperature improves the mechanical properties of these PVA-OH hydrogels (Fig. 4h and Supplementary Fig. 23), although they have the same water content. Young’s modulus of 1.9 ± 0.1 MPa, tensile strength of 7.4 ± 0.2 MPa, and fracture toughness of 12.4 ± 0.1 kJ m−2 are achieved simultaneously as the complexation temperature is 4 °C. These values are 4.75, 4.93 and 4.96 times higher than the modulus (0.4 ± 0.01 MPa), strength (1.5 ± 0.04 MPa) and fracture toughness (2.5 ± 0.4 kJ m−2) of the hydrogel obtained by complexation at 75 °C (Fig. 4i, j). We find through comparison that these improved mechanical parameters are obviously better than previous PVA hydrogels with similar water content prepared by crystal growth-dominant process (e.g., wet-annealing17, salting-out15, drying-annealing16 and solvent-exchange13) (Supplementary Fig. 24 and Table 2), suggesting the importance of the control of nucleation in improving the mechanical property of crystalline PVA hydrogels. In addition, fatigue resistance for optimized PVA-OH hydrogel is quantitatively evaluated by a single-notch method38. The fatigue threshold is measured to be ~275 J m−2 (Supplementary Fig. 25), comparable to that of annealed PVA hydrogel ( ~ 300 J m−2) with a water content of ~75% and a crystallinity of ~10%16.
Strengthening and toughening mechanism
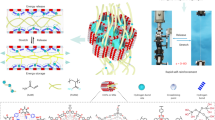
To reveal the strengthening and toughening mechanism caused by the dense and uniform nucleation, we have prepared a control sample called PVA-EX hydrogel by direct solvent exchange without usage of base. In relative to PVA-OH hydrogel, PVA-EX hydrogel without nucleation control by using alkali features as coarse crystalline morphology at nanoscale (Rg: 4.2 ± 0.5 nm Vs 3.2 ± 0.4 nm, Rd: 25.1 ± 0.5 nm Vs 15.3 ± 0.1 nm) and porous morphology at microscale (3.50 ± 0.88 μm Vs 0.83 ± 0.16 μm) (Supplementary Table 3). Given their micro-/nanoscale morphology differences, we first compare the mechanical performance of the two hydrogels, as shown in Fig. 5a and Supplementary Table 4. The refined morphology of PVA-OH hydrogel leads to a 1-fold increase in elongation at break, a 15-fold increase in strength and a 31-fold increase in toughness. Why can the refined morphology simultaneously increase strength and elongation at break? On one hand, small-size pores favor homogenization of stress distribution, while large-size pores would serve as defect with concentrated stress, and tend to cause premature fracture. On the other hand, the refinement of crystalline morphology can disperse stress more uniformly, leading to extensive plastic deformation throughout the whole sample. To verify the hypothesis, we investigate the micro deformation behavior of PVA-OH and PVA-EX hydrogel through in-situ SAXS during tension. Their 2D SAXS patterns show a geometrical change from an isotropic ring (λ = 1, d0) to an ellipse, demonstrating that average intercrystallite distance (d) in the minor axis parallel to stretch direction (d∥) increases but decreases in the major axis perpendicular to stretch direction (d⊥) during deformation (Supplementary Fig. 26). We have extracted the corresponding d∥ and d⊥ to calculate micro deformation ratio (d/d0) and then plotted against macro stretch ratio λ (Fig. 5b, Supplementary Fig. 27). Apparently, the micro deformation of PVA-OH hydrogel better follows macro deformation than PVA-EX hydrogel at the same stretch ratio, that is, refined crystalline morphology deforms more uniformly under tension. Hence, refinement of crystalline morphology imparts favorable stretchability to PVA-OH hydrogel, leading to a much larger stretch ratio. Moreover, PVA-OH hydrogel has significantly improved network chain density of 73.18 mol m-3 (Supplementary Fig. 28), which suggests the number of cross-linked polymer chains per unit volume of hydrogel sharply increases. To analyze the orientation degree of molecular chains before fracture, in-situ wide-angle X-ray scattering (WAXS) is performed. The orientation factor for PVA-OH hydrogel is 0.90 at λ = 13, higher than 0.82 at λ = 5 for PVA-EX, implying PVA-OH hydrogel has the higher orientation degree of molecular chains before fracture failure (Fig. 5c, Supplementary Fig. 29). At the same time, the scattering intensity of chains orientation is much higher for PVA-OH hydrogel, indicating far more polymer chains orient toward stretch direction before failure. Therefore, simultaneous stiffening of multiple polymer chains strengthens PVA-OH hydrogel, leading to its remarkable strength.

