
Abstract
The sluggish kinetics of Volmer step in the alkaline hydrogen evolution results in large energy consumption. The challenge that has yet well resolved is to control the water adsorption and dissociation. Here, we develop biaxially strained MoSe2 three dimensional nanoshells that exhibit enhanced catalytic performance with a low overpotential of 58.2 mV at 10 mA cm−2 in base, and long-term stable activity in membrane-electrode-assembly based electrolyser at 1 A cm−2. Compared to the flat and uniaxial-strained MoSe2, we establish that the stably adsorbed OH engineer on biaxially strained MoSe2 changes the water adsorption configuration from O-down on Mo to O-horizontal on OH* via stronger hydrogen bonds. The favorable water dissociation on 3-coordinated Mo sites and hydrogen adsorption on 4-coordinated Mo sites constitute a tandem electrolysis, resulting in thermodynamically favorable hydrogen evolution. This work deepens our understanding to the impact of strain dimensions on water dissociation and inspires the design of nanostructured catalysts for accelerating the rate-determining step in multi-electron reactions.
Similar content being viewed by others


Introduction
Water electrolysis to green hydrogen under alkaline condition is regarded as one of the most effective approaches to address the energy crisis and environmental pollution associated with fossil fuels1,2. Currently, one of the main obstacles for alkaline water electrolysis is the large reaction barrier for the water adsorption and dissociation, and adsorption/desorption of intermediates, which makes the alkaline hydrogen evolution reaction (HER) kinetics more sluggish and complex than HER in acidic solutions3,4. Especially for the Volmer step (H2O + * + e− → H* + OH−), it consumes most of input energy due to the unfavorable water adsorption configuration and dissociation energy barrier. In addition, hydroxide (OH−) has been suggested to impact activity, but the exact role of adsorbed OH- in the reaction mechanism is unclear5,6. It is thus important to gain fundamental insight into the electrolysis mechanism and develop effective means to manipulate the surface process.
An electrolyte comprises of various types of cations and anions; some originate from the precursors of the electrolyte, while others from ions precipitated from the catalyst or from surface reconstruction of the catalyst7,8,9,10,11. Metal ions, especially for the alkali-metal cations, are generally hydrated with water molecules by electrostatic attraction, which will determine the water dissociation process on active sites12,13,14. Moreover, oxygen-containing anions adsorbed onto the catalyst surface can modulate the localized electronic structure of reaction sites15,16,17. However, at present, it is not yet possible to actively control the adsorption behavior. Hydroxide is the most abundant anion in alkaline solutions and could directly participate in catalytic reactions. Yet study on its intriguing dual nature in alkaline electrolysis remains scarce. We ask a fundamental question: how does hydroxide affect alkaline water electrolysis?
Strain has been proven an effective way to enhancing the intrinsic catalytic activity18,19,20. Most strain-related research focus solely on in-plane tension and compression20,21,22,23,24 or bending tension25,26, resulting in limited control on local environment of active sites. Recently, Su et al. reported the positive influence of uniaxial bending strain on single atom catalysis, in which carbon nanotubes served the substrates for inducing optimum local geometries and electronic structures through molecular curvature18. Local strain engineering has been a potential route to control the band structure of two-dimensional transition metal dichalcogenides (2D TMDs) and tailor the optoelectronic properties on the nanoscale. For instance, bending the surface of 2D TMDs can generate vacancies and enhance electronic field, which is favorable for catalytic performance15,23,27. Moreover, it has been shown that tensile strain will lead to a higher degree of electron accumulation around the metal single atoms, thereby accelerating the electron interactions between d and s/p orbitals25. Inspired by these advancements, we attempt to investigate the impact of strain dimensions on reaction pathways and kinetics, which remains scarcely explored. However, strain in TMDs is usually inherited from deformed substrates (flexible or rigid) or carbon templates, which is difficult to control the dimensions of 2D TMDs15,23,25,27. To realize biaxial train, the first hurdle is controllable synthesis of such catalyst materials. We thus aim to overcome the current synthesis limitations to design 2D materials with various strain dimensions (zero strain, uniaxial strain, and biaxial strain), which will render a route to investigate how hydroxide affects the Volmer process of water electrolysis. This endeavor will add a new dimension to tuning the intrinsic activity of the layered semiconductor catalysts and potentially precious metal atoms.
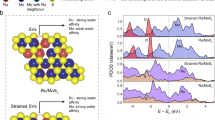
Here, we develop a template method to synthesize flat, uniaxial, and biaxial strained MoSe2, and thoroughly investigate the effects of strain dimensions on the performance of alkaline HER (Fig. 1a–c). The biaxially strained MoSe2 three-dimensional (3D) nanoshells (B-MoSe2) show superior HER catalytic performance with a low overpotential of 58.2 mV at 10 mA cm−2 in alkaline condition and enhanced reaction kinetics, as well as advanced long-term durability in membrane-electrode-assembly (MEA) electrolyser at the industrial current level (1 A cm−2). Operando Raman shows that biaxial strain can effectively induce the transition of hydrated K+ ion water (K+-H2O) to four hydrogen bonded water (4HB-H2O) through desolvation, thereby accelerating dissociation of H2O. Controlled experiments and density functional theory (DFT) calculations reveal that biaxial strain can effectively stabilize OH on the MoSe2 surface in comparison with the flat and uniaxial strained MoSe2, changing the water adsorption configuration from O-down on Mo to O-horizontal on OH* via stronger hydrogen bonds and thus promoting intrinsic HER performance (Fig. 1d). Meanwhile, the onion-like 3D nanoshells endow MoSe2 excellent structural stability. This work explicitly reveals the role of the strain dimensions on the reaction behavior of surficial hydroxide and Volmer process. It also provides a guidance to optimize rate-determining step (RDS) in complex chemical reactions.

a–c Atomic structures. d Water adsorption process on F-MoSe2, U-MoSe2, and B-MoSe2 (from left to right), and the intrinsic alkaline HER activity of MoSe2 with different strain dimensions.
