
Abstract
Constructing powerful anode/cathode interphases by modulate ion solvation structure is the principle of electrolyte design. However, the methodological and theoretical design principles of electrolyte/solvation structure and their effect on electrochemical performance are still vague. Here, we propose a cationic weakly coordinating-intervention strategy for modulating the Na+ solvation sheathes and constructing robust anode/cathode interphases in sodium-metal batteries. Unlike the local highly concentrated electrolytes, 1,2-difluorobenzene can weakly coordinate with Na+ thus transforming the solvation structure into Na+-anion-incorporated structures and strengthening anode/cathode interphases formation by combining with salt decomposition. Furthermore, the correlations between the electrode interface properties and solvation structure are revealed, which can be tuned by the weakly coordination. Ultimately, the modulated electrolyte achieves 97.5% Coulombic efficiency for 600 cycles in Na‖Cu cells at 1 mA cm−2 and a beneficial lifetime (2500 h) in Na‖Na cells. Meanwhile, Na‖PB cells have achieved long-term operation at 4.8 V, along with operation at wide temperatures.
Similar content being viewed by others
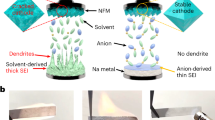


Introduction
High-voltage sodium-metal batteries (SMBs) are considered as one of the most promising energy storage systems due to the abundance of sodium metal, high theoretical capacity (~1166 mAh g−1) and low electrode potential (−2.714 V)1,2. Sodium metal matched cathodes and operation at high-voltage are critical for increasing battery energy density3. However, like lithium metal anodes, sodium metal anodes (SMAs) present a series of problems, including inhomogeneous Na plating/stripping, uncontrolled interfacial reactions triggering constant cracking of the solid electrolyte interphase (SEI), and SEI dissolution4,5. These problems directly accelerate the capacity loss and degradation of SMBs6. In addition, high active transition metal ion dissolution at the cathode side and inhomogeneous cathode-electrolyte interface (CEI) at high-voltage also induce side reactions between the highly catalytically active cathode and the electrolyte, leading to a rapid capacity degradation of the SMBs7,8,9. Stable SMBs are largely dependent on the composition and properties of the SEI and CEI, which are directly related to the solvation properties of the used electrolyte system10. However, the unfavorable solvation structure of electrolyte (unfavorable cation-solvent-anion-solvent interactions) resulting in undesired interfacial reactions, limited Na+ transport kinetics, difficulties in charge-transfer and unstable anode/cathode interfaces, etc11,12,13. Therefore, it is significant to remodel the solvation structure of electrolytes to improve performance of SMBs for meeting wider application requirements.
A large number of electrolyte engineering studies have begun to emphasize the critical role of solvation structures and their important impact on interfacial chemistry14,15. Currently, the stability of SMBs has been improved to some extent by forcing anions to participate in solvation through the formation of contact ion pairs (CIPs) and aggregated ions (AGGs) structures by increasing the salt concentration to form highly concentrated electrolytes (HCEs), thereby generating an anion-derived electrode/electrolyte interfaces16,17,18,19,20. However, the poor wettability and high cost of HCEs are not favorable for commercialization. Subsequently, fluorinated uncoordinated ether-based co-solvents (e.g., 1,1,2,2-tetrafluoroethyl-2,2,2-trifluoroethyl ether (HFE), 1,1,2,2-tetrafluoroethyl-2,2,3,3-tetrafluoropropyl ether (TTE), and bis(2,2,2-trifluoroethyl) ether (BTFE)) have been proposed for the formation of local highly concentrated electrolytes (LHCEs) from HCEs, which retain the solvation environment of the HCEs and reduce the viscosity of the electrolyte9,10,21. However, the diluents in LHCEs are expensive and not favorable for application. More importantly, the diluents are difficult to dissociate sodium salts, preventing Na+ migration and leading to a lower Na+ transference number22. The Na+ transference number is a sign of Na+ movement during battery operation, a high Na+ transference number has the advantages of inhibiting concentration polarization and promoting uniform Na deposition at the anode side during rapid charging and discharging of the battery23. In addition, hydrofluoroether diluents like TTE and BTFE not only have a low boiling point and insufficient solvation ability, making it difficult to apply LHCEs at a wide range of temperatures, but also their high density adversely affects battery energy density24. Weakly coordinated electrolytes can lead to more anion coordination due to the low coordination ability of the solvent with Na+ compared to LHCEs, leading to more anion decomposition to stabilize the SEI. Jin et al. reported a weak-solvent electrolyte design principle and utilized tris (2,2,2-trifluoroethyl) phosphate (TFP) to form a low-solvation electrolyte to stabilize the SEI and promote the formation of insoluble SEI components, which allowed for stable cycling of sodium-ion batteries5. However, the high price, high viscosity and limited temperature range also hinder its application. Therefore, despite the scarcity of reports on the preparation of stable SMBs with non-fluorinated or low-fluorinated solvents, a simple and economical method should be considered to guide the design of electrolytes with tailored solvation structures to overcome the ionic transport, desolvation, and cathodic/anodic stability problems of electrolytes25,26. In addition, performance improvements are largely attributed to cathode/anode protection, stability of electrolyte molecules, SEI dissolution inhibition, Al corrosion protection, and solvation structure, but the correlations between these key factors have rarely been elaborated, thus impeding the precise design of electrolytes27.
In this work, based on the intermolecular interactions (ion-solvent and ion-ion interactions) of electrolytes, we proposed a weakly coordinating-intervention (WCI) of cations strategy for modulating the Na+ solvation sheathes and constructing robust interphase to achieve high performance and safety of SMBs at ultra-high voltage (see Supplementary Table 1 for the specific formulation). Unlike conventional LHCEs, the 1,2-difluorobenzene (DFBn) with a slightly larger spatial site resistance is partially solvated with Na+ while also achieving a solvation structural transition of the trimethyl phosphate (TMP) electrolyte (Fig. 1a). This spatial effect and weak coordination allowed to obtain a desired solvation structure at low salt-solvent ratios. DFBn facilitated Na+ migration in the solvation sheathes, exhibiting high ion transference numbers and achieving rapid desolvation and ion transport at the interface. Importantly, WCI solvation sheathes facilitated the decomposition of Na+/salt-derived anionic clusters by adjusting the decomposition order of solvated clusters to facilitate the formation of a stable interface at the anode/cathode. In addition, we also confirmed the essential correlation between the solvation behavior and stability at the cathode/anode by means of the WCI-based electrolyte (WCIE), including redox pathways, electrode passivation behavior, SEI dissolution, and Al collector corrosion. Ultimately, the WCIE (1.7 M Sodium trifluoromethanesulfonimide (NaTFSI) in TMP- fluoroethylene carbonate (FEC)-DFBn (2: 1: 5 by mol.)) achieves a high Coulombic efficiency (CE) of 97.5% in Na‖Cu cells and a beneficial lifetime of 2500 h in Na‖Na cells. Meanwhile, WCIE can withstand long-term operation at high voltages (4.7 and 4.8 V), wide temperatures (−30 − 70 °C), and practical SMBs (N/P ratio= 4, 2 and 0). In particular, the WCI-based system significantly enhances the cyclic stability and variable-temperature storage performance of Na‖PB cells at high-voltage compared with the previously reported electrolytes based on HCEs and LHCEs, and the beneficial cells performance confirms the feasibility of strategy.

a Schematic design of electrolyte solvation structure from Conventional electrolyte to WCIE. b ESP of various solvents under vacuum conditions calculated by Density functional theory (DFT). c The calculated ESP minimum valves (eV) for various solvents are summarized. d Diagram of Donor number and dielectric constant for solvents. e Deconvolution analyses of FEC and NaTFSI Raman spectra from the solvents and electrolytes. f Summary of physical property data for different electrolytes.