PVA-EX hydrogel prepared from PVA sol in DMSO by directly solvent exchange with water is used as control sample. a Representative tensile curves of PVA-OH and PVA-EX hydrogels. The inserts are their representative SEM images, scale bar: 2 μm. b Microscopic deformation ratio (d/d0) in the parallel (∥) and vertical (⊥) directions to stretch ratio λ from SAXS measurement, where black solid-line represents affine deformation of an incompressible material. The essence of affine deformation is that micro deformation completely follows macro deformation. As a consequence, its micro deformation follows the law: d∥/d0 = λ, d⊥/d0 = λ−0.5. c Scattering intensity versus azimuthal angle curves extracted from WAXS. d–f Successive tensile loading-unloading curves without interval for PVA-OH (d) and PVA-EX (e) hydrogels, as well as the corresponding dissipated energy and dissipated ratio under different stretch ratios (f). g–i Deconvoluted FTIR spectra of PVA-OH (g) and PVE-EX (h) hydrogels in the wavenumber range of 3000 to 3700 cm−1, as well as the area ratio of the peaks located at 3209 and 3364 cm−1 (i). The two peaks are assigned to hydroxyl groups on PVA that form hydrogen bonds with PVA and water, respectively.
Energy dissipation is investigated under prolonged cyclic loads without intervals between two cycles (Figs. 5d, e). As stretch ratio increases, for PVA-OH hydrogel, the dissipation energy per unit volume sharply increases from 0.41 MJ m−3 at λ = 2 to 4.89 MJ m-3 at λ = 9 (Fig. 5f). A large amount of energy is dissipated through breaking hydrogen bonds in amorphous region. In addition, PVA-OH hydrogel shows the loading curve crosses the last unloading curve during successive loading-unloading, indicating prominent self-recovery as a result of partially reconstructed hydrogen bonds. In view of the crucial role of interchain hydrogen bonds in energy dissipation, we further quantify the content of hydrogen bonds of PVA-PVA by FTIR peak fitting. FTIR fitting is performed on the region of O–H stretching vibration from 3700 cm−1 to 3000 cm−1, where two peaks at 3209 cm−1 and 3364 cm−1 are ascribed to hydrogen bonds of PVA-PVA and PVA-water13, respectively (Figs. 5g, h). The area ratio of the peak at 3209 cm−1 to the other at 3364 cm−1 is calculated to quantify the content of hydrogen bonds of PVA-PVA in relative to PVA-water, as shown in Fig. 5i. The area ratio for PVA-OH hydrogel is much greater, as expected, suggesting that it has an enhanced density of PVA-PVA hydrogen bonds. The increased interchain interaction is consistent with aforementioned deprotonation-induced chain extension in solution that favors more interchain hydrogen bonding in resultant PVA-OH hydrogel. In a word, the nucleation-controlled refinement of crystalline morphology, along with the refinement of porous morphology, promotes a uniform deformation, simultaneous stiffing of massive polymeric chains, and intensive energy dissipation of sacrificial hydrogen bond within amorphous regions, leading to a well combination of stretchability, strength and toughness of PVA-OH hydrogel. In addition to the conventional static toughness, we also evaluate its dynamic toughness through ballistic impact test (Supplementary Fig. 30a). The bullet with diameter of 6 mm and weight of 0.22 g was shot perpendicular to the surface of hydrogels at a speed of 40 m s-1. When shooting after 1 ms, it contacted the surface of hydrogels and possessed the kinetic energy of 0.176 J appropriately. PVA-OH hydrogel with thickness of 1 mm rebounded the bullet without being perforated, showing high dynamic toughness (Supplementary Fig. 30b and Movie 1). On the contrary, PVA-EX hydrogel was easily punched under the same condition (Supplementary Fig. 30c and Movie 2).