Results
B-MoSe2 3D nanoshells were obtained by a hard template method. Au nanospheres were first synthesized by reducing gold precursors in oil bath system with ethylene glycol (EG) as both reductant and solvent (Supplementary Fig. 1). A given amount of Au nanospheres/graphene (G) mixture underwent sequentially annealing treatment by Mo(CO)6 and Se powder, forming Au@MoSe2/G composite material (Supplementary Figs. 2–4). After removing the Au core, B-MoSe2 were synthesized in high yield of approaching 100%. Transmission electron microscopy (TEM) images emphasize that the product is characterized by monodispersed hollow nanoshells with an average diameter of 56.8 ± 2.5 nm (Fig. 2a and Supplementary Figs. 5–7). High-resolution transmission electron microscopy (HRTEM) image reveals the uniform few-atomic thick layers and the atomically curved MoSe2 from the cross-sectional view (Fig. 2b, c). Aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM) confirms the existence of 2H-MoSe2, which resolves a characteristic hexagonal lattice arrangement of Mo (brighter dots) and Se (darker dots) atoms (Fig. 2d and Supplementary Fig. 8). The Fast Fourier transform (FFT) image indicates the corresponding (100) and (110) planes of 2H phase. Moreover, the biaxial strain endows MoSe2 with abundant Se vacancies evidenced by the yellow circle. A closer inspection of the cross-sectional view of the nanoshell suggests that the typical atomic arrangement of Mo and Se in (002) plane of 2H-MoSe2 with expanded lattice spacing of 0.69 nm. To investigate their unique atomic configuration, the geometric phase analysis (GPA) is adopted to detect the lattice strain changes (Fig. 2f). The obvious evenly color can be observed in the strain mapping, indicating the atomic curvature generates a uniform biaxial strain of ~1.2%. HAADF-STEM and the corresponding EDS mapping images show the uniform distribution of Mo and Se elements (Fig. 2g). For detailed comparison, we further fabricated uniaxially strained (U-MoSe2) and flat (F-MoSe2) MoSe2 without strain using similar strategies (Fig. 2h–k and Supplementary Figs. 9–12). As shown in AC-HAADF-STEM image and corresponding scheme, the U-MoSe2 with an average diameter of 57.1 ± 2.8 nm is composed of curved (002) planes with a lattice spacing of 0.69 nm (Fig. 2i and Supplementary Fig. 7). The uniaxial strain was calculated to be ~1.2%, which is similar with the biaxial one. This enables us to investigate the effect of strain dimension on the HER behaviors. The F-MoSe2 present a flat (002) planes with decreased lattice spacing (0.65 nm). Overall, all these results conclude that MoSe2 with controlled strain dimensions has been successfully prepared.

a–c TEM images under different magnifications. The inset in c is the scheme of B-MoSe2. d Aberration-corrected HAADF-STEM image of B-MoSe2. The inset is the FFT image. e Aberration-corrected HAADF-STEM image from cross-sectional view. f GPA analysis for (d). The change from blue to red in color scale indicates the gradually change from compressive to tensile strain. g STEM and corresponding EDS elemental mapping images. TEM, aberration-corrected HAADF-STEM image, and scheme of (h, i) U-MoSe2 and (j, k) F-MoSe2. The bule and yellow spheres in (d, e, i, k) indicate Mo and Se atoms, respectively. The yellow dash circle is Se vacancy.
The electronic structure and chemical environment of MoSe2 with controlled strain dimensions were investigated by Raman spectrum, X-ray photoelectron spectroscopy (XPS), X-ray absorption spectroscopy (XAS), and electron paramagnetic resonance (EPR). The Raman spectrum confirms the 2H phase of MoSe2 of samples, indicating the strain does not change the crystal phase (Fig. 3a). It is known that the A1g vibration modes are sensitive to the structural information of MoSe2. Compared to the flat MoSe2, the A1g mode shows an obvious blueshift with the increase of strain dimension, which is usually generated from the increased vibration energy27. The high-resolution XPS Mo 3d signals show the double peaks centered at ~228.8 and ~232.2 eV can be assigned to 3d5/2 (Mo4+) and 3d3/2 (Mo4+), respectively (Fig. 3b and Supplementary Fig. 13)28. With increasing the strain dimension from F-MoSe2, U-MoSe2 to B-MoSe2, the binding energy of 3d5/2 gradually blueshifts from 228.8, 228.9 to 229.1 eV, indicating a more delocalized local electric structure. Another peak at about 235.8 eV for F-MoSe2 is attributed to Mo6+ species from the surface oxidation. Moreover, the Se 3d5/2 peaks of B-MoSe2 shift to lower binding energy (54.33 eV) when compared to those of F-MoSe2 (54.54 eV) and U-MoSe2 (54.47 eV), as shown in Fig. 3c. This means Se sites are pumped into abundant electronics by the effect of biaxial strain, which is favorable for the adsorption of proton and thus accelerating HER performance29,30. Moreover, the absence of oxidation peaks in U-MoSe2 and B-MoSe2 suggests their superior anti-oxidation ability and structure stability compared to the F-MoSe2 (Fig. 3b). Possible reason is, an enhanced electron interaction is induced in the strained MoSe2 compared to the flat counterpart, leading to a stronger Mo-Se bonding in the former.

a Raman spectra, (b) Mo 3d and (c) Se 3d XPS spectra. d Mo K-edge XANES spectra. e The fitted average oxidation states and d-band hole counts of Mo from XANES spectra. f The corresponding FT-EXAFS k3χ(R) spectra. g EXAFS wavelet transform analysis. The color scale changed from blue to red indicate the increasing intensity of Mo-Se bonds. h Comparison of the mass normalized EPR spectra.
X-ray absorption near-edge spectroscopy (XANES) was conducted for the MoSe2 composites at the Mo K-edge to study intrinsic electronic structures. As shown in Fig. 3d, all the synthesized MoSe2 samples show the absorption edges between the MoSe2 and MoO3 references, implying that their average valence of Mo species between +4 and +6 coincide with the XPS result. The peaks at ~20,010 eV are mainly due to the electron transition between 1s and 4d orbital in Mo-anion configuration ([MoO6] in MoO3 and [MoSe6] in MoSe2)31,32. With increasing the strain dimension from F-MoSe2, U-MoSe2 to B-MoSe2, this peak gradually grows, indicating the enhanced electron interaction between Mo and Se. Moreover, the adsorption edge of B-MoSe2 shifts to higher energy compared to F-MoSe2 and U-MoSe2, which is due to the strong electronic delocalization by biaxial strain effect. The white-line intensity of B-MoSe2 is higher than that of F-MoSe2 and U-MoSe2, and the oxidation states can be estimated to be +4.17 for F-MoSe2, +4.34 for U-MoSe2, and +4.45 for B-MoSe2, using MoSe2 (4d2) and MoO3 (4d0) as references (Fig. 3e). Moreover, the d-band hole number for F-MoSe2, U-MoSe2, and B-MoSe2 was estimated to be 8.07, 8.34, and 8.45, respectively, indicating the variation of the unoccupied Mo 4d-orbitals.