Results
To optimize the performance of SMBs, it is crucial to understand and control the Na+ solvation environment, particularly at the interphase of SEI/CEI. This requires careful redesign of the electrolyte, considering solvent properties and applications, to ensure stable operation at high- voltage28. Electrostatic potentials (ESP), dielectric constant and donor number (DN) values are critical for theoretically designing electrolyte solvation structures and tuning the decomposition kinetics of Na clusters29,30. In this guiding direction, we first analyze the Electrostatic potential minimum (ESPmin) and Electrostatic potential maximum (ESPmax) of different solvent molecules (Fig. 1b, Supplementary table 2). Our ESP analysis shows that strongly polar solvents (e.g., TMP, TEP), with |ESPmin | > ESPmax, concentrate charges on oxygen and nitrogen atoms, leading to strong coordination with Na+ and reduced Na+-anion coordination capacity. On the contrary, most diluents of LHCEs (e.g., HFE, BTFE, TTE) exhibit the opposite property; they have |ESPmin | <ESPmax and the negative charge is well dispersed. As a weak electron donor, they hinder sodium salt dissolution and affect Na+ migration. Solvents like EC and AN, where |ESPmin | ≈ ESPmax, also exhibit strong polarity with pronounced positive and negative surface electrostatic potentials. These solvents are visually categorized based on their ESPmin (Fig. 1c). Obviously, there exists a region of weakly coordinating solvents among the strongly coordinating and non-coordinating solvents. They have a significantly lower ESPmin and a more concentrated negative charge than non-coordinating solvents but are weaker than strongly coordinating solvents31,32. They promote both the association of anions with Na+, which can form different solvation sheaths at low concentrations, and partial coordination with Na+32. We further categorized these solvents using dielectric constant and donor number (DN) to further inform our selection (Fig. 1d). Most of the polar solvents with high salt dissociation capacity have high DN values (>10) (regions with DN values > 10), whereas solvents with low DN values and low dielectric constant have poor salt dissociation capacity (lower-left region) and represent uncoordinated diluents33,34,35. However, there appears to be an equilibrium region (yellow region in the lower right corner) that includes solvents with low DN values and having moderate dielectric constant, leading to moderate sodium salt dissociation that can effectively reduce the affinity of Na+ for the solvent without affecting kinetic transport. Therefore, we choose TMP, FEC, and DFBn as solvents based on the design needs of safety and high-voltage SMBs for the following reasons: (1) TMP with high |ESPmin | , dielectric constant and DN value has favorable Na+ coordination ability, wide operating temperature, and electrochemical window13. (2) FEC with the highest ESPmax has a large polarity change ability, and a small amount of FEC can achieve precise coordination with Na+ to ensure a certain FEC solvation number32. In addition, FEC has appropriate dielectric constant and DN value, as a cosolvent it has both high ionic conductivity and good SEI formation ability, as well as high antioxidant capacity11. (3) Importantly, The presence of fluorine atoms in DFBn can compete with the strongly coordinated TMP, weakening the coordination of TMP with Na+ and promoting the desolvation of Na+. In addition, the large spatial effect of DFBn enhances the coordination of Na+-TFSI−, which will play a key role in the decomposition of TFSI−. Hence, we use them for electrolyte design. Firstly, we determined the optimal ratio and concentration of NaTFSI, TMP, and FEC for WCIE. The HCEs-TMP electrolyte and the electrolyte with NaPF6 as salt had poor Na | |Cu cells performance (Supplementary Figs. 1–4). WCIE also exhibit favorable flame retardancy and beneficial physical and chemical properties (Supplementary Figs. 5–7, Videos. 1–3 and Note 1). We further performed Raman spectroscopy on Base (1.7 M NaTFSI in EMC-FEC (3.5: 1 by mol.)), LF (1.7 M NaTFSI in TMP-FEC (3.5: 1 by mol.)), WCIE and pure solvents to determine the role of DFBn (Fig. 1e). The peak at 725 cm−1 and 768 cm−1 appeared in FEC and DFBn solvents32,36. In all electrolytes, the peak at 740.3 cm−1 corresponds to the S-N stretching vibrational peak of TFSI−37. This peak keeps moving to higher wave numbers and divides into three peaks: free TFSI− at 740.3 cm−1, the two solvated peaks were at 743.5 cm−1 (CIPs) and 747.9 cm−1 (AGGs), respectively. The proportions of these solvation structures are quantified and summarized in Supplementary Fig. 8. It can be found that the solvation structure with one Na+-TFSI− coordination (CIPs) dominates in Base and LF electrolytes (45% for Base and 53% for LF). In the WCIE system, solvation structure of two or more Na+- TFSI− coordination (AGGs) were much stronger than the other two systems (77%). In addition, we can find that the DFBn solvent peak is shifted to the right in the WCIE, suggesting that DFBn and Na+ may be weakly coordinated. DFBn will participate in the competitive coordination of the first solvation shell layer, reducing the solvation capacity of TMP. In addition, the low solvation capacity of DFBn will be knocked down by the TFSI− (More Na-OTFSI- coordination), allowing more Na+ to coordinate with TFSI−. This can likewise be demonstrated in the solubility test where a small portion of NaTFSI is dissolved in DFBn (Supplementary Fig. 9). The solvation structure of electrolytes was further analyzed by 13C-NMR (Supplementary Fig. 10). With the addition of electrolyte components, the chemical shifts of 13C in the solvent and anion gradually moved to lower ppm values, indicating a decrease in the density of extranuclear electron cloud on the C atom36. These results confirm the ability of TMP and DFBn to change the solvent state, implying the intervention of DFBn create a tightly integrated environment for Na+ and TFSI−. In addition, we measured ionic conductivity and Na+ transference numbers of different electrolytes (Supplementary Figs. 11–13 and Note 2). In the LSV test of Na | |SS cells, WCIE demonstrated oxidation stability up to 5.5 V, which has favorable potential for high-voltage battery (Supplementary Fig. 14). Matching Na with PB for a more stringent electrochemical float test (Supplementary Fig. 15). Base electrolyte fails quickly, and WCIE anode leakage current is minimized to 2.1 µA at 5.0 V. WCIE also achieves the most stable cycling in 4.5 V Na | |PB cells (1 C, 400 cycles) (Supplementary Fig. 16 and Note 3). Based on these results, WCIE combines the beneficial performance of Base and LF electrolytes, and its physicochemical properties are better than LHCEs, obtaining an overall improvement. (Fig. 1f and Supplementary Fig. 17). Overall, there is a strong correlation between WCIE stability and solvent solvation ability. Therefore, further determination of their solvation structures is needed to help explain the differences in performance.
Solvation structure and its solvation ability
The solvation structures of various electrolytes were further explored using ab initio molecular dynamics (AIMD) simulations. Snapshots of solvation boxes for three different electrolytes are displayed in Fig. 2a–c. The results distinctly indicate that with the addition of NaTFSI, both Base and LF electrolytes form solvation clusters predominantly composed of SSIPs and CIPs, in agreement with Raman. Interestingly, DFBn incorporation directly compresses the distance between Na+- Na+ (Supplementary Fig. 18). From the radial distribution functions (RDFs), the Na-OEMC peak at 2.25 Å in Base electrolyte suggests that EMC will preferentially enter the first solvation sheath of Na+, which would be expected to constitute an organic-rich SEI/CEI (Fig. 2d)14,36. When the electrolyte is LF, the Na-O of TMP, TFSI− and FEC are almost all at about 2.25 Å, they all preferentially enter the first solvation sheath of Na+ (Fig. 2e). Thus, the formation of organic/inorganic SEI/CEI is expected. The distance of Na-OTFSI- peaks in WCIE becomes smaller (2.03 Å) and the coordination numbers of Na-OTFSI- increases to 2.31, implying that TFSI− is more likely to enter the first solvation sheath of Na+, especially with the intervention of DFBn (Fig. 2f, g). Meanwhile, the coordination numbers show that Na-O solvent interaction is the smallest in WCIE, with only 0.2 and 1.1 for Na-OFEC and Na-OTMP, respectively. In addition, the main solvation structure of WCIE, as depicted in Fig. 2h, shows a highly compressed WCI solvation structure under the influence of DFBn. In this WCI solvation configuration, all TFSI− and Na+ are tightly bound. Meanwhile, Base and LF electrolytes predominantly exhibit solvation structures characterized by SSIPs and CIPs.