Application advantages
Robust PVA-OH hydrogels with high water content have application advantages in wet adhesion and diverse biomedical applications. Excellent mechanical properties impart wet adhesion advantage to PVA-OH hydrogels. Glued PET hoses by PVA-OH hydrogel lift a weight of 1 kg (Supplementary Fig. 31a), indicating high adhesive properties. Moreover, the adhesion strength of PVA-OH hydrogel on different substrates is evaluated by lap shear adhesion test. The values of their adhesion strength are 210.1 ± 13 kPa for aluminum, 251.1 ± 14 kPa for mica, 375.0 ± 14 kPa for glass, 420.4 ± 14 kPa for PET and 506.9 ± 17 kPa for Al2O3, respectively (Supplementary Fig. 31b). Excellent mechanical properties coupled with desired water content confer the advantage in biomedical application. It is a key problem that hydrogels with good mechanical properties should be non-toxic and biocompatible for biomedical applications and can be used as excellent carriers for cell proliferation. Hence, cell viability assay was carried out to evaluate the biocompatibility of PVA-OH hydrogel. In spite of the involvements of DMSO and C4H9OK in the preparation process, PVA-OH hydrogel shows good biocompatibility with negligible cellular toxicity (Supplementary Fig. 32).
Discussion
To conclude, we proposed a deprotonation-complexation-reprotonation strategy to refine crystalline morphology, thereby obtaining a water-laden ( ~ 80%), yet strong and tough PVA crystalline hydrogel with fracture strength of 7.4 ± 0.2 MPa and fracture toughness of 12.4 ± 0.1 kJ m-2. In the crystallization process, alkali content manipulated the number of crystal nuclei by providing nucleation sites, and complexation temperature manipulated the nucleation uniformity by stabilizing the crystal nucleus, thus synergistically determining the refined degree of crystalline morphology of resultant hydrogels. Refinement of crystalline morphology combined high strength with toughness in the hydrogel, and its comprehensive mechanical performance is well dictated by the refined degree. Considering these special advantages, it is believed that our strategy would open up an avenue for developing water-laden, yet strong and tough hydrogels for diverse biomedical applications, such as tissue engineering, soft robots, and bioelectronics.
Methods
Materials
Polyvinyl alcohol (PVA, Mw = 146000–186000, Sigma Aldrich) and potassium tert-butoxide (C4H9OK, ≥ 98%, Shanghai Macklin Biochemical Co., Ltd.) were used as received. Dimethyl sulfoxide (DMSO, ≥ 99.0%), glutaraldehyde (25 vol%), hydrochloric acid (36.5 wt%), sodium hydroxide, sodium ethoxide, sodium methoxide, sodium trifluoroethanol, N-Methylpyrrolidone (NMP) and N,N-Dimethylacetamide (DMAc) were purchased from Sinopharm Chemical Reagent Co., Ltd.
Fabrication of PVA-OH hydrogel
PVA (3 g) was dissolved in DMSO (12 g), forming a solution with mass concentration of 20%. A certain amount of C4H9OK (0.075, 0.15, 0.3, 0.45 g) was added into the PVA solution and stirred at 90 °C for 2 h. Then, the solutions were cooled down at a certain temperature (75, 50, 25, 4 °C) for 12 h to become organagels. The organogels were immersed in ice-water mixture for 24 h to thoroughly remove DMSO and C4H9OK, forming PVA-OH hydrogels. PVA-OH hydrogel for strengthening and toughening mechanism was prepared by using 0.45 g C4H9OK and cooling down at 4 °C.
Fabrication of PVA-EX hydrogel
PVA (3 g) was dissolved in DMSO (12 g), forming a solution with mass concentration of 20%. Afterwards, PVA solution that does not contain C4H9OK was immersed directly in ice-water mixture for 24 h, forming a solvent-exchanged hydrogel as control sample, denoted as PVA-EX.