In addition, the extended X-ray adsorption fine structure (EXAFS) (Fig. 3f) and the corresponding wavelet transform (Fig. 3g) both show gradual decrease in intensity. As listed in Supplementary Table 1, the Mo-Se coordination numbers of F-MoSe2, U-MoSe2, and B-MoSe2 are 5.9, 4.4, and 4.1, respectively, which are lower than that (6) of MoSe2 foil (Supplementary Fig. 14). Such a trend is mainly due to the increased strain dimension of MoSe2, resulting in the breaking of Mo-Se bonds and thus creating more Se vacancies. Moreover, compared with F-MoSe2, there appears a significant shift of Mo-Mo bonds to longer location in R-space for U-MoSe2 and B-MoSe2, which can be attributed to the tensile strain on the surface and the enlarged Mo-Mo bond length. Se vacancies are important factors for the HER activity of strained MoSe2. Hence, electron paramagnetic resonance (EPR) was further performed to investigate the condition of Se vacancies. The EPR signal intensity at g = 2.009 of F-MoSe2 approaches 0, while those of U-MoSe2 and B-MoSe2 are drastically enhanced. Moreover, even the tensile strain degree is similar, B-MoSe2 exhibits the higher concentration of Se vacancies than U-MoSe2 (Fig. 3h). Based on the above fine structure analysis, the tensile strain effect could vary the oxidation state, d band holes, and vacancies of atoms as well as the Mo-Mo bond distance. At the same strain degree, this effect is more immense in case of biaxial strain.
To investigate the stabilized OH engineer for accelerating alkaline hydrogen evolution, three types of MoSe2 were initially evaluated to explore the underlying influence of tensile strain dimension on the catalytic activity by a three-electrode cell in 1 M KOH aqueous electrolyte. All the polarization curves were calibrated against reversible hydrogen electrode (RHE) as shown in Supplementary Fig. 16. Linear sweep voltammograms (LSV) obtained for F-MoSe2, U-MoSe2, B-MoSe2, and commercial Pt/C (20 wt%) are presented in Fig. 4a. B-MoSe2 shows the highest HER catalytic activity, as evident from the early onset potential and lowest overpotentials of only 58.2 and 190.5 mV to deliver current densities of 10 and 100 mA cm−2, respectively, superior to F-MoSe2 (144.3 and 330.1 mV), and U-MoSe2 (101.1 and 231.2 mV) (Fig. 4b). The overpotential of B-MoSe2 at 10 mA cm−2 is much lower than that of the reported work (most of them are higher than 100 mV), indicating superior activity (Supplementary Table 2). The reaction kinetics were further investigated by Tafel analysis (Fig. 4c). B-MoSe2 demonstrates the fastest HER reaction kinetics confirmed by the smallest Tafel slope value of 97.1 mV dec−1, much lower than that of F-MoSe2 (131.8 mV dec−1) and U-MoSe2 (122.4 mV dec−1). Even three catalysts all show the Volmer-Heyrovsky mechanism according to their Tafel slope values, the HER kinetics exhibited by F-MoSe2 and U-MoSe2 are more sluggish compared to that of B-MoSe2, revealing favorable surface electronic modulation effects via the biaxial stain. The lower Tafel slope value of B-MoSe2 indicates a fast water dissociation (Volmer step) behavior (H2O + * + e− → H* + OH−). The decreased energy barrier of the RDS may have substantially accelerated the overall hydrogen evolution rates. The double-layer capacitance (Cdl) measurement was carried out to evaluate the electrochemically active surface areas (ECSAs) of catalysts (Fig. 4d and Supplementary Fig. 17). The B-MoSe2 catalyst displays the highest Cdl value of 34.3 mF cm−2 among the other two samples, implying the highest ECSA and more accessible active sites. Moreover, the catalytic performance was normalized by ECSA, which shows the best intrinsic activity of B-MoSe2 (Supplementary Fig. 18). To further evaluate the intrinsic activity, the turnover frequency (TOF) value for B-MoSe2 at the overpotential of 200 mV was calculated to be 8.7 s−1, which is an order of magnitude higher than that of the U-MoSe2 catalysts (0.79 s−1) and ~2 order of magnitude higher than that of F-MoSe2 (0.09 s−1), showing the most favorable HER kinetics. The electrochemical impedance spectroscopy (EIS) measurements were then performed to track the charge transfer kinetics between H2O and catalyst surface (Fig. 4e). As expected, all MoSe2 samples exhibit the similar uncompensated solution resistance (Rs) value of ~3.0 Ω. Furthermore, the charge-transfer resistance (Rct) of the B-MoSe2 electrode was measured to be 11.6 Ω in Nyquist plot, significantly lower than those of F-MoSe2 (45.2 Ω) and U-MoSe2 (30.5 Ω), suggesting the faster charge transfer kinetics between biaxial strained surface and H2O molecules. The adsorption behavior of OH− on the catalyst surface could govern the thermodynamics and kinetics of alkaline HER33. The CO-stripping measurement was thus applied to assess the electrocatalyst’s capability for adsorbing OH, owing to the CO-oxidation process ignites by the reactive OH* 6,34. It means that the lower the onset potential, the stronger the OH− adsorption capability. By measuring the LSV curves in CO-saturated electrolyte (Fig. 4f), one can find that compared with those on U-MoSe2 and F-MoSe2, the onset potential of CO oxidization on B-MoSe2 has negatively shifted ~40 and ~190 mV, respectively. These significant shifts caused by the strain effect indicate the easier adsorption of OH− on the curved surface of MoSe2 than flat one. More importantly, biaxial strain could generate much lower OH− adsorption energy compared to uniaxial strain. Based on the above comparison results, we have successfully verified that the biaxial strain from B-MoSe2 plays a more significant role in accelerating the kinetics of Volmer step and decreasing OH− adsorption energy in alkaline HER activity. Moreover, we investigated the effect of the magnitude of biaxial strain on HER activity by obtaining MoSe2 nanoshells with other diameters (35.7 ± 3.6 and 115.0 ± 6.0 nm) (see Method and Supplementary Figs. 19, 20). LSV curves show that decreasing the B-MoSe2 diameters results in higher HER activity and reaction kinetics (Supplementary Fig. 21). The B-MoSe2 with diameter of 35.7 ± 3.6 nm displays the highest Cdl compared to the other two samples (Fig. 4d and Supplementary Fig. 21). Hence, a higher magnitude of biaxial strain is beneficial to the HER activity.

