
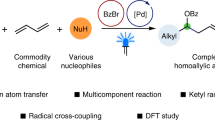
Abstract
Catalytic methods to couple alcohol and alkene feedstocks are highly valuable in synthetic chemistry. The direct oxidative coupling of primary alcohols and alkenes offers a streamlined approach to ketone synthesis. Currently, available methods are based on transition metal-catalyzed alkene hydroacylation, which involves the generation of an electrophilic aldehyde intermediate from primary alcohol dehydrogenation. These methods generally require high reaction temperatures and a high loading of precious metal catalysts and are predominantly effective for branch-selective reactions with electron-rich alkenes. Herein, we designed a dual photo/cobalt-catalytic method to manipulate the reactivity of nucleophilic ketyl radicals for the synthesis of ketones from primary alcohols and alkenes in complementary reactivity and selectivity. This protocol exhibits exceptional scope across both primary alcohols and alkenes with high chemo- and regio-selectivity under mild reaction conditions. Mechanism investigations reveal the essential role of cobalt catalysis in enabling efficient catalysis and broad substrate scope.
Similar content being viewed by others
Introduction
Primary alcohols are among the most frequently encountered functional groups in natural products1 and commodity chemicals2, which makes them ideal starting materials in organic synthesis to approach complex structures. Therefore, they serve as important precursors for the convergent construction of ketones, which occur abundantly in natural and man-made chemicals, and also function as versatile intermediates to construct diverse functionalities1,3,4. Classical convergent ketone synthesis generally requires multistep protocols to manipulate the oxidation state to activate primary alcohol and to react with large amounts of nucleophilic organometallic reagents (Fig. 1, A)5,6,7,8. In this context, direct catalytic methods to convert primary alcohols to ketones are highly desirable.

A Classic method for convergent ketone construction from primary alcohol and alkene. B Transition metal-catalyzed ketone synthesis from primary alcohol and alkene via dehydrogenative activation of primary alcohol. C Photocatalytic HAT enabled coupling of primary alcohols with activated alkenes to form high alcohols and dehydrogenation of secondary alcohols to form ketones. D Design of this work: a dual photo/cobalt-catalyzed hydrogen atom transfer enabled ketone assembly from primary alcohol and alkene via secondary alcohol intermediate.
The advancement of transition metal catalysis has enabled the catalytic activation of primary alcohols through dehydrogenation to be integrated into ketone synthesis in a one-step manner9,10,11,12,13,14,15,16,17,18,19. Among these methods, assembling ketone from primary alcohols and alkenes represents the most straightforward and cost-effective approach, given the ready availability of both starting materials (Fig. 1B)20,21,22,23,24,25. In these processes, a metal hydride complex is the key catalytic species merging the dehydrogenation of alcohol and alkene hydroacylation via an aldehyde intermediate. Consequently, branched adducts from electron-rich alkenes are dominant unless directing groups are installed22,23. Nevertheless, the majority of existing methods require high loading of precious metal catalysts and elevated temperatures, which narrows the substrate scopes and induces side reactions. Therefore, it is imperative to develop mild and efficient strategies for the conversion of these feedstocks into valuable ketones.
Photo-mediated hydrogen atom transfer (HAT) has been recognized as a powerful tool for activating C(sp3)-H bonds under mild conditions, which provides radical pathways for bond formation with distinct reactivity and selectivity compared to closed-shell mechanisms26,27,28. Selective HAT of α-hydroxy C–H bonds in alcohols by photocatalysis generates nucleophilic ketyl radicals (I), which readily participate in Giese-type addition to electron-deficient alkenes to form higher alcohols29,30,31,32. While ketones can be derived from the dehydrogenation of secondary alcohols via photocatalyzed HAT, direct access to ketones from the dehydrogenative coupling of primary alcohols and alkenes remains elusive in photocatalysis (Fig. 1C)33,34,35,36. We thought that the challenge lies in discriminating the reactivities of primary and secondary ketyl radicals to selectively undergo alkene addition and dehydrogenation, respectively. To address this, we hypothesized a photocatalyzed cascade reaction, involving a secondary alcohol intermediate to directly synthesize ketones from primary alcohols and alkenes (Fig. 1D). The process initiates with the photo-mediated selective HAT of primary alcohol to form primary ketyl radical I, which then undergoes nucleophilic addition to an electrophilic alkene, producing a secondary alcohol intermediate. Subsequent photo-mediated HAT of this intermediate, coupled with cobalt-catalyzed hydrogen transfer using alkenes as the acceptor, leads to the desired ketone product via secondary ketyl radical II. We here describe the development of a dual photo/Co catalytic system that enables the efficient and selective synthesis of ketones from primary alcohols and alkenes under mild conditions. This system accommodates a wide range of activated alkenes and styrenes, complementing existing transition metal-catalyzed protocols. The cobalt catalyst is demonstrated critical for promoting efficient photocatalysis and overcoming the scope limitation of activated alkenes in Giese-type addition.