Snapshots of the MD simulation boxes of (a) Base, (b) LF and (c) WCIE at 25 °C. RDFs of Na+ and solvent interactions with TFSI- from MD simulations of (d) Base, (e) LF and (f) WCIE. g Atomic coordination numbers of the three electrolytes. h The corresponding main solvation structure of each electrolyte from MD simulations.
Overall, the experimental results correlate well with MD simulations. Low-polar fluorinated solvents, such as DFBn and fluorobenzene (FBn), have a weak coordination ability that favors the formation of WCI solvation structure from a low concentration (Supplementary Fig. 19 and Note 4)38. The coordination of Na+-TFSI− in WCIE is stronger than in Base and LF electrolytes, even higher than in HCEs-TMP electrolyte and LHCEs. Meanwhile, WCIE is significantly different from LHCEs (Supplementary Figs. 20, 21 and Note 5). Although anionic coordination to Na+ also implies the presence of more abundant free solvent molecules (e.g., EMC, TMP), which is mainly considered as a key factor for the low stability39. The free solvent molecules are essentially DFBn for WCIE, and their decomposition can likewise aid in the formation of inorganic interface40. With these interesting results, the following sections will endeavor to explain this improved anodic and cathodic stability, especially considering the role of solvation structure in the most stable WCIE. We consider that the improved performance may be due to the following reasons: (1) SEI/CEI formed in the super compressed electrolyte may be robust to prevent parasitic reactions of phosphate molecules on the electrode. (2) Oxygen-chelated phosphate molecules more beneficial than carbonate molecules, thereby increasing the ion conductivity, electrochemical window, etc. (3) The solvation behavior of WCIE leads to different decomposition pathways and interfacial behavior on the electrode surface, which improves stability.
Passivation behavior of anodic SEI
Due to SEl formation, anode passivation should be investigated first as it has been reported to be the main cause of anode stability. Inorganic-rich SEI usually reduces parasitic reactions and dendrite growth to guide uniform deposition of Na+. The interfacial chemistry was characterized using X-ray photoelectron spectroscopy (XPS) (Fig. 3a and Supplementary Figs. 22, 23). C-C/C-H (284.8 eV), C-O (285.7 eV), C = O (289.3 eV), CO32− (290.3 eV), and -CFx (292.9 eV) existed in C 1 s spectra of all electrolytes23,24. As sputtering proceeds, the C 1 s intensity of WCIE plummets, while Base and LF electrolytes show little change. The presence of CFx is confirmed in F 1 s spectra, with another peak corresponding to Na-F (684.8 eV)25,26. The decomposition of TFSI− was subsequently confirmed by the detection of two deconvolution peaks in N 1 s spectrum attributed to Na3N (397.2 eV) and N-SOx (399.5 eV), respectively27,36. The low electronic conductivity of SEI leads to the preferential formation of fully decomposed TFSI− components internally. For O 1 s and S 2p, the intensity of Na2O and Na2S components in the SEI is higher in any etching state for WCIE. SEI with inorganic components such as NaF, Na3N, Na2O and Na2S not only improves its own mechanical strength and inhibits dendrite penetration, but also enhances Na+ transport flux and facilitates highly reversible Na plating/stripping5,20,21,22,23,24,25,26,27,28. The elemental content distributions at different sputtering depths were investigated (Fig. 3b). As the sputtering depth increases, WCIE produces inorganic SEI, resulting in a significant decrease in elemental C (3.32% at 50 nm) and P (0.81% at 50 nm), whereas F (31.76% at 50 nm), N (4.10% at 50 nm), and S (5.21% at 50 nm) increased. In contrast, Base and LF electrolytes form solvent-derived SEI due to insufficient TFSI− decomposition, with the presence of very abundant C and O (12.90% and 33.13% for Base and 8.45% and 30.56% for LF at 50 nm). Meanwhile, the high activity TMP of LF electrolyte decomposes more, the organic P content is still 7.55% at 50 nm. The organic-rich SEI layer is porous, has low mechanical strength and cannot protect sodium metal from electrolytes16. Further summarizing the information on the species content of different SEI (Fig. 3c and Supplementary Figs. 24, 25), the content of inorganic species (e.g., NaF, Na3N, Na2O, and Na2S) in the WCIE continuously with increasing sputtering depth, whereas the content of organic species, and undecomposed TFSI− (e.g., RONa, ROCOONa, CFX, and N-SOx) content decreases continuously. This organic to inorganic gradient distribution in WCIE promotes the formation of multilayer SEI41,42.

a C 1 s, F 1 s, O 1 s and N 1 s XPS depth profiles of the Cu electrode after 50 cycles at a current density of 1 mA cm−2 and a capacity of 1 mAh cm−2 in WCIE. b Corresponding to the atomic concentration of different electrolytes at different sputtering. c Normalized ratios of different species in the SEI formed in the WCIE. d Electrolyte exchange study between WCIE and Base electrolyte within Na | |Cu cells. The projected density of states (PDOS) profiles of (e) Base, (f) LF and (g) WCIE.
To evaluate the role of SEI in preventing ester solvent decomposition, we performed electrolyte exchange studies after 100 cycles in WCIE (Supplementary Fig. 26)38,43. Na electrodes after cycling were removed and then coupled with Base and LF electrolytes to continue cycling (Fig. 3d). Interestingly, Base electrolyte allowed stable cycling and produces a more stable CE (94.2% vs. 86.5%). However, the contribution of SEI still does not allow it to achieve the same results as WCIE (97.3%), and WCIE pre-passivation does not completely prevent the decomposition of the carbonate solvent. This is also reflected in the exchange experiments of LF electrolyte, despite the enhanced CE (Supplementary Fig. 27). Meanwhile, XPS analyses of Na electrodes after electrolyte exchange experiments showed differences in the SEI (Supplementary Fig. 28, Note 6). Therefore, the improved anodic stability of WCIE should be the result of synergistic action of the stable SEI and the WCI solvation structure. The performance improvement of WCIE, in other words, cannot simply be attributed to SEI. If SEI can largely prevent the degradation of solvents, then the decomposition of EMC and TMP molecules after electrolyte exchange should also be prevented and the CE should improve more40. It is also interesting to consider how the solvation structure of WCIE could form an anion-derived SEI, even though the free ester solvent is an unstable species.
We have investigated the decomposition behavior of three electrolytes in terms of the electrochemical stability of solvation structures. Existing DFT studies are often based on simple solvation models, where the stability of electrolytes is explained by the lowest unoccupied molecular orbital (LUMO) and highest occupied molecular orbital (HOMO) energy levels of individual electrolyte components or coordination44. However, complex coordination scenarios often alter the stability of electrolyte species, leading to significant redox shifts. We investigate the projected density of states (PDOS) directly from the solvation structure of electrolyte in MD simulations, rather than relying on subjective intuition to construct them. Here, PDOS spectra allow more accurate prediction of redox processes for different electrolytes. Considering the electrochemical compatibility with SMA, the conductive band of PDOS is of great interest. In Fig. 3e, the LUMO of Base electrolytes are located at FEC and EMC, thus it can be surmised that their reductive decomposition is much earlier than TFSI−. With the replacement of TMP, the energy level of TMP conduction band is the highest in LF electrolyte, and its LUMO is still located in the FEC, the decomposition of FEC is still dominant, followed by TFSI− (Fig. 3f). Thus, the decomposition of Base and LF electrolytes is solvent-dominated and will eventually lead to the formation of organic SEI, which is consistent with XPS results. WCIE shows the lowest energy levels for TFSI− conduction band, followed closely by DFBn (Fig. 3g), and high energy levels for the conduction bands of FEC and TMP. The advantage of TFSI− reduction will be greatly enhanced. Further DFT calculations of the HOMO/LUMO energy levels of solvation structures exhibit mutually corresponding results (Supplementary Fig. 29, Note 7). Therefore, the Na+-TFSI− WCI solvation structure is favorable over the solvation structure of other electrolytes. During SEI formation, the decomposition of WCI solvation structure dominates the passivation reaction, resulting in the formation of anion-derived inorganic SEI that completely passivate anode12,27. In other hand, Base and LF electrolytes cannot be stabilized because most of EMC and TMP molecules compete for coordination with Na+ to form solvent-dominated solvation structures. PDOS and DFT indicate that the LUMO of their solvation structures are much lower than solvent molecules themselves and the conduction band for decomposition are located on the solvent. It is more susceptible to reduction reactions and the formation of solvent-derived organic SEI. The unfavorable solvation sheaths formed during cell cycling produce organic products in the ongoing reduction reaction that are unable to passivate the anodic reaction sites or prevent the ongoing parasitic reaction of the electrolyte45. For the electrolyte exchange experiments, although WCIE formed a well-structured SEI through cycling, the degradation of solvent-rich structure in Base and LF electrolyte after reaching the interface layer could not be inhibited, thus further caused a new disruption of the already stabilized SEI46,47. Na+ transport of SEI became difficult, leading to a decrease in the reversibility of Na plating/stripping. Thus, there is a strong correlation between the solvation structure of electrolyte and the formed SEI in improving anode stability.