a HER polarization curves of F-MoSe2, U-MoSe2, B-MoSe2, and 20 wt% Pt/C. The potential is corrected by an automatic 90% of iR compensation. b Comparison of overpotentials at 10 and 100 mA cm−2, c Tafel plots, (d) half of current density differences (Ja-Jc) plotted against scan rates, and (e) Nyquist plot of electrochemical impedance spectra. charge-transfer resistance (Rct) of the B-MoSe2, U-MoSe2, and F-MoSe2electrode was measured to be 11.6, 30.5, and 45.2 Ω. f LSV curves in CO-saturated 1 M KOH. g Chronopotentiometric test at two current densities. h Durability test of the MEA electrolyser at the current density of 1 A cm−2. The insets in (h) are scheme of MEA electrolyser (left) and LSV curves operated at 25 and 60 °C (right).
The B-MoSe2 catalyst exhibits advanced stability in the alkaline HER. We evaluated the durability through the chronopotentiometric method up to 200 h, during which only a slight decrease in potential occurred at a set current density of 200 mA cm−2 (Fig. 4g). Even at a higher current density of 500 mA cm−2, it still maintained 94.3% after more than 70 h continuous test. After the durability test, the hollow morphology, composition, and electronic structure of B-MoSe2 were evaluated in detail. There are no apparent shifts for the Mo 3d and Se 3d XPS spectra compared to the initial ones (Supplementary Fig. 22). Inductively coupled plasma-mass spectrometry (ICP-MS) indicates the molar ratio of Mo/Se was still approaching 1/2, verifying the stable structures (Supplementary Table 3). In addition, the unchanged hollow structures were confirmed by TEM and HRTEM images (Supplementary Fig. 23). Upon closer examination, it can be observed that the partial surface of B-MoSe2 has become thinner by stripping away certain layers, which leads us to conclude the reason of superior durability. We further conducted the aberration-corrected HAADF-STEM image of a single B-MoSe2 after durability test (Supplementary Fig. 24). The peeling off area is clearly observed. The lattice strain was determined to be ~1.2% by GPA, which is comparable to that of the pristine sample (Supplementary Fig. 24d). In addition to the stronger Mo-Se bonds, the onion-like shell of B-MoSe2 ensures the remarkable stability during catalysis. Catalysis primarily occurs on the external surface of the catalyst. For layered B-MoSe2, it possesses a nearly perfect spherical hollow structure, and each layer can function as an active layer due to the uniformity of biaxial strain. The outermost layer of B-MoSe2 can be stripped away due to structural damage during catalysis. The exposed surface from the inner layers becomes new catalytic active sites. This self-protection process will boost the stability of active structures. To test B-MoSe2 under more practical conditions, we constructed a membrane-electrode-assembly (MEA) based electrolyser, using anion exchange membrane (AEM) as the solid electrolyte and commercial IrO2/Ti as the anodic catalyst (in set in Fig. 4h and Supplementary Fig. 25). A higher temperature could enhance the MEA performance than that at 25 °C. Specifically, cell voltages of only 2.13 and 2.37 V are needed to reach current densities of 1 and 1.5 A cm−2 at an operation temperature of 60 °C, respectively. When the B-MoSe2-based AEM electrolyser runs 100 h at 1 A cm−2, only slight activity loss was observed, better than Pt/C60 and other advanced catalysts (Fig. 4h and Supplementary Table 4)35.
The behavior of interfacial water on electrocatalysts is extremely important for water adsorption/dissociation during alkaline HER. The Operando Raman spectra under different applied potentials was performed to reveal the effects of strain dimensions on the reaction of interfacial water (Fig. 5 and Supplementary Fig. 26). The broad band in the region of 3000–3750 cm−1 can be ascribed to the O-H stretching mode (νO–H) of H2O36. It can be further deconvoluted into three different peaks: four hydrogen bonded water (4HB-H2O), two hydrogen bonded water (2HB-H2O), and hydrated K+ ion water (K+-H2O) (Fig. 5a, b and Supplementary Fig. 27)37. As the applied potentials negatively increases from 0 to −0.5 V, the content of K+-H2O on the surface of B-MoSe2 decreases from 8.9 % to 2.1%, while that of 4HB-H2O increases from 45.7 % to 56.0 %. For the U-MoSe2 and F-MoSe2, the contents of 4HB-H2O and K+-H2O shows negligible changes (Fig. 5c). It can be deduced that the biaxial strain can effectively induce the transition of K+-H2O to 4HB-H2O through desolvation, thereby confirming the accelerated dissociation of H2O and the occurrence of water reduction38. The relationship of the vibrational frequency of adsorbate as a function of applied potentials has been considered to be vibrational Stark effect, which can describe the sensitivity between intermediates and catalyst surfaces12. As shown in Fig. 5d, the Stark tuning rate of 4HB-H2O for B-MoSe2 was evaluated to be 23.2 cm−1/V, which is much higher than those of U-MoSe2 (6.4 cm−1/V) and F-MoSe2 (5.9 cm−1/V). Moreover, the vibrational frequency at −0.3 V of 4HB-H2O on B-MoSe2 (3252.0 cm−1) is lower than that of U-MoSe2 (3258.3 cm−1) and F-MoSe2 (3257.7 cm−1). This result can be explained by the stronger electrostatic interaction and closer distance between the 4HB-H2O and the B-MoSe2 surfaces at negative potentials, compared to the other two surfaces. Such an strong interaction is conducive to the breaking of the O-H bond of surficial water and protons transfer36. Therefore, we may conclude that the biaxial strain can promote the transformation of K+-H2O to 4HB-H2O via desolvation at negative potentials. This mechanism and its effect to alkaline HER performance is schematically illustrated in Fig. 5e, f.

Operando Raman spectra of interfacial water at (a) B-MoSe2 and (b) F-MoSe2. The 4HB-H2O, 2HB-H2O, and K+-H2O present three kinds of O-H stretching modes for H2O. c Stark tuning rates of the νO-H in Raman spectra for 4HB-H2O. d Comparison of the contents for 4HB-H2O and K+-H2O during alkaline HER. Scheme of interfacial water on (e) F-MoSe2 and (f) B-MoSe2. 4HB-H2O, 2HB-H2O, and K+-H2O indicate four hydrogen bonded water, two hydrogen bonded water, and hydrated K+ ion water, respectively. The blue (e) and red (f) shadings present sluggish and enhanced reaction between water and catalysts.