Results
We commenced exploration of reaction conditions using n-butanol (1a) and n-butyl acrylate (2a) as the model substrates (Table 1). 60% of the desired ketone 3a was obtained from the reaction using tetrabutylammonium decatungstate (TBADT) (2 mol%) as the photocatalyst, and the combination of CoCl2 (1 mol%) and PPh3 (2 mol%) as cocatalyst after 20 hours of violet light irradiation in acetonitrile at ambient temperature (entry 1). Further evaluation of the reaction parameters revealed that the cobalt catalyst precursor and ligand have significant influences on catalytic efficiency (entries 2–12, Supplementary Table 1 and Supplementary Fig. 1). The electron-rich and less steric-demanding triarylphosphine outperformed the electron-deficient and bulky ones and diphosophines (entries 2–9, and Supplementary Fig. 1). Notably, the combination of CoCl2 and tri(p-anisyl)phosphine exhibited the highest catalytic efficiency (entry 2). Moreover, replacing TBADT with the commonly used organic HAT catalyst benzophenone (BP), only 9% of 3a was detected (entry 13)28. The solvent evaluation showed that acetone was comparable to acetonitrile (entry 14, and Supplementary Table 2). Finally, a superior level of product formation was achieved using 4 mol% of TBADT, 1 mol% of CoCl2, and 2 mol% of tri(p-anisyl)phosphine as the catalytic system in acetonitrile (entry 15). Performing the reaction in the presence of water or under air, leads to severely diminished yields (entries 16 and 17). Control experiments verified the essential roles of TBADT, the cobalt catalyst, and photoirradiation (entries 18–20, and Supplementary Table 5).
With optimized reaction conditions in hand, we sought to determine the generality of this protocol with respect to the alkene component 1 (Fig. 2A). With 3-phenylpropanol (1b) as the representative primary alcohol partner, a variety of alkyl and aryl acrylates reacted efficiently and furnished the desired ketone products (3b–3m) in moderate to excellent yields (49–93%). Notably, this protocol showed good compatibility with small rings (3f and 3g), heterocycle (3k) and tertiary alcohol (3l). In addition, high chemoselectivity was evidenced by the preference for activating α-C–H bonds of alcohol in the presence of α-oxy C–H bonds in esters (3b–3k) and benzylic C–H bonds (3h–3k), despite that such C–H bonds are readily susceptible to TBADT-mediated HAT27. Other carbonyl activated alkenes, acrylamides and ethyl vinyl ketone, are also viable substrates to produce β-keto-amides (3m and 3n) and 1,4-diketone (3p) in good yields. Besides, acrylonitrile, phenyl vinyl sulfone, diethyl vinylphosophonate, and pinacol vinylboronate were converted to diversely functionalized ketones (3q–3t) in good yields, providing convenient handles for down-stream transformations. Remarkably, multi-substituted alkenes in different substitution patterns were demonstrated to be suitable substrates (3u–3z), which highlights a particular strength of this strategy for its relative insensitivity to steric encumbrance compared to state-of-the-art methods30,32.

A activated alkenes; B aryl alkenes; C primary alcohol; D polyols. aConditions: 1 (0.25 mmol), 2 (0.6 mmol), TBADT (4 mol%), CoCl2 (1 mol%), (4-MeO-C6H4)3P (2 mol%), acetonitrile (0.5 mL), LEDs (2 W, 385 nm) irradiation for 20 h. isolated yields. bNMR yield using mesitylene as the internal standard. cCoCl2 (2 mol%), (4-MeO-C6H4)3P (4 mol%). d2 (0.85 mmol). eCoCl2 (1 mol%), dppe (1 mol%). f40 h. gCo2(CO)8 (0.5 mol%), PPh3 (4 mol%). h48 h. i2 (0.88 mmol), Co2(CO)8 (0.5 mol%), PCy3 (3 mol%). j2 (1.13 mmol), TBADT (4 mol%). k2 (1.38 mmol), TBADT (6 mol%).