Water content measurement
First, PVA-OH hydrogels were weighed in the air, and their weights were recorded as mwet. Then, the hydrogels were dried thoroughly at 55 °C for 48 h. The obtained solids were weighed in the air, and their weights were recorded as mdry. Finally, the water contents (W) of PVA-OH hydrogels were calculated from the equation:
Crystallinity measurement
PVA-OH hydrogels were soaked in a solution containing glutaraldehyde (8 ml), hydrochloric acid (0.4 ml) and water (92 ml). After 2 h, the hydrogels were washed with deionized water to remove excessive glutaraldehyde and hydrochloric acid, followed by drying for crystallinity measurement. In this way, amorphous chains within the hydrogels were fixed by glutaraldehyde to minimize the additional formation of the crystalline domain during the drying process.
Differential scanning calorimetry (DSC) was carried out by TA Q2000 to measure the crystallinity16. We first weighed the total mass of the dried sample (containing a little residual water), recorded as m. Then, DSC test was performed under a nitrogen atmosphere with a flow rate of 30 mL min−1, and the temperature was increased from 50 to 250 °C with a heating rate of 10 °C min−1. The heat flow-time curves presented two peaks. One was a broad peak ranging from 60 °C to 180 °C, corresponding to the evaporation of residual water. The other was a narrow peak ranging from 200 °C to 250 °C, corresponding to the melting of crystalline domains. The enthalpy for the evaporation of residual water per unit mass of the dried sample Hresidual was obtained from the integration of the broad peak. The enthalpy for the melting of crystalline domains per unit mass of the dried sample Hcrystalline was obtained from the integration of the narrow peak. The mass of the residual water mresidual was calculated as:
where H0water is the evaporation heat (2260 J g-1) of pure water39. The mass of the crystalline domains mcrystalline was calculated as:
where H0crystalline is the melting enthalpy (138.6 J g-1) of 100 wt% crystalline PVA40. The crystallinity in the ideally dry sample Xdry was calculated as:
The crystallinity in PVA-OH hydrogels Xwet was calculated as:
SAXS measurement
SAXS was conducted on a Xenocs Xeuss 2.0 System by using an incident Cu-Kα X-ray beam that was perpendicular to the sample plane. X-ray wavelength, spot size and distance between specimen and detector were 0.154 nm, 172 × 172 μm2 and 1185 mm, respectively. Samples were square slices with a side length of 10 mm. Scattering patterns were collected by a Pilatus 300 k detector. The distance distribution function, P(r), was analyzed by SASfit software.
(1) The inter-crystallite distance (Rd) was obtained from the Bragg expression41:
Where qmax is the scattering vector at which Iq2 shows a maximum value.
(2) The radius of gyration (Rg) could be calculated at low q according to Guinier equation35:
Where I(q) was the scattering intensity and I0 was the zero angle scattering intensity. Rg was used to represent the average size of crystallites.
(3) The pair-distribution function P(r) could be calculated through a Fourier transform of the scattering curve35:
(4) The orientation order parameter (f) was calculated to describe the orientation degree of crystal domains. Its value ranges from 0 to 1, where the former corresponds to an isotropic structure and the latter corresponds to a perfect orientation structure along the director. A Maier-Saupe distribution function was used to fit the azimuthal angle profile42:
where I0 is the free baseline intensity, φ is the azimuthal angle, φ0 is the azimuth at the position of maximal intensity, and ω is a parameter that determines the width of the distribution. After the fitting, parameter ω was obtained, and the orientation factor f can be determined using the following formula:
where the function P2(cosφ) is the second-order Legendre polynomial of cosφ and often referred to as the Hermans orientation function:
Tensile test
Mechanical properties were measured on a Shimadzu AGS-X Tester with a gauge length of 10 mm and a load speed of 20 mm min-1.
(1) Young’s modulus (E) was calculated from the stress-strain (σ-ε) curve within the initial linear region. The toughness, a parameter that characterizes the energy required to fracture the sample per unit volume, was calculated by the integral area from ε = 0 to the fracture point (εf) under the stress-strain (σ-ε) curve. Specifically, the toughness (U) was calculated as follows:
(2) The cyclic loading-unloading tensile test was performed to investigate the energy dissipated mechanism. The dissipated energy (W) was calculated from the hysteresis area:
(3) The dissipated ratio was the ratio of the dissipated energy (W) to toughness (U) at a specific strain, i.e., dissipated ratio = W/U.