The stability of SEI
Reversible Na plating/stripping is critical to facilitate stable cycling of SMBs and is also an important manifestation of stable SEI. The sodium-deposited Cu foils were characterized using scanning electron microscopy (SEM) (Fig. 4a–c). Numerous mossy deposits, overall porous and loose with many flocculent deposit particles in Base and LF electrolytes, organic SEI hindering the uniform diffusion of Na+37,38. In contrast, sodium was uniformly deposited on the Cu foil without any dendrites, and the connection between the deposits was smooth in WCIE. The cross-sectional image shows that WCIE achieved uniform deposition with a thin deposition surface (24.5 µm). The evolution of deposition morphology was explored by further increasing the capacity (Supplementary Fig. 30), WCIE showed Na adaptation at high capacity. The dynamic sodium plating process was monitored by optical microscopy to illustrate the morphological evolution. As the preferred nucleation site for sodium deposition, the SMA surfaces of Base and LF electrolytes ranged from numerous spikes appearing at 5 min of deposition to being covered with flocculated sodium dendrites at 20 min (Fig. 4d1, d2, Supplementary Videos 4, 5). In contrast, the SMA of WCIE showed a smooth and dense morphology without dendrites throughout the plating process (Fig. 4d3, Supplementary Video 6). In addition, the mechanical stability of the SEI for WCIE is higher than the Base and LF electrolytes, this is consistent with the atomic force microscope (AFM) (Fig. 4e1-e3). Therefore, the compact SEI formed by WCI solvation structure are dense, and the presence of abundant grain boundaries for the inorganic components allows efficient Na+ migration, which is not hindered by the internal structure of SEI, achieving rapid and uniform deposition31. In addition, cyclic voltammetry (CV), exchange current density and Temperature-dependent electrochemical impedance spectroscopy (EIS) showed the benefits of WCIE in electrochemical kinetics (Supplementary Figs. 31, 32, tables 3–5, and Note 8). The battery capacity loss was further quantified to investigate the dissolution of SEI by the electrolyte in real cases (See Supplementary Fig. 33 and Note 9 for specific details)5,48. The capacity growth after each pause indicates that the anode electrolyte decomposition produces a parasitic reduction reaction to compensate for the dissolution of the SEI formed during the long open circuit pause (Fig. 4f). The capacity loss after a xh long pause (the difference between capacity after and before the pause) quantifies SEI dissolution in the electrolyte (Fig. 4g). The average capacity loss of Base electrolyte after 50 h pause (5.82 µAh) was 1.1 times for LF electrolyte (5.38 µAh) and 3.3 times for WCIE (1.77 µAh). Notably, the actual dissolution of SEI is much more severe on the inhomogeneous anode surface exposed with porous after cycling. In addition, the dissolution of SEI in LF electrolyte remains slow and persistent, with an average capacity loss after the last 5 h pause (0.773 µAh) that is 1.9 times for WCIE (0.403 µAh). The dissolution of SEI was largely suppressed in WCIE, as evidenced by the lowest capacity loss, the least variable SEI fraction, and the most stable long-cycle performance (Supplementary Fig. 34 and Note 9). This highlights the strengths of WCIE: a solvation structure with less active free solvent, low polarity DFBn and more salt decomposition derived SEI with lower solubility, further demonstrating the contribution of beneficial solvation structure to stable SEI even a good anode, paving the way for subsequent electrochemical performance.

Top and Side-view SEM images of Cu foils in (a1, a2) Base, (b1, b2) LF and (c1, c2) WCIE cycled for 100 cycles at 1 mA cm−2 and 1 mAh cm−2. In situ optical microscopy images showing cross-sections of symmetric cells at (d1) Base, (d2) LF, and (d3) WCIE plating Na electrodes. AFM surface and 3D topography analysis of Na electrodeposits on Cu foils in (e1) Base, (e2) LF and (e3) WCIE at 1 mA cm−2 and 1 mAh cm−2. f Capacity from electrolyte reduction reactions (at 1 µA cm−2) before and after each pause as a function of the number of cycles. g Capacity loss of three parallel cells with different pause times in different electrolytes.
Compatibility with sodium anodes
After analyzing the effects of different electrolyte solvation structures on the structural composition, deposition morphology and kinetics of the SEI, the electrochemical reversibility of Na metal was assessed by various electrochemical performances. For the Na | |Cu cells (Fig. 5a, b and Supplementary Fig. 35), the reversibility of CE was about 86.5% and 91% with low cycle life at 1 mA cm−2 and 1 mAh cm−2 in Base and LF electrolytes. The SEI produced by the action of solvation clusters in both electrolytes were fragile and poorly tolerant to high currents49. Surprisingly, WCIE-formed SEI achieved stable Na+ plating/stripping over 600 cycles with an average CE of 97.5% and a low voltage hysteresis (224 mV). The CE values derived from the Aurbach50 method also indicate the advantages of the WCIE (Supplementary Fig. 36 and Note 10). Fixed capacity CE measurements were performed at different current densities to examine the adaptability of Na plating/stripping (Fig. 5c, d and Supplementary Fig. 37). Maintaining a capacity of 1 mAh cm−2, the CE of Base and LF electrolytes changed dramatically when the current density was increased from 0.5 mA cm−2 – 3 mA cm−2. In contrast, WCIE showed good current suitability, returning to 0.5 mA cm−2 still cycling stably for 600 cycles, along with a low overpotential at high current densities. The low nucleation and growth overpotentials of Na deposition for the first cycle discharge curve at different current densities (Supplementary Fig. 38) reveal the WCIE imparts highly reversible Na plating/stripping51. Summarizing recent studies on Na | |Cu cells (Fig. 5e, Supplementary table 6), the results show useful progress in our work, demonstrating the beneficial effects of the solvation structure and SEI of WCIE compared to conventional ester electrolytes. Na | |Na cells were further evaluated for long-term plating/stripping stability of sodium metal (Fig. 5f). Operating at 1 mA cm−2 and 1 mAh cm−2 for <1500 h, Base and LF electrolyte rapidly failed due to dendrite formation and continued electrolyte depletion. Impressively, WCIE exhibited low voltage hysteresis and regular voltage distribution for up to 2500 h. The low Rint from EIS curves (Supplementary Fig. 39) also suggests that SEI is well suited for Na+ migration. In addition, after changing the capacity to 2 mA cm−2–4 mAh cm−2, 5 mA cm−2–1 mAh cm−2, and rate (Supplementary Fig. 40), the WCIE still have the capability of cycling for >1000 h, 350 h, and a stable rate cycling ability, respectively. Na | |Na cells also show better performance than most ester electrolytes (Supplementary Fig. 41, Supplementary table 7) in the literature. The above cell performance results further indicate that WCI solvation clusters greatly improve the cycling stability of sodium metal in phosphate ester electrolytes.