Aryl alkenes are challenging substrates for the nucleophilic ketyl radicals, due to the slow transformation of the adduct radicals stemming from their low electrophilicty29,30,31,32,37,38. Cobalt hydride has proven effective for hydrogen transfer to alkyl carbon radicals39. Therefore, through careful selection of cobalt catalyst precursors and ligands, the conversion of primary alcohols and aryl alkenes to ketones can be facilitated. By utilizing the combination of Co2CO8 and PPh3, we successfully extended the alkene scope to styrenes to produce β-aryl ketones (4a–4q) in satisfactory to high yields (47–93%) with the tolerance of various functionalities (Fig. 2B). The enhanced reactivity is presumably due to the proficiency of cobalt carbonyl complexes with phosphine ligands in HAT processes40. In general, electron-rich styrenes gave lower yields of ketones than the electron-neutral and -poor counterparts. Vinylpyridines were also suitable substrates despite their coordinating character giving high yields of ketones (4i and 4j). Moreover, α-methyl and -phenyl styrenes performed well and provided β-disubstituted ketones (4k and 4l) in high yields. However, α-trifluoromethyl styrene gave a moderate yield of 4m, possibly due to competitive C-F bond cleavage41. β-Methylstyrene also successfully engaged in reaction to furnish 4n in moderate yield. A pair of regioisomers (4o, rr = 4.9:1) were obtained from methyl cinnamate with preferred radical attack at the α-position of cinnamate. Simple primary alcohols, ethanol and propanol, were demonstrated as good reaction partners with styrene and delivered 4q and 4q in excellent yields.
Next, we turned to investigate the scope of the alcohol component (Fig. 2C). A wide range of primary alcohols were competent coupling partners with methyl acrylate affording desired γ-keto-ester (5a–5u) in acceptable to high yields (33–85%), which is not obviously influenced by increasing steric congestion. Primary alcohols with a variety of functional groups, such as esters (5h and 5i), tertiary alcohol (5j), ketone (5k), aryl halides (5n and 5o), ether (5m), trifluoromethyl (5p), thioether (5q), and pyridyl (5r) were transferred to the corresponding ketones in satisfactory yields. The reaction with 2-aryl-ethanols furnished 5s and 5t in moderate yields even in the presence of more excess alkene, presumably owing to the high tendency of the radical intermediate to undergo dehydroformylation42. Primary alcohols derived from the natural product geraniol and the anti-inflammatory drug oxaprozin could be transformed into ketones (5e and 5u) in high yields.
α,ω-Diols are renewable high-value commodity chemicals in food, pharmaceutical, and polymer industries, such as 1,4-butadiol, 1,6-hexadiol, 1,8-octadiol, and so on43,44. These diols could be transformed into diketones by this protocol in satisfactory yields (6a–6d). Secondary alcohols are susceptible to TBADT-catalyzed HAT to react with activated alkenes to provide tertiary alcohols or to underdo dehydrogenation to produce ketones29,31,34. Notably, the combination of Co2CO8 and PCy3 as cobalt catalyst could selectively catalyze dehydrogenation of secondary alcohol without influence on the reaction of primary alcohol and alkene, which transformed diols to diketones (6e–6o) in moderate to high yields (49–86%). The diol from jasmone was converted to 6l in 86% yield. Even triketone 6p could be obtained from 1,3,5-pentanetriol though in diminished yield, which showed the great potential of this method in functionalizing polyols.
To emphasize the synthetic utility of this method, we conducted scale-up reactions with representative substrates, which basically maintained their performance (4p, 5l, and 6g) (Fig. 3A). Moreover, the versatility of β-functionalized ketones as building blocks in organic synthesis was highlighted by a series of derivatization reactions (Fig. 3B). 5l could be reduced to 1,4-diol (7) in quantitative yield and subsequently cyclized to 2-phenethyltetrahydrofuran (8) in 83% yield45. Hydrolysis of 5l delivered 91% of β-keto-acid (9). Condensation of β-keto-acid (9) with D- or L-phenylglycinol gave a pair of bicyclic lactam enantiomers 10 and 10’ in high yields and ees, respectively46. 9 could undergo cyclization with hydrazine hydrate to give dihydropyridazinones (11) in 79% yield. Next, 11 underwent halogenation and HCl elimination readily generating pyridazinone (12) in 79%47. In addition, β-keto-ester 3d was converted into the versatile γ-vinyl-γ-lactone (13) skeleton48. Furthermore, a copper-catalyzed [3 + 3] annulation was applied to 4i giving the pyrimidine 1449.