Fracture toughness test
The fracture toughness was determined by a single-notch test43. Two identical hydrogels were fabricated with the same length 20 mm, width 5 mm and thickness about 1 mm for tensile test at a rate of 10 mm min-1 on the Shimadzu AGS-X material testing machine with a load cell of 500 N. A notch 1 mm was introduced across the width of one sample. A critical extension λc was defined when the notch turned into a running crack. Another sample without notch was uniformly stretched to measure the relationship between nominal stress s and strain λ. The work done to reach λc for an unnotched sample was defined as U(λc), and the fracture toughness Γ was calculated as:
where c was the crack length.
Fatigue threshold of the hydrogel samples
Fatigue threshold of the hydrogel samples was quantified through the single-notch method, following previous protocol16,38. All fatigue tests in this study were performed on fully swollen hydrogel samples within a water bath using a mechanical stretcher (Cellscale), in order to avoid dehydration-induced crack propagation. All cyclic tensile tests were conducted on notched and unnotched samples with identical dog-bone shapes. A notched sample with pre-cut crack (length less than 1/5 of the overall width) was exposed to a cyclic tensile test at a strain of λmax, and we recorded the crack length at the undeformed state cover cycles using a digital microscope (AM4815ZT, 20 mm pixel−1). Meanwhile, the same stretch λmax was also applied on a unnotched sample, and the strain energy density of under the Nth cycle estimated following this formula:
The applied energy release rate G in the notched sample under the Nth cycle with a maximal applied stretch of λmax can be calculated following this formula:
where k is a slowly varying function of the applied stretch as k = 3·λmax−1/2. By varying the applied stretch of λmax, we acquired the curve of crack extension per cycle dc/dN versus the applied energy release rate G. The fatigue threshold can be obtained by linearly extrapolating the curve of dc/dN versus G to the intercept with the abscissa. To validate that the energy release rate value obtained from the linear extrapolation is equal to the real fatigue threshold of the hydrogel sample, a notched hydrogel sample was exposed to a single-notch tensile at this energy release rate for 30000 cycles, and crack propagation was further monitored with the camera.
Lap shear adhesion test
The adhesion strength was evaluated by lap shear adhesion test. PVA-O- solution was evenly coated on the surface of one substrate with an overlap area of 1 cm2. After that, it was quickly covered with the other substrate, followed by complexation for adhesion and immersed in DI water for removing extra reagent. Lap shear adhesion tests were performed on a Shimadzu AGS-X Tester with a 500 N load cell at a loading speed of 10 mm min-1. The adhesion strength was calculated as the failure force divided by the overlap area.
Rabbit Chondrocytes Isolation and Culture
All animal experiments in this research were approved by the Animal Care and Ethics Committee of the College of Biology, Hunan University. The isolation and culture of rabbit chondrocytes from articular cartilage of 1 week-old New Zealand white rabbits were following previous method44. Concretely, rabbit legs were collected and immersed in PBS in the biosafety cabinet. The surrounding tissues around the cartilage were removed, and the cartilage was washed twice with PBS containing 1% antibiotics. The washed articular cartilage was cut into small pieces ( < 1 mm3) and transferred into centrifugal tubes. Adequate amounts of the DMEM medium (HyClone, USA) containing 15% (w/v) type II collagenase (300 u/mg, Worthington, USA) was added to the centrifuge tube to digest the ECM. Chondrocytes were collected every 4 h, and the collection was repeated once. The collected digestive fluids were filtrated and centrifuged at 3000 rpm for 5 min, and then, the collected chondrocytes were resuspended in the DMEM medium containing 10% fetal bovine serum (Gibco, USA) and cultured in cell culture dishes. The cell culture medium was changed every 2 days. After two passage culture, chondrocytes were used for this study.