a CE test in Na | |Cu cells using different electrolytes at 1 mA cm−2 and 1 mAh cm−2. b Capacity-Voltage curves for corresponding WCIE. c Fixed 1 mAh cm−2 capacity, Na | |Cu cells CE test at different current densities. d Na plating/stripping voltage hysteresis corresponding to different electrolytes. e Comparison of CE between recently reported work and the present work. f Voltage profiles of Na | |Na symmetric cells in different electrolytes at 1 mA cm−2 and 1 mAh cm−2.
High-Voltage Na | |PB cells testing and cathodic passivation
From the DFT and PDOS results, the solvation clusters have high HOMO values, and the oxidation of Base and LF electrolyte solvation clusters dominates the cathodic passivation reaction, with the HOMO of the preferentially oxidized conduction bands being located on EMC and TMP, resulting in the formation of organic, inhomogeneous CEI27,38. WCI solvation clusters formed by WCIE likewise have high HOMO values. On the contrary, the HOMO of the preferentially oxidized conduction bands of WCI solvation clusters are located on TFSI−, favoring the formation of inorganic CEI to cathodic passivation. As we predicted, WCIE showed beneficial cycling stability at high-voltage 4.7 V (Fig. 6a). The capacity of cells with Base and LF electrolytes decayed rapidly during cycling, and CE was chaotic. Although EMC was replaced with TMP to increase the oxidation voltage of LF electrolyte to about 5.0 V in the LSV, the full cells still showed poor stability during cycling. One, due to the catalytic effect of PB cathode, the LF electrolyte started to decompose far below the tested oxidation voltage in the actual full cells. The second is due to the formation of solvent-rich clusters leading to the formation of brittle SEI/CEI at 4.7 V for long cycles52,53. The capacity retention after 200 cycles was 84.3% and the average CE was 99.5% for WCIE. Corresponding capacity-voltage curves (Fig. 6b and Supplementary Fig. 42) also show low overpotential. Rate performance showed that WCIE could discharge at a higher rate (Fig. 6c). The discharge capacity of WCIE was still 118.7 mAh g−1 and 105.1 mAh g−1 at 10 C and 20 C, respectively, with high CE and narrow voltage gap (Supplementary Figs. 43, 44). EIS investigated the charge transfer kinetics of different electrolytes after cycling (Fig. 6d). The low CEI and charge transfer impedance of WCIE suggests beneficial CEI and a low Na+ embedding/detachment impedance at the electrode54. Self-discharge tests were performed to further probe the high-voltage resistance of different electrolytes (Supplementary Fig. 45 and Note 11). In addition, WCIE has much less parasitic reactions to the cathode at high voltage, inhibiting side reactions and Al corrosion (Supplementary Figs. 46–49 and Note 12). WCIE also has some promising applications in hard carbon anodes and sodium ion batteries. (Supplementary Figs. 50, 51 and Note 13).

a Long-term cycling of different electrolytes at 4.7 V (PB loading: ~3 mAh cm−2). b Capacity-Voltage curves for corresponding WCIE. c Rate performances of different electrolytes at 4.7 V. d EIS curves for different electrolytes at 4.7 V after 50 cycles. e1, f1 Base, (e2, f2) LF, (e3, f3) WCIE of PB cathode at 4.7 V after 80 cycles SEM and HRTEM. XPS analysis (g) and Corresponding atomic concentration contents (h) of PB cathode after 80 cycles.
We observed morphology of electrode material and CEI after cycling in different systems using SEM and high-resolution transmission electron microscopy (HRTEM) (Fig. 6e, f). In the Base electrolyte, particles on the electrode surface were severely agglomerated and formed a thick and inhomogeneous CEI. This is due to a series of complex chain reactions such as oxidative decomposition of electrolyte on high-voltage, corrosion of Al collector, and dissolution of transition metals52,53,55. Despite the improvement of LF electrolyte, it still does not achieve good protection. The material has a smooth surface with well-defined edges, forming a thin and uniform CEI (7 nm) in WCIE. Stable and homogeneous CEI is closely related to the structural composition, the chemical composition of CEI was investigated using XPS (Fig. 6g). In F 1 s spectra, the major peaks correspond to NaF and C-F, respectively. 160.2 eV in the S 2p spectra corresponds to Na2S, and 397.3 eV in the N 1 s spectra is Na3N. NaF, Na2S and Na3N are the main inorganic components that are very effective in the construction of thin and homogeneous CEI, are decomposition products of TFSI− and FEC, inhibit electrode/electrolyte side-reactions and facilitate Na+ transport25,26,27,33. The same C = O, C-O and S-O peaks are present in O 1 s spectra. However, the peaks at 530.3 eV for Base and LF electrolytes are attributed to M-O (M = Fe, Mn, etc.), showing significant dissolution of transition metals and the formation of CEI that have broken down and become inhomogeneous56. Organic products such as RONa and ROCONa accumulated on cathode surface are mainly decomposition products of electrolyte. In C 1 s spectra, the strongest peak at 284.8 eV corresponds to C-C/C-H, which is mainly related to conductive agent26. The elemental contents of different CEI were compared by quantitative XPS analysis (Fig. 6h). WCIE had low C, O and P contents and high inorganic F, N and S contents. Therefore, the NaF/Na3N/Na2S-rich CEI layer was denser and more homogeneous, the smaller thickness and beneficial composition ensured rapid Na+ migration across CEI. These results indicate that WCI solvation clusters formed by WCIE not only provide a robust CEI, but also inhibit solvent oxidation and Al collector corrosion, improving the stability of full cells during charge/discharge37,57.
Special and practical conditions for NallPB cells
Finally, the electrochemical performance of three electrolytes was investigated in various SMBs. NallPB cells performance was firstly investigated at 60 °C with a highly loaded PB cathode (3 mAh cm−2) (Fig. 7a, b and Supplementary Fig. 52). Base and LF electrolytes fail equally quickly, with WCIE showing significantly better cycling stability at high temperature, with 85% capacity retention after 200 cycles and an average CE of 99.5%. As a result, WCIE also show high temperature application possibilities, with the advantage of solvation minimizing unfavorable overcharging. In addition, considering wide temperature application ranges of phosphate and DFBn, the electrochemical performance of Na | |PB cells was tested from −30 °C to 70 °C at 4.7 V (Fig. 7c). At 30 °C, the discharge capacity of WCIE is close to LF electrolyte. However, the difference in capacity begins to appear when the temperature is lowered to 10 °C. When the temperature dropped to −30 °C, WCIE still had a capacity of 110.6 mA h g−1, Base electrolyte appeared to be in disarray and LF electrolyte was left with only 72.5 mA h g−1. Capacity-voltage curves further proved the advantages of WCIE at different temperatures (Supplementary Fig. 53). To investigate WCIE at a higher voltage, we tested Na | |PB cells at a cut-off voltage of 4.8 V (Fig. 7d, e). Na | |PB cells could not be charged up to 4.8 V due to the coupling between carbonate solvent and TFSI− in Base electrolyte and the unfavorable solvation structure leading to easy decomposition under high-voltage (Supplementary Fig. 54)58,59. In contrast, WCIE has beneficial cycling stability and boasts a high-capacity retention after cycling with an average CE of 99.3%. Similarly, Na | |PB cells shows beneficial performance at 20 C (Fig. 7f and Supplementary Fig. 55). Only slight voltage polarization and degradation were observed at an ultra-high charging voltage, suggesting that the solvated layer of the WCIE has good compatibility with SEI/CEI formed at such high voltage. Based on the beneficial electrochemical performance of WCIE at the anode/cathode, a full Na | |PB cells with an N/P ratio of 4:1 was matched and cycled at 4.7 V (Fig. 7g, h and Supplementary Fig. 56) WCIE not only has an average CE of 99.5% and capacity retention of 85% over 150 cycles with a narrow charge/discharge gap, but also has fast kinetic properties with capacities of 98.1 mAh g−1 and 76.7 Ah g−1 at rates of 10 C and 20 C, respectively (Fig. 7i and Supplementary Fig. 57). Similarly, stable cycling was achieved with N/P ratio of 2:1 and anode-free condition (Supplementary Figs. 58–61 and Notes 14, 15). Therefore, our design strategy using WCI has good application prospects.