Scale-up reactions (A), Derivatizations (B), mechanistic experiments (C) and proposed reaction mechanism (D). (See supplementary information for detailed information. a1H-NMR yield using mesitylene as the internal standard. bGC yields with dodecane as the internal standard).
To evaluate the validity of our hypothesis, we performed a series of mechanism investigations. The reaction with 1b and acrylonitrile (2q) under standard conditions was halted before full conversion (Fig. 3C, 1). 1H-NMR analysis of the reaction mixture recognized a secondary alcohol (int, 28%) and a tertiary alcohol (3q’, 11%) besides the ketone product (3q, 26%). In a control experiment under otherwise standard conditions without cobalt catalyst, a comparable yield of int (22%) and a lower yield of 3q’ (6%) with a much lower yield of 3q (6%) were detected (Fig. 3C, 1). These results disclose the important role of the cobalt catalyst in ketone production and the existence of a competing alkylation of the secondary alcohol intermediate (3q’)31,38. In addition, the absence of detectable aldehyde and related byproducts implies that a chemoselective α-hydroxyl alkyl C–H bond addition to alkene is in charge in this protocol. To get insight into the reaction process, a time profile of the reaction with 1b and methyl acrylate was obtained, which showed the secondary alcohol intermediated accumulated at the early stage and consumed as the reaction approaches completion (Supplementary Fig. 10). When int was subjected to standard reaction conditions, only 10% of 3q was formed, revealing a difficult direct dehydrogenation (Fig. 3C, 2)29,34. On the other hand, adding alkene 2p to the reaction led to 67% of ketone 3q and 16% of 3q’, suggesting an essential role of alkene in promoting dehydrogenation of the secondary alcohol (Fig. 3C, 2). While, removal of the cobalt catalyst from the reaction of int and 2p resulted in a dramatically decreased yield of 3p and an increased yield of 3p’, which again emphasizes the importance of cobalt in ketone formation (Fig. 3C, 2). Careful analysis of the template reaction with 1a and 2a showed that n-butyl propionate (2a’) was produced in 68% yield, which indicates the excess alkene functioning as the hydrogen acceptor (Fig. 3C, 3). Furthermore, analysis of the gas phase revealed dihydrogen, suggesting a hydrogen evolution also involved (Supplementary Fig. 9). By varying the loading of catalysts, we found that cobalt is more efficient in facilitating the alkene hydrogenation, while TBADT is more likely to promote dihydrogen formation, presumably via the disproportionation of the reduced photocatalyst33.
(Fig. 3C, 2)33. The deuterium scrambling experiment with 1b and 2j in the presence of D2O transferred deuterium equally to β-C–H bond in 3j and α- and β-C–H bonds in alkene hydrogenation product 2j’ (Fig. 3C, 4). When the primary alcohol deuterated on the hydroxyl group (1c-D) or on the α-carbon of the hydroxyl group (1b-D) was subjected to standard conditions with 2j, the same deuterium distribution with unbiased deuteration levels was also detected in the corresponding products (Fig. 3C, 5, 6). The deuterium scrambling may arise from the acidic nature of the protonated TBADT50. Moreover, compared to 3j (73%) from the reaction with 1b, a dramatically reduced yield of 3j-D (28%) from that with 1b-D, indicating the C–H bond cleavage may be related to the rate-limiting step. The reaction with cyclopropylmethanol (1v) and t-butyl acylate (2c) provided both ring-retaining (5v) and ring-opening (5v’) coupling products, implying the existence of α-hydroxyl carbon radical (Fig. 3E). In addition, light on-off experiment showed the necessity of continuous photoirradiation to drive the reaction (Supplementary Fig. 8).