In vitro cell viability study
The live/dead cell staining experiment was applied following the manufacturer’s protocol. To obtain the hydrogel extracts, 1 g PVA-OH was added to 10 mL DMEM and incubated in incubator for 24 h at 37 °C. Then the supernatant was collected and filtered through a 0.22 μm filter (Millipore, USA) to obtain hydrogel extracts for the following cell experiments. Rabbit chondrocytes inoculated with 20,000 cells per well in a 12-well plate (Corning, USA). After the cells were attached to the plate, extracts were used instead of media. The cells with AM/PI staining at day 1, 3 and 5 were observed and photographed by fluorescence microscope (BX53; Olympus, Tokyo, Japan). In cell proliferation experiments, chondrocytes were inoculated into 96-well plates (Corning, USA) with a density of 5000 cells per well and cultured with PVA-OH hydrogel extracts or DMEM (control). Cells were counted using the Cell Counting Kit-8 (CCK-8; Beyotime, China) after 1day, 3 days and 5 days of culture. Optical density at 450 nm (OD450) was measured with a microplate reader (BioTek, ELX808, USA). The experiment was repeated at least three times.
Material characterizations
FTIR spectra were collected using a Nicolet IS50 spectrometer (Thermo Fisher Scientific) with an attenuated total reflectance mode in the wavenumber range from 4000 cm-1 to 400 cm-1. WAXD was carried out on a MiniFlex X-ray diffractometer (Rigaku) in a scanning range from 10° to 60° under a scanning rate of 10° min-1. A rheological test was carried out on a MARS60 rheometer (HAAKE). Unless otherwise stated, the temperature for test is 90 °C for solution and 25 °C for gels. 13C-NMR spectra were recorded by a 400 MHz nuclear magnetic resonance spectrometer (Bruker) at 25 °C. DMSO-d6 was used as a solvent. The concentration of samples was set as 30 mg ml-1. WAXS was conducted on a Xenocs Xeuss 2.0 System by using an incident Cu-Kα X-ray (λ = 0.154 nm) beam that was perpendicular to the sample plane. The distance between specimen and detector was 100 mm.
Molecular dynamics simulation
Molecular simulation was used to explore atomic-level details of PVA and PVA-O- solution. Models were constructed using Material Studio, mainly including MS Visualizer, Amorphous Cell and Forcite modules. Molecular models were constructed by MS Visualizer. The polymerization degree of PVA molecules was set to be 20, larger than its value of Characteristic ratio, C∞, which was calculated to be 6.945. PVA-O- molecular model was constructed by deleting four H atoms of hydroxyl groups that occupy 5%.
Solution models were constructed by Amorphous Cells. Molecules constructed by MS Visualizer were filled into the empty cell. Two PVA or PVA-O- molecules were put into the empty cell, and the filling density of DMSO was set to 1.1 g cm−3 (the experimental value of DMSO density). Filling with different numbers of cations (K+ ions) to balance the charge of the system, the modeling process is illustrated in the figure below. The box was a cube with a side length of about 40 Å. The volume ratio of solvent DMSO to PVA in the model was about 20:1, in which the amount of solvent was sufficient to reflect the solvent environment around PVA molecules.
For molecular dynamics simulation, the ensemble was set to NVT, the initial velocity was adopted as Maxwell-Boltzmann distribution, the simulated temperature was 298 K (25 °C), the Nose-Hoover thermostat was adopted, the time step was 1 fs, and the total simulation time was 5000 ps.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data that support the plots within this article and other findings of this study are available from the corresponding author upon request. Supplementary data are provided with the manuscript. Source data are provided with this paper.
References
Lu, X. L. & Mow, V. C. Biomechanics of articular cartilage and determination of material properties. Med. Sci. Sports Exerc. 40, 193–199 (2008).
Zhang, Y. S. & Khademhosseini, A. Advances in engineering hydrogels. Science 356, eaaf3627 (2017).
Lin, W. et al. Cartilage-inspired, lipid-based boundary-lubricated hydrogels. Science 370, 335–338 (2020).
Wang, X. L. et al. Nanostructured dense collagen-polyester composite hydrogels as amphiphilic platforms for drug delivery. Adv. Sci. 8, 2004213 (2021).
Zhang, Y. Y. et al. Reactive oxygen species-responsive and Raman-traceable hydrogel combining photodynamic and immune therapy for postsurgical cancer treatment. Nat. Commun. 13, 4553 (2022).