Density functional theory (DFT) calculations were conducted to deep insight to the reaction mechanism. Three strain dimensionalities of 2H-MoSe2 (without strain, with uniaxial strain and biaxial strain) were constructed as the theoretical models, in which the strain was applied to be 1.2% (Supplementary Fig. 28). Considering the practical condition, the Mo coordination numbers of 2H-MoSe2 were specified as 6 (6Mo), 5 (5Mo), 4 (4Mo), and 3 (3Mo) (Supplementary Fig. 29). The OH- species prefer to stabilize at the Se top site of MoSe2 (Supplementary Fig. 30). As the ignition step for HER, the H2O adsorption (\({\Delta {{{\rm{G}}}}}_{{{{{\rm{H}}}}}_{2}{{{\rm{O}}}}}\)), was first systematically investigated (Fig. 6a and Supplementary Fig. 31). The optimization of MoSe2 surface with HO- modification shows the enhanced water adsorption, which is consistent with the experimental results. Moreover, in case of the biaxially strained MoSe2 with the modification of OH- adsorption, the adsorption of H2O strengthens as the coordination number decreases. To be more specific, the \({\Delta {{{\rm{G}}}}}_{{{{{\rm{H}}}}}_{2}{{{\rm{O}}}}}\) of H2O on B-3Mo-OH is as low as −0.10 eV, indicating a spontaneous exothermic reaction (Fig. 6b). Notably, H2O prefers to adsorb onto OH* via the formation of hydrogen bond between OH* and H2O (Fig. 6c). The change of adsorption configuration of H2O from O-down on Mo (B-3Mo) to O-horizontal mode on OH* (B-3Mo-OH) may be responsible for the negative \({\Delta {{{\rm{G}}}}}_{{{{{\rm{H}}}}}_{2}{{{\rm{O}}}}}\).

a Calculated \({\Delta {{{\rm{G}}}}}_{{{{{\rm{H}}}}}_{2}{{{\rm{O}}}}}\) values of various Mo coordination numbers. b Comparation of \({\Delta {{{\rm{G}}}}}_{{{{{\rm{H}}}}}_{2}{{{\rm{O}}}}}\) values between different Mo configurations and strain dimension with (with OH) and without OH− (without OH) adsorption. c Model of H2O adsorption on B-3Mo-OH. d Calculated \({\Delta {{{\rm{G}}}}}_{{{{{\rm{H}}}}}^{*}}\) for U-5Mo-OH, B-5Mo-OH, B-4Mo-OH, and B-3Mo-OH. e, f The H* adsorbed optimized structures of B-4Mo and B-4Mo-OH, respectively. g The charge density difference plot of B-4Mo-OH. h Calculated pDOS of Mo-3d band and H-1s band. i Schematic of the tandem process (coexistence of 3Mo and 4Mo) for alkaline HER on the B-MoSe2. For the name of sample, the number before Mo is the coordination number of Mo; U and B presents uniaxial and biaxial strain, respectively; the OH after Mo indicates the OH adsorption on MoSe2.
As one of the crucial descriptors for HER activity, the \({\Delta {{{\rm{G}}}}}_{{{{{\rm{H}}}}}^{*}}\) values for series of MoSe2 configurations are further screened (Supplementary Fig. 33 and Table 5). One can find that strain, Se vacancies, and OH− modification will influence the hydrogen binding. The \({\Delta {{{\rm{G}}}}}_{{{{{\rm{H}}}}}^{*}}\) of B-4Mo-OH was calculated to be −0.004 eV, which is better than compared samples (Fig. 6d)39. As shown in Fig. 6e, the H species prefer to adsorb and stabilize on the hollow sites bonded with three Mo atoms in B-4Mo. When the OH- ions stabilize on Se sites in B-4Mo-OH, H will preferentially adsorb onto the unsaturated 4Mo sites closest to OH* with a reduced Mo-H bond length of 1.72 Å (Fig. 6f), indicating an enhanced binding energy of H. Additionally, the electronic structures of the Mo species connected to the OH* adsorbed Se sites all undergo alterations (Fig. 6g). The Se site transforms into electron donor to stabilize OH- adsorbates. More electrons are thus transferred from the bridging Mo sites to maintain the charge balance of B-4Mo-OH, leading to substantial changes for electronic structure (Supplementary Table 6). From the projected density of states (pDOS, Fig. 6h), the B-4Mo-OH structure exhibits an stronger hybridization of Mo-3d and H-1s orbitals, thus leading to the strengthened Mo-H bond.
By integrating the results of water and hydrogen adsorption, the B-3Mo-OH structure exhibits remarkable water adsorption capability via O-horizontal configuration, while the B-4Mo-OH structure displays the highest intrinsic hydrogen evolution activity. Considering that both 4Mo and 3Mo structures should coexist in our B-MoSe2, a collaborative promotion mechanism may occur during HER (Fig. 6i). 3Mo sites may accelerate the water adsorption, and the generated H* then preferentially spillover and adsorb onto the 4Mo sites, expediting the hydrogen adsorption/desorption. This tandem process within the B-MoSe2 structure is thus expected to facilitate the catalytic kinetics for alkaline HER.
Discussion
We have developed a series of MoSe2 with tailored strain dimensions to investigate the effect of OH engineer on accelerating alkaline hydrogen evolution, especially on the water dissociation process. Experimentally, compared to the flat or uniaxial MoSe2, the Volmer process of B-MoSe2 is largely accelerated by lower water adsorption and dissociation energy. Our B-MoSe2 thus shows a superior alkaline HER activity with a low overpotential of 58.2 mV at 10 mA cm−2. Meanwhile, the onion-like 3D nanoshells endow MoSe2 with excellent structural stability for the long-term performance in MEA based electrolyser at 1 A cm−2. Systematic Operando Raman and theoretical calculations reveal a highly efficient tandem catalysis mechanism: the stably OH* on B-MoSe2 can change the water adsorption configuration, lower the energy barrier of water dissociation on 3Mo sites, and then accelerates the hydrogen adsorption on 4Mo sites, resulting in thermodynamically favorable alkaline HER process. This work enhances our understanding of the relationship between strain dimensions and alkaline water splitting, which may guide the design of efficient catalysts for multi-electron chemical reactions.
Methods
Chemicals
Chloroauric acid (HAuCl4, 99.9%), silver nitrate (AgNO3, ≥99.8%), ethylene glycol (EG, ACS reagent), poly-(diallyldimethylammonium) chloride (PDDA, Mw = 100,000–200,000, 20 wt % in water), Hexadecyltrimethylammonium chloride (CTAC, >95%) graphene, Molybdenum hexacarbonyl (Mo(CO)6, 98%), Selenium powder (Se, 99.95%), iodine (I, 99.8%), potassium iodide (KI, 99%), Nafion®117 solution (5%), and commercial Iridium Oxide (IrO2, 99.9%) were purchased from Sigma-Aldrich. Potassium hydroxide (KOH, 99.9%) was purchased from Sinopharm Chemical Reagent Co., Ltd. The Carbon fiber paper (CFP, TGP-H-060) was obtained Toray Industries, Inc. and titanium fiber felt (Ti) with a thickness of 0.4 mm was obtained from Zhejiang Jiuyu Technology Co., Ltd. Deionized (DI) water with a resistivity of 18.25 MΩ was obtained by a Milli-Q water purification system. All chemicals were used without further purification.