Based on the above results, a plausible reaction mechanism is described in Fig. 3–F. Initially, the decatunstate photocatalyst ([W]4−) is excited by violet light forming the relaxed excited state of triplet multiplicity (wO) after intersystem crossing51. Then, the α-hydroxyl C–H bond of the primary alcohol (1) is activated by wO (E(wO/[W]5−) = 2.44 V vs SCE) via selective HAT, affording the nucleophilic ketyl radical (R1) and reduced decatungstate (H+[W]5−)31,33,38. Subsequently, R1 undergoes nucleophilic addition to alkene 2 leads to γ-hydroxyl carbon radical (R2)30,31,32,38. With an electron-withdrawing group in the α-position, R2 possesses a relatively low reduction potential (E(·CH2(CH3)CO2Et/–CH2(CH3)CO2Et)= −0.66 V vs SCE in MeCN) and can be reduced to carbon anion by H+[W]5− (E([W]4−/[W]5−) = −0.96 V vs SCE), following by protonation to generate the secondary alcohol intermediate (int) (path a)51,52,53,54. Alternatively, when styene is applied, the reductive potential of benzyl carbon radical (int, R=Aryl) is relatively high (E(PhCH2·/PhCH2−) = −1.43 V vs SCE in MeCN) and difficult to be directly reduced by H+[W]5-55. In this case, the HAT from Co-H (A) to R2 could lead to int (path b). Next, the HAT between wO and int forms H+[W]5− and the ketyl radical R3 (E(Me2C·OH/Me2C+OH) = −0.61 V vs SCE), which can be oxidized by [W]4− producing ketone 329,31,34,38. Meanwhile, the accumulation of H+[W]5− could hinder ketone production and facilitate the formation of tertiary alcohol 3’ from R3 addition to 2. On the other hand, H+[W]5− could be oxidized by cobalt catalyst (B) regenerating the ground state decatungstate [W]4− and producing Co-H (A) after protonation56. A would coordinate with 2 and afford alkyl cobalt (C) via migratory insertion. Protonation of C furnishes the alkene hydrogenation product 2’ and Co(D), which can be reduced by H+[W]5− completing the cobalt-catalytic cycle and regenerate the photocatalyst. It is also possible that cobalt catalyst B could abstract hydrogen atom from R3 and deliver ketone 3 and Co-H (A). The hydrogen evolution may attribute to the disproportionation of H+[W]5-33.
In conclusion, this dual photo/Co catalysis enables selective manipulation of the reactivity of ketyl radicals, which allows the assembly of ketone functionality from broadly available primary alcohols and alkenes. The cobalt catalyst is crucial to efficient catalysis and broad substrate scope across both alcohols and alkenes. We expect the extension of this catalytic system will enable new and streamlined synthetic pathways and unlock a variety of transformations of primary alcohols.
Methods
General procedure for ketone synthesis from primary alcohols and alkenes
The stock solution of cobalt catalyst was prepared according to the following procedure: in an argon-filled glovebox, a 20 mL vial was charged with CoCl2 (64.9 mg, 0.5 mmol) and tris(4-methoxyphenyl) phosphine (352.4 mg, 1.0 mmol), and 10 mL of acetonitrile. The resulting mixture was stirred at ambient temperature. A clear solution was obtained after stirring for several hours. The solution was stored in the glovebox at ambient temperature.
In an argon-filled glovebox, an oven-dried reaction tube equipped with a magnetic stir-bar was added TBADT (33.2 mg, 0.01 mmol, 4 mol%), alcohol (0.25 mmol, 1.0 equiv), alkene (0.6 mmol, 2.4 equiv), 0.45 mL acetonitrile as solvent, and 50 μL of the stock solution of cobalt and ligand. The reaction tube was sealed with a cap with PTFE lining, fixed on a stir plate, and irradiated with 385 nm light for 20 hours at ambient temperature. Then, the reaction mixture was diluted with ethyl acetate, filtered through celite, and concentrated under reduced pressure. The resulting residue was purified by flash column chromatography on silica gel using hexane/ethyl acetate to give the desired products.
Data availability
All data are available in the manuscript or Supplementary Information. Correspondence and requests for materials should be addressed to Jing Zhang.
References
Ertl, P. & Schuhmann, T. A systematic cheminformatics analysis of functional groups occurring in natural products. J. Nat. Prod. 82, 1258–1263 (2019).