a Long-term cycling of different electrolytes in the 3 mAh cm−2 cathode-load at 4.7 V, 60 °C. b Capacity-Voltage curves for corresponding WCIE. c Electrochemical performances testing using different electrolytes at temperatures from −30 °C–70 °C. d Long-term cycling of different electrolytes at 4.8 V. e Capacity-Voltage curves for corresponding WCIE at 4.8 V. f Rate performances of different electrolytes at 4.8 V. g–i Electrochemical performances testing of Na | |PB full cells with an N/P ratio of 4: 1 at 4.7 V.
Discussion
In conclusion, we have reconfigured the solvation behavior of phosphate-based electrolytes by exploiting intermolecular interactions to improve the performance and the compatibility of SMBs. Our WCIE system is shown to extend the operation of SMBs at high-voltage, which exceeds the currently reported cell performances by feasible cells configurations (Na‖Cu cells, Na‖Na cells, variable temperature high-load cathode, and controlled N/P ratio). The relative LUMO and HOMO of the solvated clusters are modulated due to the weakly coordinating-intervention of Na+ by DFBn and the solvation behavior enriched with Na+, which directs the decomposition sequence of electrolyte on the anode/cathode surface to form a strong anion-dominated interfacial layer. More importantly, this work not only proposes an effective theoretical design principle to overcome the poor stability of electrolytes for SMBs, but also elucidates the correlation between the solvation structure and the redox behavior of electrolyte, SEI dissolution, and anodic/cathodic protection. Meanwhile, it is determined that the SEI/CEI chemical changes in SMBs are due to differences in solvation rather than differences in the mechanism of SEI formation, which is a useful advance for SMBs. The design and beneficial performance of WCIE provide a reliable approach to the study of existing SMAs in Na plating/stripping reversibility, long-term operation of high-voltage SMBs. Our results also provide an insight into understanding the process of interface formation and tailoring its chemical composition.
Methods
Materials
Sodium metal was obtained from Alfa Aesar Reagent Co. (99.9%) (Supplier: Shanghai Mern Chemical Technology Co.). The salts, solvents, including Sodium trifluoromethanesulfonimide (NaTFSI) (battery-grade purity), Ethyl Methyl Carbonate (EMC) (battery-grade purity), Fluoroethylene carbonate (FEC) (battery-grade purity) were purchased from Duoduo Chemical Reagent Co., Ltd. The Trimethyl phosphate (TMP) was purchased from ALFA (Supplier: Shanghai Titan Technology Co.) with purity of >98%. 1,2-difluorobenzene (DFBn) was purchased from TCl (Shanghai) Kasei Industry Development Co., with purity of >98%. Before preparing the electrolyte, the solvent was dried by 4 Å zeolite to ensure that the water content was below 10 ppm. Prussian Blue (PB) cathodes were purchased from Naba Co., Ltd. Chemical reagents. Coin chute (stainless steel, SS-316), collector (aluminum and copper foil with purity of >99%), conductive carbon, and polyvinylidene fluoride (PVDF) were purchased from TOB New Energy Technology Co. Glass fiber separator (GF/C) was purchased from Whatman Company. Copper and aluminum foils (thickness 10 µm, diameter 15 mm) were cleaned of surface dust and impurities with ethanol before use. The copper foil did not have any surface treatment before use. Electrolytes were prepared by mixing the required amount of salt, solvent. The usage of electrolyte in button cell was 90 µL. The preparation of electrolytes was all carried out in a glove box with O2, and H2O content < 1 ppm.
Material characterization
The viscosity was collected on TA Instruments DHR-2 rotational rheometer with accurate temperature control system. The contact angle was collected on the instrument DSA-30 Droplet Shape Analyser. The flammability tests of electrolytes were carried out in the fume hood. The morphological of cycles studies were obtained by HITACHI S-4800 field emission scanning electron microscopy (FESEM) and Bruker Dimension Icon Atomic Force Microscope (AFM). To analyze the chemical composition of SEI and CEI, characterization of electrodes after cycling using XPS measurements (Thermo Scientific ESCALAB Xi+, American). It is accompanied with depth profiling, which is performed by Ar+ sputtering. High resolution transmission electron microscope (HRTEM) images were obtained from a TECNAI F30 at 300 kV. The cycled cells were disassembled in the glove box filled with argon (the contents of O2 and H2O were kept below 0.1 ppm) and the electrodes were washed with dimethyl ether (DME) before morphology and composition characterizations. The in-situ dynamic Na deposition behaviors were observed at 0.5 mA cm−2 for 30 min by ZOOM-0850C (SHANGHAI PUQIAN OPTICAL INSTRUMENT CO., LTD.) using Na as working electrode. Raman results were captured on XploRA with an excitation laser wavelength of 532 nm. 13C NMR spectra were recorded at room temperature (295 K) with a Bruker Avance III HD 850 MHz spectrometer. For NMR measurements, 600 µL of electrolyte sample was placed in the outer tube of a 5 mm NMR Wilmad coaxial tube.
Electrochemical measurements
The ionic conductivities (σ) and Electrochemical impedance spectroscopy (EIS) of different electrolytes were tested by electrochemical impedance spectroscopy (IM6, Zahner Elektrik, Germany) tests of button cells in a frequency range from 0.1 Hz – 105 Hz with an amplitude voltage of 5 mV. The electrodes were assembled with two stainless steels (SS | | SS). The ionic conductivity (σNa) was calculated using the following equation:
Where R is the bulk resistance, L is the thickness of the membrane, S is the contact area of inert electrode (stainless steel). In this work, the thickness of membrane is 0.13 mm, and the diameter is 19 mm.
The Na+ transference number (tNa+) of different electrolytes were measured by DC polarization (POL) combined with EIS tests of Na | | Na symmetrical coin cells on Zaher and calculated from the following equation:
where ΔV is the POL voltage (10 mV), I0 and Is are the response current at the initial and after stabilization, respectively. R10 and R20 are the bulk ohmic resistance and interfacial charge transfer resistance of the Na | |Na cells before POL, respectively. R1s and R2s are the bulk ohmic resistance and interfacial charge transfer resistance of the Na | |Na cells after stabilization.