Synthesis of Au nanospheres, Au@Ag nanorods, and Au nanoplates. A 60 mL EG solution containing HAuCl4, PDDA, AgNO3, and HCl with concentration of 0.5 mM, 25 mM, 0.01 mM, and 1 mM, respectively, was vigorously stirred and then reacted at 220 °C for 2 h in an oil bath to synthesize Au nanospheres with a diameter of 56.8 ± 2.5 nm. For smaller Au nanospheres (37.7 ± 3.6 nm), the synthesis was conducted with the same method above except that 2.5 mM HCl and saturated O2 was introduced into the initial solution before reaction. Au nanospheres with an average diameter of 115.0 ± 6.0 nm were synthesized by decreasing the reaction temperature to 200 °C.
To synthesize Au@Ag nanorods, a 60 mL EG solution containing HAuCl4, PDDA, and AgNO3 with concentration of 0.5 mM, 25 mM, and 1.6 mM, respectively, was vigorously stirred and then reacted at 220 °C for 72 h in oil bath.
To synthesize the Au nanoplates, 8 mL DI water, 1.6 mL of 0.1 M CTAC, 75 μL of 0.01 M KI, 80 μL of 25 mM HAuCl4, and 20 μL of 0.1 M NaOH were mixed to obtain a yellowish solution. 100 μL of 0.05 M ascorbic acid (AA) was then injected into the solution. After that, 10 μL of 0.1 M KOH was injected to the solution. The reaction completed in 10 min. The final products (Au nanoplates) were collected by centrifugation at 14,100 × ɡ and washed repeatedly with deionized water and ultrasound for further use.
Synthesis of B-MSe2, U-MSe2, and F-MSe2. In a typical synthesis, 5 mg of Au based nanoparticles (Au nanospheres, Au@Ag nanorods, or Au nanoplates) were first mixed with 20 mg of graphene in 10 mL of DI water, followed by 1 h of sonication. The mixture was then heated in an oven to completely remove DI water. 20 mg of dried mixture and 10 mg of Mo(CO)6 underwent further annealing treatment in a tube furnace at 550 °C for 0.5 h with the protection of N2 atmosphere to obtained AuMo alloy/graphene. Then the AuMo alloy/graphene was treated with Se powder as selenium source upstream in tube furnace at 550 °C for another 0.5 h in N2 atmosphere, forming Au@MoSe2/graphene structures. After removing Au core of Au@MoSe2 by etching agent of I/KI for 24 h treatment, B-MSe2, U-MSe2, or F-MSe2 was obtained.
Characterizations
Field emission scanning electron microscopy (FESEM) images were taken by a Sirion 200 device. For the transmission electron microscopy (TEM), high-resolution TEM (HRTEM), and energy-dispersive X-ray spectroscopy (EDS) elemental mapping, a transmission electron microscope (FEI, Tecnai G2 F20) was applied with an accelerating voltage of 200 kV. Philips X’pert Pro X-ray diffractometer equipped with Cu Kα radiation (λ = 0.15419 nm) was used to obtain powder X-ray diffraction (XRD) data. An ESCALAB MK II X-ray photoelectron spectrometer was used to measure X-ray photoelectron spectroscopy (XPS) spectra. The elemental composition of the samples was determined using inductively coupled plasma mass spectrometry (ICP-MS). Electron spin resonance (ESR) spectra were obtained using a Bruker EMX plus 10/12 instrument equipped with an Oxford ESR910 Liquid Helium cryostat at the Steady High Magnetic Field Facilities. X-ray absorption spectroscopy (XAS) measurements for the Mo K-edge were carried out at BL14b2, SPring-8 Japan. Data reduction and analysis for XANES and EXAFS were performed using Athena and Artemis software. Operando Raman spectra were conducted on inVia QONTOR with a laser wavelength of 532 nm, a 20× objective lens, and 10 s acquisition time. The three-electrode Operando Raman cell consists of a catalyst supported carbon fiber electrode (working electrode), a saturated Ag/AgCl electrode (reference electrode), and a platinum wire (counter electrode).
Electrochemical Measurements in a three-electrode cell. Electrocatalytic measurements were performed at ambient temperature using an electrochemical workstation equipped with a standard three-electrode system (CHI 760e, Shanghai Chenhua). All the electrochemical tests were conducted at a room temperature of about 23 °C. Initially, 5 mg of obtained MoSe2/G sample was combined with a mixture of 500 μL of DI water, 450 μL of ethanol, and 50 μL of Nafion®117 to create the catalyst ink, and then underwent ultrasonication in ice water for 1 h. Subsequently, 500 μL of this ink was evenly applied to a CFP working electrode (typically 1 cm × 1 cm). The mass loading of the MoSe2 catalyst on the electrode was calculated to be 0.5 mg cm−2. The counter electrode (CE) was a graphite rod with a diameter of 6 mm, and the reference electrode (RE) was a KCl-saturated Ag/AgCl electrode with a salt bridge. For the preparation of 1 M KOH electrolyte, 56.1 g of KOH was added into a 1-liter volumetric flask and then add DI water up to the 1-liter mark. After that, the 1 M KOH electrolyte was transferred from volumetric flask to the plastic container for storing. The electrolyte was an N2-saturated 60 mL of 1.0 M KOH aqueous solution. Long-term stability was assessed using a Hg/HgO reference electrode (RE). Linear sweep voltammetry (LSV) measurements were performed at a scan rate of 2 mV s−1. The electrochemical impedance spectroscopy (EIS) was collected at with a frequency range from 0.01 Hz to 100 kHz with A.C. amplitude of 5 mV. The electrochemical double layer capacitance (Cdl) was conducted with cyclic voltammetry (CV) measurements at various scan rates from 20 to 200 mV s−1 at an interval of 40 mV s−1 in nonreactive region. The durability was evaluated by the constant current chronopotentiometry. The reference electrode was calibrated by a three-electrode electrochemical cell with working electrode (Pt foil), counter electrode (Pt foil), reference electrode (Ag/AgCl), and gas (H2) inlet. In this work, the calibration of reference electrode against reversible hydrogen electrode (RHE) was conducted in H2-saturated 1 M KOH. The cyclic voltammetry (CV) in the voltage range of hydrogen electrocatalysis with the scan rate of 1 mV s−1. The HER CV has two voltage intercepts at zero current. The calibrated E is the average value of two voltages, which was estimated to be −1.004 V for Ag/AgCl electrode and −0.923 for Hg/HgO electrode (Supplementary Fig. 15). The RHE was thus determined by using the following equation:
Electrochemical measurement of MEA electrolyser
The ion-exchange-resin membrane (SELEMION AMN/N Type1, 0.05 cm × 2 cm × 2 cm) was first activated by immersing into 1.0 M KOH for 18 h at 60 °C. The cathode catalyst ink for B-MoSe2/G was prepared by mixing 5 mg of B-MoSe2/G, 400 μL of DI water, 450 μL of ethanol, and 150 μL of Nafion®117 by ultrasonication in ice water for 1 h. Subsequently, 5 mg of B-MoSe2/G was deposited onto a CFP with geometric surface area of 1 × 1 cm2. The catalyst mass loading is thus calculated to be 5 mg cm−2. For the anode, commercial IrO2 (4 mg) was combined with a mixture of 400 μL of DI water, 450 μL of ethanol, and 150 μL of Nafion®117 to create the catalyst ink and followed by ultrasonication in ice water for 1 h. Then, the 500 μL of commercial IrO2 ink was loaded on titanium fiber felt with a geometric surface area of 1 × 1 cm2 (2 mg cm−2) as the anode for the electrolyser. Then the catalyst-side of anode and cathode attached onto to two sides of SELEMION AMN/N Type1 membrane, followed by hot press treatment under 8 Mpa at 120 °C for 15 min. The AEM electrolyser was evaluated at 60 °C, using 1.0 M KOH as the electrolyte, which was circulated through a peristaltic pump with a flowing rate of 40 mL/min. The polarization curve of the MEA electrolyzer was collected at the cell voltage window from 1.4 to 3.0 V at 25 °C and 60 °C, and the stability was tested by chronopotentiometry at a current density of 1 A cm−2.