Falbe, J., Bahrmann, H., Lipps, W., Mayer, D. & Frey, G. D. In: Ullmann’s encyclopedia of industrial chemistry (2012).
Siegel, H. & Eggersdorfer, M. In: Ullmann’s Encyclopedia of industrial chemistry (2012).
Lawrence, J.N. Aldehydes and ketones. J. Chem. Soc. Perkin Trans. 1, 1739–1750 (1998).
Martín, R., Romea, P., Tey, C., Urpí, F. & Vilarrasa, J. Simple and efficient preparation of ketones from morpholine amides. Synlett 12, 1414–1416 (1997).
Dieter, R. K. Reaction of acyl chlorides with organometallic reagents: a banquet table of metals for ketone synthesis. Tetrahedron 55, 4177–4236 (1999).
Davies, S. G., Fletcher, A. M. & Thomson, J. E. Direct asymmetric syntheses of chiral aldehydes and ketones via N-acyl chiral auxiliary derivatives including chiral Weinreb amide equivalents. Chem. Commun. 49, 8586–8598 (2013).
Douchez, A., Geranurimi, A. & Lubell, W. D. Applications of γ,δ-unsaturated ketones synthesized by copper-catalyzed cascade addition of vinyl grignard reagents to esters. Acc. Chem. Res. 51, 2574–2588 (2018).
Dobereiner, G. E. & Crabtree, R. H. Dehydrogenation as a substrate-activating strategy in homogeneous transition-metal catalysis. Chem. Rev. 110, 681–703 (2010).
Watson, A. J. A. & Williams, J. M. J. The give and take of alcohol activation. Science 329, 635–636 (2010).
Verheyen, T. et al. Ketone synthesis by a nickel-catalyzed dehydrogenative cross-coupling of primary alcohols. J. Am. Chem. Soc. 141, 6869–6874 (2019).
Isbrandt, E. S., Nasim, A., Zhao, K. & Newman, S. G. Catalytic aldehyde and alcohol arylation reactions facilitated by a 1,5-diaza-3,7-diphosphacyclooctane ligand. J. Am. Chem. Soc. 143, 14646–14656 (2021).
Suchand, B., Sreenivasulu, C. & Satyanarayana, G. Palladium-catalyzed direct oxidative coupling of iodoarenes with primary alcohols leading to ketones: application to the synthesis of benzofuranones and indenones. Eur. J. Org. Chem. 2019, 4832–4843 (2019).
Williams, V. M., Leung, J. C., Patman, R. L. & Krische, M. J. Hydroacylation of 2-butyne from the alcohol or aldehyde oxidation level via ruthenium catalyzed C–C bond forming transfer hydrogenation. Tetrahedron 65, 5024–5029 (2009).
Zhao, X., Li, B. & Xia, W. Visible-light-promoted photocatalyst-free hydroacylation and diacylation of alkenes tuned by NiCl2·DME. Org. Lett. 22, 1056–1061 (2020).
Hatanaka, S., Obora, Y. & Ishii, Y. Iridium-catalyzed coupling reaction of primary alcohols with 2-alkynes leading to hydroacylation products. Chem. Eur. J. 16, 1883–1888 (2010).
Shibahara, F., Bower, J. F. & Krische, M. J. Diene hydroacylation from the alcohol or aldehyde oxidation level via ruthenium-catalyzed C−C bond-forming transfer hydrogenation: synthesis of β,γ-unsaturated ketones. J. Am. Chem. Soc. 130, 14120–14122, (2008).
Spinello, B. J., Wu, J., Cho, Y. & Krische, M. J. Conversion of primary alcohols and butadiene to branched ketones via merged transfer hydrogenative carbonyl addition–redox isomerization catalyzed by rhodium. J. Am. Chem. Soc. 143, 13507–13512 (2021).
Cook, A. & Newman, S. G. Alcohols as substrates in transition-metal-catalyzed arylation, alkylation, and related reactions. Chem. Rev. 124, 6078–6144 (2024).
Guo, X., Wu, Y., Li, G. & Xia, J.-B. Redox-triggered ruthenium-catalyzed remote C–H acylation with primary alcohols. ACS Catal. 10, 12987–12995 (2020).
Yang, P.-F. & Shu, W. Direct synthesis of mono-α-arylated ketones from alcohols and olefins via Ni-catalyzed oxidative cross-coupling. Org. Lett. 22, 6203–6208 (2020).