Pan, Z. et al. Designing nanohesives for rapid, universal, and robust hydrogel adhesion. Nat. Commun. 14, 5378 (2023).
Liang, X. Y. et al. Bioinspired 2D isotropically fatigue-resistant hydrogels. Adv. Mater. 34, 2107106 (2022).
Jin, S. et al. Injectable tissue prosthesis for instantaneous closed-loop rehabilitation. Nature 623, 58–65 (2023).
Darabi, M. A. et al. An alkaline based method for generating crystalline, strong, and shape memory polyvinyl alcohol biomaterials. Adv. Sci. 7, 1902740 (2020).
Zhao, X. H. et al. Soft materials by design: unconventional polymer networks give extreme properties. Chem. Rev. 121, 4309–4372 (2021).
Taylor, D. et al. The fracture toughness of soft tissues. J. Mech. Behav. Biomed. Mater. 6, 139–147 (2012).
Hua, M. T. et al. Strong tough hydrogels via the synergy of freeze-casting and salting out. Nature 590, 594–599 (2021).
Xu, L. et al. A solvent-exchange strategy to regulate noncovalent interactions for strong and antiswelling hydrogels. Adv. Mater. 32, 2004579 (2020).
Wu, S. W. et al. Poly(vinyl alcohol) hydrogels with broad-range tunable mechanical properties via the Hofmeister effect. Adv. Mater. 33, 2007829 (2021).
Cui, W. et al. Strong tough conductive hydrogels via the synergy of ion-induced cross-linking and salting-out. Adv. Funct. Mater. 32, 2204823 (2022).
Lin, S. et al. Anti-fatigue-fracture hydrogels. Sci. Adv. 5, eaau8528 (2019).
Wu, Y. et al. Solvent-exchange-assisted wet annealing: a new strategy for superstrong, tough, stretchable, and anti-fatigue hydrogels. Adv. Mater. 35, 2210624 (2023).
Xu, L. J., Qiao, Y. & Qiu, D. Coordinatively stiffen and toughen hydrogels with adaptable crystal-domain cross-linking. Adv. Mater. 35, 2209913 (2023).
Cranford, S. W., Tarakanova, A., Pugno, N. M. & Buehler, M. J. Nonlinear material behaviour of spider silk yields robust webs. Nature 482, 72–76 (2012).
Keten, S., Xu, Z., Ihle, B. & Buehler, M. J. Nanoconfinement controls stiffness, strength and mechanical toughness of β-sheet crystals in silk. Nat. Mater. 9, 359–367 (2010).
Du, N. et al. Structural origin of the strain-hardening of spider silk. Adv. Funct. Mater. 21, 772–778 (2011).
Mahendia, S. et al. Determination of glass transition temperature of reduced graphene oxide-poly(vinyl alcohol) composites using temperature dependent Fourier transform infrared spectroscopy. J. Mol. Struct. 1111, 46–54 (2016).
Wang, Y. M. et al. 1H NMR and 13C NMR investigations of sequence distribution and tacticity in poly(vinyl alcohol-co-vinyl levulinate). Polymer 47, 7827–7834 (2006).
Zhang, M. Y. et al. Toughening double-network hydrogels by polyelectrolytes. Adv. Mater. 35, 2301551 (2023).
Burch, R. R., Sweeny, W., Schmidt, H. W. & Kim, Y. H. Preparation of aromatic polyamide polyanions: a novel processing strategy for aromatic polyamides. Macromolecules 23, 1065–1072 (1990).
Olmstead, W. N., Margolin, Z. & Bordwell, F. G. Acidities of water and simple alcohols in dimethyl sulfoxide solution. J. Org. Chem. 45, 3295–3299 (1980).
Chen, B. et al. Giant negative thermopower of ionic hydrogel by synergistic coordination and hydration interactions. Sci. Adv. 7, eabi7233 (2021).
Mansur, H. S., Sadahira, C. M., Souza, A. N. & Mansur, A. A. P. FTIR spectroscopy characterization of poly (vinyl alcohol) hydrogel with different hydrolysis degree and chemically crosslinked with glutaraldehyde. Mater. Sci. Eng., C. 28, 539–548 (2008).