The relationship between ECSA and C dl
The value of electrochemical active surface area (ECSA) of electrocatalysts was calculated using the following equation:
where Cdl was given by the half slope of (Δj = janodic − jcathodic) plotted against the scan rate, Cs refer to a general surface specific capacitance, S is the geometric surface area of work electrode.
The value of turnover frequency (TOF) of the electrocatalytic reaction was calculated based on the amount of metal atoms in the catalysts by the follow equation:
where J, A, F, and n refers to the current density at a given overpotential, geometric surface area of work electrode (1 cm2), Faradic constant (96485 C mol−1), and the mole numbers of atoms in the electrode given by ICP-MS, respectively.
DFT calculations
All the DFT calculations were conducted by using the Vienna Ab initio Simulation Package (VASP)40. The projector augmented wave (PAW) basis set was used to calculate the ion-electron interactions41, and the exchange-correlation energy was treated by the generalized gradient approximation (GGA) with the Perdew-Burke-Ernzerhof (PBE) functional42. The cutoff energy is set to 500 eV, and the convergence criterion of energy and force was set to 10−5 eV and 0.02 eV/Å, respectively. For the calculations on the 2H-MoSe2 slab models, a vacuum space of 15 Å was added to avoid interlayer interactions. The Brillouin zones were sampled by the Monkhorst–Pack 3 × 3 × 1 k-point grids for optimization and electronic structure calculations43. The lattice coefficients of MoSe2 (001) surfaces were increased by 1.2% to obtain MoS2 (001) surfaces with 1.2% strain. Biaxial strain refers to application of strains along both x and y axes, while uniaxial strain was applied solely along the x axis. In addition, the DFT-D3 method is included to improve the description of the long-range weak van der Waals (vdW) interaction for all DFT calculations44.
To evaluate the theoretical HER performance of corresponding catalytic structures, the free-energy of H* adsorption (ΔGH*) of the studied 2H-MoSe2 were calculated. Briefly, the ΔGH*could be obtained by using the following equation:
where ΔEH*, ΔZPE and ΔS are the binding energy, zero point energy change and entropy change of H* adsorption, respectively. In this work, TΔS and ΔZPE were compared according to the scheme proposed by Norskov et al.34.
Data availability
All relevant data generated in this study are provided in the Supplementary Information/Source Data file. Source data are provided with this paper.
References
Glenk, G. & Reichelstein, S. Economics of converting renewable power to hydrogen. Nat. Energy 4, 216–222 (2019).
Kong, S. et al. Delocalization state-induced selective bond breaking for efficient methanol electrosynthesis from CO2. Nat. Catal. 6, 6–15 (2023).
Wan, C. et al. Amorphous nickel hydroxide shell tailors local chemical environment on platinum surface for alkaline hydrogen evolution reaction. Nat. Mater. 22, 1022–1029 (2023).
Staszak-Jirkovský, J. et al. Design of active and stable Co–Mo–Sx chalcogels as pH-universal catalysts for the hydrogen evolution reaction. Nat. Mater. 15, 197–203 (2016).
McCrum, I. T. & Koper, M. T. M. The role of adsorbed hydroxide in hydrogen evolution reaction kinetics on modified platinum. Nat. Energy 5, 891–899 (2020).
Zhang, J. et al. OH spectator at IrMo intermetallic narrowing activity gap between alkaline and acidic hydrogen evolution reaction. Nat. Commun. 13, 5497 (2022).
Zhang, T. et al. Oxidation state engineering in octahedral Ni by anchored sulfate to boost intrinsic oxygen evolution activity. ACS Nano 17, 6770–6780 (2023).
Guo, M. et al. Synergy in Au−CuO Janus structure for catalytic isopropanol oxidative dehydrogenation to acetone. Angew. Chem. Int. Ed. 61, e202203827 (2022).
Zhang, Z., Wang, M., Zhou, H. & Wang, F. Surface sulfate ion on CdS catalyst enhances syngas generation from biopolyols. J. Am. Chem. Soc. 143, 6533–6541 (2021).
Li, T. et al. Sulfate-enabled nitrate synthesis from nitrogen electrooxidation on a rhodium electrocatalyst. Angew. Chem. Int. Ed. 61, e202204541 (2022).
Hausmann, J. N. & Menezes, P. W. Effect of surface-adsorbed and intercalated (Oxy) anions on the oxygen evolution reaction. Angew. Chem. Int. Ed. 61, e202207279 (2022).
Wang, Y.-H. et al. In situ Raman spectroscopy reveals the structure and dissociation of interfacial water. Nature 600, 81–85 (2021).
Wang, P. et al. Precise tuning in platinum-nickel/nickel sulfide interface nanowires for synergistic hydrogen evolution catalysis. Nat. Commun. 8, 14580 (2017).