Denichoux, A., Fukuyama, T., Doi, T., Horiguchi, J. & Ryu, I. Synthesis of 2-hydroxymethyl ketones by ruthenium hydride-catalyzed cross-coupling reaction of α,β-unsaturated aldehydes with primary alcohols. Org. Lett. 12, 1–3 (2010).
Jun, C.-H., Huh, C.-W. & Na, S.-J. Direct synthesis of ketones from primary alcohols and 1-alkenes. Angew. Chem. Int. Ed. 37, 145–147 (1998).
Li, H.-S., Guo, G., Zhang, R.-Z. & Li, F. Rhodium-catalyzed synthesis of α,β-unsaturated ketones through sequential C–C coupling and redox isomerization. Org. Lett. 20, 5040–5043 (2018).
Spinello, B. J., Strong, Z. H., Ortiz, E., Evarts, M. M. & Krische, M. J. Intermolecular metal-catalyzed C–C coupling of unactivated alcohols or aldehydes for convergent ketone construction beyond premetalated reagents. ACS Catal. 13, 10976–10987 (2023).
Protti, S., Fagnoni, M. & Ravelli, D. Photocatalytic C-H activation by hydrogen-atom transfer in synthesis. ChemCatChem 7, 1516–1523 (2015).
Ravelli, D. et al. functionalization by decatungstate anion photocatalysis: synergistic control by polar and steric effects expands the reaction scope. ACS Catal. 8, 701–713 (2018).
Capaldo, L., Ravelli, D. & Fagnoni, M. Direct photocatalyzed hydrogen atom transfer (HAT) for aliphatic C–H bonds elaboration. Chem. Rev. 122, 1875–1924 (2022).
Dondi, D., Fagnoni, M. & Albini, A. Tetrabutylammonium decatungstate-photosensitized alkylation of electrophilic alkenes: convenient functionalization of aliphatic C-H bonds. Chem. Eur. J. 12, 4153–4163 (2006).
Jeffrey, J. L., Terrett, J. A. & MacMillan, D. W. C. O-H hydrogen bonding promotes H-atom transfer from C-H bonds for C-alkylation of alcohols. Science 349, 1532–1536, (2015).
Fukuyama, T. et al. Site-selectivity in TBADT-photocatalyzed C(sp3)–H functionalization of saturated alcohols and alkanes. Chem. Lett. 47, 207–209 (2018).
Merkens, K., Sanosa, N., Funes-Ardoiz, I. & Gómez-Suárez, A. Accessing α-amino ketyl radicals from β-amino alcohols via chemoselective hydrogen atom transfer catalysis. ACS Catal. 12, 13186–13192 (2022).
Yamase, T., Takabayashi, N. & Kaji, M. Solution photochemistry of tetrakis(tetrabutylammonium) decatungstate(VI) and catalytic hydrogen evolution from alcohols. J. Chem. Soc. Dalton Trans. 793–799 (1984).
West, J. G., Huang, D. & Sorensen, E. J. Acceptorless dehydrogenation of small molecules through cooperative base metal catalysis. Nat. Commun. 6, 10093–10100 (2015).
Zhong, J.-J., To, W.-P., Liu, Y., Lu, W. & Che, C.-M. Efficient acceptorless photo-dehydrogenation of alcohols and N-heterocycles with binuclear platinum(II) diphosphite complexes. Chem. Sci. 10, 4883–4889 (2019).
Fuse, H., Mitsunuma, H. & Kanai, M. Catalytic acceptorless dehydrogenation of aliphatic alcohols. J. Am. Chem. Soc. 142, 4493–4499 (2020).
Prieto, A. & Taillefer, M. Visible-light decatungstate/disulfide dual catalysis for the hydro-functionalization of styrenes. Org. Lett. 23, 1484–1488 (2021).
Yamada, K. et al. Cooperative polar/steric strategy in achieving site-selective photocatalyzed C(sp3)−H functionalization. Chem. Eur. J. 23, 8615–8618 (2017).
Ai, W., Zhong, R., Liu, X. & Liu, Q. Hydride transfer reactions catalyzed by cobalt complexes. Chem. Rev. 119, 2876–2953 (2019).