Sodium metal anodes were obtained after extruding sodium metal foil (~1 mm) into discs (Ф 14.0, ≥99.9%) using a stainless-steel punch with a diameter of 14.0 mm. Prior to preparation, white oxide powder was removed from the surface using scissors to obtain sodium with a silvery-white metallic luster. Afterwards, the lumps of sodium metal were pressed into sodium foils with a thickness of about 1 mm using a roller press, and then pressed into sodium anodes with a diameter of 14 mm using a punch. To prevent deterioration of the sodium metal due to excessive storage in the glove box, it is cut and used now before assembling the battery. The oxygen and water content of the glove box was <1 ppm and the ambient temperature was 25 °C during the whole operation. Electrochemical cycling and rate tests were assembled by 2032-type coin batteries with glass fiber (Whatman GF-C) and tested by using a battery test system (Shenzhen NEWARE Co. LTD, China). The Na | |Cu cells were using Cu foil as working electrode, Na metal disk as counter electrode. The coulombic efficiency (CE) of the Na | |Cu cells was calculated by charge/discharge capacity, and the stripping cut-off potential was set to 1 V. Na | |Na symmetric cells were assembled by using two piece of Na disks as working and counter electrodes. Stainless steel (SS-316) discs and springs were added to the Na piece to ensure even pressure distribution. The battery was allowed to stand for 6 h before testing to allow the electrodes and membrane to wet completely. EIS in frequency ranges from 0.1 Hz – 105 Hz with amplitude of 5 mV and we used Zview software to fit the temperature-dependent EIS. To perform the impedance test at different temperatures, the cells were first set aside for 2 h at different temperatures. Cyclic voltammetry (CV, Squidstat prime) tests within potential range of −0.3–3 V vs. Na/Na+ at scan rate of 1 mV s−1. The oxidation potential of the electrolytes was tested by linear sweep voltammetry (LSV, Squidstat prime) of Na | |SS cells. The chronoamperometry (CA) measurements was tested by Squidstat prime. The charge/discharge performance of electrolytes were obtained by assembled full cells using PB as cathode, Na metal as anode and a piece of GF-C as separator in an Ar-filled glove box (glove box with O2 and H2O content <1 ppm). Prussian blue electrodes were prepared by mixing Prussian blue powder, conductive carbon, and PVDF binder in N Methyl-2-Pyrrolidone (NMP) in the ratio of 80:10:10 by weight to form a homogeneous paste. Round electrodes of 13 mm diameter were then cut, pressed, dried in a vacuum oven at 120 °C for a further 12 h and weighed. Cycle and rate tests were performed on the NEWARE battery test system at 25 °C. All Na | |PB cells were tested in a voltage range between 2.4 – 4.5 V, 2.4 – 4.7 V or 2.4 – 4.8 V vs. Na/Na+. All full cells were activated for 2 cycles at 0.2 C. The PB loading was around 3 mAh cm−2 for both cycling and rate tests for Na | |PB cells with cut-off voltages of 4.5 V and 4.7 V. The PB loading was around 5 mg cm−2 for both cycling and rate tests for Na | |PB cells with cut-off voltages of 4.8 V. Low-temperature and change-temperature full cells test was performed by connect with NEWARE battery test system. The electrochemical performance and EIS at different temperatures was carried out within an RTP-80CT high and low temperature test chamber (Guangdong Huan Rui Test Equipment Co., Ltd, China). All cells were maintained at specified temperature for 2 h prior to testing. Final specific capacity was determined to the active mass of PB. The Na | |PB full cells were fabricated with negative/positive capacity (N/P) ratio of 4 (For the N/P negative electrode fabrication, 6 mAh cm−2 of sodium metal was deposited onto the smooth copper foil to form a composite electrode and the areal capacity of cathode is 1.5 mAh cm−2). The Na | |PB cells contains practical loading of anode (3 mAh cm−2) and PB cathode (1.5 mAh cm−2) in N/P ratio = 2. The Na | |PB cells contains practical loading of PB cathode (3 mAh cm−2) in N/P ratio = 0.
Computational details
Ab initio Molecular Dynamics (AIMD) simulations were performed using the Vienna Ab initio Simulation Package (VASP 5.4) based on the pseudopotential plane-wave approach. The Perdew-Burke-Ernzerhof generalized gradient approximation (GGA-PBE) was utilized to represent the exchange-correlation functional, with a cutoff energy of 400 eV. The initial configurations of NaTFSI, FEC, EMC, TMP, TTE, FBn and DFBn were optimized using Gaussian09 program with M062X/6-311 + G (d, p). The NaTFSI salt/solvent mixtures (base, LF, LHCEs, FBn-based electrolyte and WCIE) were prepared by randomly placing the molecules in the simulation box based on their experimental densities and molar ratios. A cubic-shaped simulation box of length 1.357 nm was used for all dimensions. We performed DFT relaxation to equilibrate the structure after randomly packing the box. Then AIMD simulations were performed at 300 K using the NVT ensemble with a time step of 1.0 fs. Temperature oscillations were controlled using a Nose thermostat with a Nose-mass parameter of 1.0. A Monkhorst-Pack k-point mesh grid scheme with a 1 × 1 × 1 grid was used. The systems were equilibrated for at least 60 ps before the production run of 10 ps. The projected density of states (PDOS) was calculated and averaged over five different configurations (extracted from AIMD simulation snapshots) to represent an ensemble average. The HOMO, LUMO energy levels of salt/solvent molecules were directly calculated and optimized by a basis set of B3LYP/6-311 G + + (d, p). The HOMO and LUMO energy levels for solvation complex were calculated using the B3LYP/6-31 G (d, p) basis set for structure optimization, while the single point energies were calculated using the larger B3LYP/6-311 G + + (d, p) basis set. Radial distribution functions were obtained by the Visual Molecular Dynamics (VMD) software. The VESTA program was used to sample the most probable solvation shells from the simulation trajectory. In addition, ESPs of molecules were calculated with Gaussian09 program at the B3LYP/def2tzvp level and visualized by GaussView.
Data availability
The authors declare that all the relevant data within this paper and its Supplementary Information file are available from the corresponding author upon request.
References
Yang, C., Xin, S., Mai, L. & You, Y. Materials design for high‐safety sodium‐ion battery. Adv. Energy Mater. 11, 2000974 (2020).
Zhao, C. et al. Rational design of layered oxide materials for sodium-ion batteries. Science 370, 708–711 (2020).
Syali, M. S., Kumar, D., Mishra, K. & Kanchan, D. K. Recent advances in electrolytes for room-temperature sodium-sulfur batteries: a review. Energy Storage Mater. 31, 352–372 (2020).
Zheng, X. et al. Critical effects of electrolyte recipes for Li and Na metal batteries. Chem 7, 2312–2346 (2021).
Jin, Y. et al. Low-solvation electrolytes for high-voltage sodium-ion batteries. Nat. Energy 7, 718–725 (2022).
Yang, T. et al. Mega high utilization of sodium metal anodes enabled by single zinc atom sites. Nano Lett. 19, 7827–7835 (2019).
Pham, T. D., Bin Faheem, A., Kim, J., Kwak, K. & Lee, K.-K. Non-flammable electrolytes based on a fluorine-free salt for safe and high-voltage lithium metal batteries. Electrochim. Acta 458, 142496 (2023).
Liao, C. et al. Non‐flammable electrolyte with lithium nitrate as the only lithium salt for boosting ultra‐stable cycling and fire‐safety lithium metal batteries. Adv. Funct. Mater. 33, 1–13 (2023).
Liu, X. et al. Fluoride‐rich solid‐electrolyte‐interface enabling stable sodium metal batteries in high‐safe electrolytes. Adv. Funct. Mater. 31, 1–8 (2021).
Yang, Z. et al. Fire‐retardant, stable‐cycling and high‐safety sodium ion battery. Angew. Chem. Int. Ed. 60, 27086–27094 (2021).
Zheng, Q. et al. A cyclic phosphate-based battery electrolyte for high voltage and safe operation. Nat. Energy 5, 291–298 (2020).
Chen, S. et al. High-efficiency lithium metal batteries with fire-retardant electrolytes. Joule 2, 1548–1558 (2018).
Jiang, G., Liu, J., Wang, Z. & Ma, J. Stable non‐flammable phosphate electrolyte for lithium metal batteries via solvation regulation by the additive. Adv. Funct. Mater. 33, 1–9 (2023).
Cheng, H. et al. Emerging era of electrolyte solvation structure and interfacial model in batteries. ACS Energy Lett. 7, 490–513 (2022).
Yu, Z. et al. Molecular design for electrolyte solvents enabling energy-dense and long-cycling lithium metal batteries. Nat. Energy 5, 526–533 (2020).
Wu, H., Jia, H., Wang, C., Zhang, J. G. & Xu, W. Recent progress in understanding solid electrolyte interphase on lithium metal anodes. Adv. Energy Mater. 11, 2003092 (2020).
Zhou, J. et al. Low-temperature and high-rate sodium metal batteries enabled by electrolyte chemistry. Energy Storage Mater. 50, 47–54 (2022).
Ji, Y. et al. In situ probing the origin of interfacial instability of Na metal anode. Chem 9, 2943–2955 (2023).
Wang, J. et al. Superconcentrated electrolytes for a high-voltage lithium-ion battery. Nat. Commun. 7, 12032 (2016).
Yamada, Y., Wang, J., Ko, S., Watanabe, E. & Yamada, A. Advances and issues in developing salt-concentrated battery electrolytes. Nat. Energy 4, 269–280 (2019).
Wang, Y. et al. Enhanced sodium metal/electrolyte interface by a localized high-concentration electrolyte for sodium metal batteries: first-principles calculations and experimental studies. ACS Appl. Energy Mater. 4, 7376–7384 (2021).