Chen, X., McCrum, I. T., Schwarz, K. A., Janik, M. J. & Koper, M. T. M. Co-adsorption of cations as the cause of the apparent pH dependence of hydrogen adsorption on a stepped platinum single-crystal electrode. Angew. Chem. Int. Ed. 56, 15025–15029 (2017).
Yang, C. et al. A Generalized surface chalcogenation strategy for boosting the electrochemical N2 fixation of metal nanocrystals. Adv. Mater. 32, 2001267 (2020).
Shi, Y. et al. Unveiling the promotion of surface-adsorbed chalcogenate on the electrocatalytic oxygen evolution reaction. Angew. Chem. Int. Ed. 59, 22470–22474 (2020).
Li, S. et al. A Glass-ceramic with accelerated surface reconstruction toward the efficient oxygen evolution reaction. Angew. Chem. Int. Ed. 60, 3773–3780 (2021).
Su, J. et al. Strain enhances the activity of molecular electrocatalysts via carbon nanotube supports. Nat. Catal. 6, 818–828 (2023).
Yao, Y. et al. Engineering the electronic structure of single atom Ru sites via compressive strain boosts acidic water oxidation electrocatalysis. Nat. Catal. 2, 304–313 (2019).
Liu, G. et al. Hydrogen-intercalation-induced lattice expansion of Pd@Pt Core–Shell nanoparticles for highly efficient electrocatalytic alcohol oxidation. J. Am. Chem. Soc. 143, 11262–11270 (2021).
He, T. et al. Mastering the surface strain of platinum catalysts for efficient electrocatalysis. Nature 598, 76–81 (2021).
Han, S., Wang, C., Wang, Y., Yu, Y. & Zhang, B. Electrosynthesis of nitrate via the oxidation of nitrogen on tensile-strained palladium porous nanosheets. Angew. Chem. Int. Ed. 60, 4474–4478 (2021).
Li, H. et al. Activating and optimizing MoS2 basal planes for hydrogen evolution through the formation of strained sulphur vacancies. Nat. Mater. 15, 48–53 (2016).
Podjaski, F. et al. Rational strain engineering in delafossite oxides for highly efficient hydrogen evolution catalysis in acidic media. Nat. Catal. 3, 55–63 (2020).
Jiang, K. et al. Rational strain engineering of single-atom ruthenium on nanoporous MoS2 for highly efficient hydrogen evolution. Nat. Commun. 12, 1687 (2021).
Jiang, Z. et al. Interlayer-confined NiFe dual atoms within MoS2 electrocatalyst for ultra-efficient acidic overall water splitting. Adv. Mater. 35, 2300505 (2023).
Zhang, T. et al. Biaxially strained MoS2 nanoshells with controllable layers boost alkaline hydrogen evolution. Adv. Mater. 34, 2202195 (2022).
Deng, S. et al. Directional construction of vertical nitrogen-doped 1T-2H MoSe2/Graphene Shell/Core nanoflake arrays for efficient hydrogen evolution reaction. Adv. Mater. 29, 1700748 (2017).
Chang, Y. S. et al. Surface electron accumulation and enhanced hydrogen evolution reaction in MoSe2 basal planes. Nano Energy 84, 105922 (2021).
Tian, Y. et al. Strain tunable electronic states of MoSe2 monolayer. Chem. Phy. Lett. 765, 138286 (2021).
Guda, A. A. et al. Understanding X-ray absorption spectra by means of descriptors and machine learning algorithms. npj Comput. Mater. 7, 203 (2021).
Henderson, G. S., de Groot, F. M. F. & Moulton, B. J. A. X-ray absorption near-edge structure (XANES) spectroscopy. Rev. Mineral. Geochem. 78, 75–138 (2014).
Subbaraman, R. et al. Trends in activity for the water electrolyser reactions on 3d M(Ni,Co,Fe,Mn) hydr(oxy)oxide catalysts. Nat. Mater. 11, 550–557 (2012).
Nørskov, J. K. et al. Trends in the exchange current for hydrogen evolution. J. Electrochem. Soc. 152, J23 (2005).
Chen, J. et al. Diversity of platinum-sites at platinum/fullerene interface accelerates alkaline hydrogen evolution. Nat. Commun. 14, 1711 (2023).
Chen, X. et al. Revealing the role of interfacial water and key intermediates at ruthenium surfaces in the alkaline hydrogen evolution reaction. Nat. Commun. 14, 5289 (2023).
Wang, K. et al. Kinetically accelerating elementary steps via bridged Ru-H state for the hydrogen-evolution in anion-exchange membrane electrolyzer. Adv. Funct. Mater. 33, 2212321 (2023).
Yan, Y. et al. Tensile strain-mediated spinel ferrites enable superior oxygen evolution activity. J. Am. Chem. Soc. 145, 24218–24229 (2023).
Hinnemann, B. et al. Biomimetic hydrogen evolution: MoS2 nanoparticles as catalyst for hydrogen evolution. J. Am. Chem. Soc. 127, 5308–5309 (2005).
Kresse, G. et al. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B Condens Matter 54, 11169 (1996).
Blöchl, P. E. et al. Projector augmented wave method: ab initio molecular dynamics with full wave functions. Bull. Mater. Sci. 26, 33 (2003).
Perdew, J. P. et al. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77, 3865 (1996).
Monkhorst, H. J. et al. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188 (1976).
Grimme, S. et al. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Acknowledgements
This study was financially supported by Tier 1 grant from Singapore Ministry of Education (RG80/22, H.J.F), Tier 2 grant from Singapore Ministry of Education (MOE-T2EP50121-0006, H.J.F.), National Natural Science Foundation of China (Grant No. 22369003, Y.L.), Hainan Provincial Natural Science Foundation of China (Grant No. 223QN185, Y.L.) and the specific research fund of the Innovation Platform for Academicians of Hainan Province (YSPTZX202123, Y.L.), Tier 1 grant from Singapore Ministry of Education (RG81/22, D.W), and NAP-SUG startup grand from NTU (D.W.).
Author information
Authors and Affiliations
Contributions
T.Z. and H.J.F. supervised and conceived this project. T.Z. designed the experiments, conducted the characterization and electrical performance measurement, and analyzed the experimental and XAS data. Q.L., Z.H. and D.W. collected the XAS data. Y.L., T.Z. and Q.Y. performed and analyzed the theoretical calculations. T.Z., Y.L. and H.J.F. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Huilong Fei, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, T., Ye, Q., Han, Z. et al. Biaxial strain induced OH engineer for accelerating alkaline hydrogen evolution. Nat Commun 15, 6508 (2024). https://doi.org/10.1038/s41467-024-50942-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-50942-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