Hebrard, F. & Kalck, P. Cobalt-catalyzed hydroformylation of alkenes: generation and recycling of the carbonyl species, and catalytic cycle. Chem. Rev. 109, 4272–4282, (2009).
Tian, F., Yan, G. & Yu, J. Recent advances in the synthesis and applications of α-(trifluoromethyl)styrenes in organic synthesis. Chem. Commun. 55, 13486–13505 (2019).
Kolb, D. et al. Photocatalytic dehydroformylation of benzyl alcohols to arenes. ChemPhotoChem 7, e202300167 (2023).
Vivek, N. et al. Recent advances in microbial biosynthesis of C3 – C5 diols: Genetics and process engineering approaches. Bioresour. Technol. 322, 124527 (2021).
Kruis, A. J. et al. Microbial production of short and medium chain esters: enzymes, pathways, and applications. Biotechnol. Adv. 37, 107407 (2019).
Ramachandran, P. V., Nair, H. N. G. & Gagare, P. D. One-pot asymmetric synthesis of 2- and 2,3-disubstituted tetrahydrofuran derivatives. J. Org. Chem. 77, 5394–5398 (2012).
Li, S., Zhang, J., Li, H., Feng, L. & Jiao, P. Preparation and application of amino phosphine ligands bearing spiro[indane-1,2’-pyrrolidine] backbone. J. Org. Chem. 84, 9460–9473 (2019).
Abouzid, K. & Bekhit, S. A. Novel anti-inflammatory agents based on pyridazinone scaffold; design, synthesis and in vivo activity. Bioorg. Med. Chem. 16, 5547–5556, (2008).
Nallasivam, J. L. & Fernandes, R. A. Pd-catalyzed site-selective mono-allylic substitution and bis-arylation by directed allylic C–H activation: synthesis of anti-γ-(Aryl,Styryl)-β-hydroxy acids and highly substituted tetrahydrofurans. J. Am. Chem. Soc. 138, 13238–13245 (2016).
Zhan, J.-L., Wu, M.-W., Chen, F. & Han, B. Cu-catalyzed [3+3] annulation for the synthesis of pyrimidines via β-C(sp3)–H functionalization of saturated ketones. J. Org. Chem. 81, 11994–12000 (2016).
Yamase, T. & Usami, T. Photocatalytic dimerization of olefins by decatungstate(VI), [W10O32]4–, in acetonitrile and magnetic resonance studies of photoreduced species. J. Chem. Soc. Dalton Trans. 1988, 183–190 (1988).
Waele, V. D., Poizat, O., Fagnoni, M., Bagno, A. & Ravelli, D. Unraveling the key features of the reactive state of decatungstate anion in hydrogen atom transfer (HAT) photocatalysis. ACS Catal. 6, 7174–7182 (2016).
Dondi, D., Fagnoni, M., Molinari, A., Maldotti, A. & Albini, A. Polyoxotungstate photoinduced alkylation of electrophilic alkenes by cycloalkanes. Chem. Eur. J. 10, 142–148 (2004).
Merrill, G. N., Dahlke, G. D. & Kass, S. R. β-cyanoethyl anion: lusus naturae. J. Am. Chem. Soc. 118, 4462–4468 (1996).
Bortolamei, N., Isse, A. A. & Gennaro, A. Estimation of standard reduction potentials of alkyl radicals involved in atom transfer radical polymerization. Electrochim. Acta 55, 8312–8318 (2010).
Wayner, D. D. M., McPhee, D. J. & Griller, D. Oxidation and reduction potentials of transient free radicals. J. Am. Chem. Soc. 110, 132–137 (1988).
Cao, H. et al. Photoinduced site-selective alkenylation of alkanes and aldehydes with aryl alkenes. Nat. Commun. 11, 1956–1966, (2020).
Acknowledgements
We thank the National Natural Science Foundation of China (22071185; 22271224), the Fundamental Research Funds for the Central Universities (2042019kf0008), and Wuhan University startup funding for financial support.
Author information
Authors and Affiliations
Contributions
G.J. and J.Z. designed and directed the project. G.J. and X.C. performed and analyzed the experiments. G.J. and J.Z. co-wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ji, G., Chen, X. & Zhang, J. Direct ketone synthesis from primary alcohols and alkenes enabled by a dual photo/cobalt catalysis. Nat Commun 15, 6816 (2024). https://doi.org/10.1038/s41467-024-51190-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-51190-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