Crist, B. V. Handbook Of Monochromatic Xps Spectra, The Elements Of Native Oxides. Handbook of monochromatic XPS spectra, (2000).
Yang, J. & Ying, J. Y. A general phase-transfer protocol for metal ions and its application in nanocrystal synthesis. Nat. Mater. 8, 683–689 (2009).
Tretinnikov, O. N. & Zagorskaya, S. A. Determination of the degree of crystallinity of poly(vinyl alcohol) by FTIR spectroscopy. J. Appl. Spectrosc. 79, 538–543 (2012).
M., S. K. & P., N. A. Dissolution mechanism of semicrystalline poly(vinyl alcohol) in water. J. Polym. Sci., Part B: Polym. Phys. 34, 1339–1346 (1996).
Otsuka, E. et al. Effects of preparation temperature on swelling and mechanical properties of PVA cast gels. Soft Matter 8, 8129–8136 (2012).
Otsuka, E. & Suzuki, A. A simple method to obtain a swollen PVA gel crosslinked by hydrogen bonds. J. Appl. Polym. Sci. 114, 10–16 (2009).
Tainer & John, A. X-ray scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Biophys. J. 100, 193–285 (2011).
Wan, H. B., Wu, B. H., Hou, L. & Wu, P. Y. Amphibious polymer materials with high strength and superb toughness in various aquatic and atmospheric environments. Adv. Mater. 36, 2307290 (2023).
Li, H. T. et al. Polychrome photonic crystal stickers with thermochromic switchable colors for anti-counterfeiting and information encryption. Chem. Eng. J. 426, 130683 (2021).
Liang, X., et al. Anisotropically fatigue‐resistant hydrogels. Adv. Mater. 33, e2102011(2021).
Lin, S., Liu, J., Liu, X. & Zhao, X. Muscle-like fatigue-resistant hydrogels by mechanical training. PNAS 116, 10244–10249 (2019).
Peppas, N. A. & Merrill, E. W. Differential scanning calorimetry of crystallized PVA hydrogels. J. Appl. Polym. Sci. 20, 1457 (1976).
Zhang, R. et al. Stretch-induced complexation reaction between poly(vinyl alcohol) and iodine: an in situ synchrotron radiation small- and wide-angle X-ray scattering study. Soft Matter 14, 2535–2546 (2018).
Zhao, C. et al. Layered nanocomposites by shear-flow-induced alignment of nanosheets. Nature 580, 210–215 (2020).
Ducrot, E. et al. Toughening elastomers with sacrificial bonds and watching them break. Science 344, 186–189 (2014).
Chen, W. et al. Tanshinone IIA delivery silk fibroin scaffolds significantly enhance articular cartilage defect repairing via promoting cartilage regeneration. ACS Appl. Mater. Interfaces 12, 21470–21480 (2020).
Bicerno, J. et al. Prediction of Polymer Properties. (Marcel Dekker Inc., New York, 2002).
Acknowledgements
The authors acknowledge the financial support from the National Natural Science Foundation of China (51973054, J.F.W.), Natural Science Funds of Hunan Province for Distinguished Young Scholar (2021JJ10018, J.F.W.), Technology Breakthrough Plan of Henan Province (242102230002, H.L.), High-level Innovative Talent Project in Henan Province (241823006, H.L.) and Guangdong Basic and Applied Basic Research Foundation (2023A1515110532, J.L.). And the authors acknowledge the Analytical Instrumentation Center of Hunan University for diverse material characterizations.
Author information
Authors and Affiliations
Contributions
L.M.H. performed major experiments and characterizations and wrote the initial manuscript. H.L. performed partial experiments and characterizations. S.X.W. participated in data analysis. P.H.X. participated in drawing. F.Z.Z. participated in experiments. C.Y.P. and J.Y. contributed to the data discussion. Y.T. and J.L. performed a fatigue test and analysis. L.J. guided experimental design. J.F.W. conceived and supervised the project, revised and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Tongqing Lu, Peiyi Wu and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Huang, L., Li, H., Wen, S. et al. Control nucleation for strong and tough crystalline hydrogels with high water content. Nat Commun 15, 7777 (2024). https://doi.org/10.1038/s41467-024-52264-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-52264-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