Watanabe, Y., Ugata, Y., Ueno, K., Watanabe, M. & Dokko, K. Does Li-ion transport occur rapidly in localized high-concentration electrolytes? Phys. Chem. Chem. Phys. 25, 3092–3099 (2023).
Diederichsen, K. M., McShane, E. J. & McCloskey, B. D. Promising routes to a high Li+ transference number electrolyte for lithium ion batteries. ACS Energy Lett. 2, 2563–2575 (2017).
Peng, X., Lin, Y., Wang, Y., Li, Y. & Zhao, T. A lightweight localized high-concentration ether electrolyte for high-voltage Li-Ion and Li-metal batteries. Nano Energy 96, 107102 (2022).
Luo, C. et al. Synergistic-effect of diluent to reinforce anion-solvation-derived interfacial chemisty for 4.5 V-class Li||LiCoO2 batteries. Nano Energy 109, 108323 (2023).
Yoo, D. J., Yang, S., Kim, K. J. & Choi, J. W. Fluorinated aromatic diluent for high-performance lithium metal batteries. Angew. Chem. Int. Ed. 59, 14869–14876 (2020).
Zhang, J. et al. Weakly solvating cyclic ether electrolyte for high-voltage lithium metal batteries. ACS Energy Lett. 8, 1752–1761 (2023).
Tian, Z. et al. Electrolyte solvation structure design for sodium ion batteries. Adv. Sci. 9, e2201207 (2022).
Murray, J. S. & Politzer, P. Molecular electrostatic potentials and noncovalent interactions. WIREs Comput. Mol. Sci. 7, 1–10 (2017).
Lu, T. & Chen, F. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592 (2012).
Wu, Y. et al. Electrostatic potential as solvent descriptor to enable rational electrolyte design for lithium batteries. Adv. Energy Mater. 13, 2300259 (2023).
Zhao, Y. et al. Electrolyte engineering for highly inorganic solid electrolyte interphase in high-performance lithium metal batteries. Chem. 9, 682–697 (2023).
Xu, J. et al. Electrolyte design for Li-ion batteries under extreme operating conditions. Nature 614, 694–700 (2023).
Chen, J. et al. Design of localized high-concentration electrolytes via donor number. ACS Energy Lett. 8, 1723–1734 (2023).
Christian, L., Jean-François, G. Lewis Basicity and Affinity Scales: Data and Measurement, Vol. 476 (John Wiley& Sons, 2010).
Tu, H. et al. Tailoring electrolyte solvation for LiF-rich solid electrolyte interphase toward a stable Li anode. ACS Nano 16, 16898–16908 (2022).
Lee, S. H. et al. Toward the sustainable lithium metal batteries with a new electrolyte solvation chemistry. Adv. Energy Mater. 10, 2000567 (2020).
Li, Z. et al. Non-polar ether-based electrolyte solutions for stable high-voltage non-aqueous lithium metal batteries. Nat. Commun. 14, 868 (2023).
Zhang, H. et al. 1,3,5-Trifluorobenzene and fluorobenzene co-assisted electrolyte with thermodynamic and interfacial stabilities for high-voltage lithium metal battery. Energy Storage Mater. 48, 393–402 (2022).
Jiao, S. et al. Stable cycling of high-voltage lithium metal batteries in ether electrolytes. Nat. Energy 3, 739–746 (2018).
Kim, S., Lee, T. K., Kwak, S. K. & Choi, N.-S. Solid electrolyte interphase layers by using lithiophilic and electrochemically active ionic additives for lithium metal anodes. ACS Energy Lett. 7, 67–69 (2021).
Han, B. et al. Probing the Na metal solid electrolyte interphase via cryo-transmission electron microscopy. Nat. Commun. 12, 3066 (2021).
Wang, C. et al. A rooted interphase on sodium via in-situ pre-implanting atom fluorine for high-performance sodium metal batteries. Energy Environ. Sci. 16, 3098–3109 (2023).
Chen, X., Li, H. R., Shen, X. & Zhang, Q. The origin of the reduced reductive stability of ion-solvent complexes on alkali and alkaline earth metal anodes. Angew. Chem. Int. Ed. 57, 16643–16647 (2018).
Zhou, C. et al. Rational design of a carbonate-glyme hybrid electrolyte for practical anode-free lithium metal batteries. Energy Storage Mater. 42, 295–306 (2021).
Xu, R. et al. Identifying the critical anion-cation coordination to regulate the electric double layer for an efficient lithium-metal anode. Angew. Chem. Int. Ed. 60, 4215–4220 (2021).
Wu, Q., McDowell, M. T. & Qi, Y. Effect of the electric double layer (EDL) in multicomponent electrolyte reduction and solid electrolyte interphase (SEI) formation in lithium batteries. J. Am. Chem. Soc. 145, 2473–2484 (2023).
Ma, L. A., Naylor, A. J., Nyholm, L. & Younesi, R. Strategies for mitigating dissolution of solid electrolyte interphases in sodium-ion batteries. Interface Angew. Chem. Int. Ed. 60, 4855–4863 (2021).
Zhou, P. et al. Rationally designed fluorinated amide additive enables the stable operation of lithium metal batteries by regulating the interfacial chemistry. Nano Lett. 22, 5936–5943 (2022).
Adams, B. D., Zheng, J., Ren, X., Xu, W. & Zhang, J.-G. Accurate determination of coulombic efficiency for lithium metal anodes and lithium metal batteries. Adv. Energy Mater. 8, 1702097 (2018).
Pei, A., Zheng, G., Shi, F., Li, Y. & Cui, Y. Nanoscale nucleation and growth of electrodeposited lithium metal. Nano Lett. 17, 1132–1139 (2017).
Bai, P. et al. Formation of LiF-rich cathode-electrolyte interphase by electrolyte reduction. Angew. Chem. Int. Ed. 61, e202202731 (2022).
Rinkel, B. L. D., Vivek, J. P., Garcia-Araez, N. & Grey, C. P. Two electrolyte decomposition pathways at nickel-rich cathode surfaces in lithium-ion batteries. Energy Environ. Sci. 15, 3416–3438 (2022).
Kim, S. C. et al. High-entropy electrolytes for practical lithium metal batteries. Nat. Energy 8, 814–826 (2023).
Li, S. et al. Synergistic dual-additive electrolyte enables practical lithium-metal batteries. Angew. Chem. Int. Ed. 59, 14935–14941 (2020).
Hu, P. et al. Concentration-gradient prussian blue cathodes for Na-ion batteries. ACS Energy Lett. 5, 100–108 (2019).
Fan, X. et al. Non-flammable electrolyte enables Li-metal batteries with aggressive cathode chemistries. Nat. Nanotechnol. 13, 715–722 (2018).
Chen, Z. et al. Anion solvation reconfiguration enables high-voltage carbonate electrolytes for stable Zn/graphite cells. Angew. Chem. Int. Ed. 59, 21769–21777 (2020).
Borodin, O., Behl, W. & Jow, T. R. Oxidative stability and initial decomposition reactions of carbonate, sulfone, and alkyl phosphate-based. Electrolytes J. Phys. Chem. C. 117, 8661–8682 (2013).
Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (NSFC) projects (22179112 [J.F.], 22072117 [R.Y.]), innovative research group project of NSFC (22021001) [Q. D.].
Author information
Authors and Affiliations
Contributions
C.W. conceived the project. C.W. designed the experiments and analyzed the results. C.W., Z.S., Y.L., L.L., J.F., and R.Y. analyzed and interpreted the experimental data. X.Y. and J.Y. carried out AFM measurements. R.Y. performed the theoretical calculations. Q.H. performed the Raman experiments. C.W. and Z.S. drafted the paper with support from M.Z. and Q.D. All authors discussed the results and commented on the manuscript. The work was supervised by M.Z. and Q.D.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Nav Nidhi Rajput, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, C., Sun, Z., Liu, Y. et al. A weakly coordinating-intervention strategy for modulating Na+ solvation sheathes and constructing robust interphase in sodium-metal batteries. Nat Commun 15, 6292 (2024). https://doi.org/10.1038/s41467-024-50751-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-50751-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
