
Abstract
Deuterium labeling compounds play a crucial role in organic and pharmaceutical chemistry. The synthesis of such compounds typically involves deuterated building blocks, allowing for the incorporation of deuterium atoms and functional groups into a target molecule in a single step. Unfortunately, the limited availability of synthetic approaches to deuterated synthons has impeded progress in this field. Here, we present an approach utilizing alkyl-substituted thianthrenium salts that efficiently and selectively introduce deuterium at the α position of alkyl chains through a pH-dependent HIE process, using D2O as the deuterium source. The resulting α-deuterated alkyl thianthrenium salts, which bear two deuterium atoms, exhibit excellent selectivity and deuterium incorporation in electrophilic substitution reactions. Through in situ formation of isotopically labelled alkyl halides, these thianthrenium salts demonstrate excellent compatibility in a series of metallaphotoredox cross-electrophile coupling with (hetero)aryl, alkenyl, alkyl bromides, and other alkyl thianthrenium salts. Our technique allows for a wide range of substrates, high deuterium incorporation, and precise control over the site of deuterium insertion within a molecule such as the benzyl position, allylic position, or any alkyl chain in between, as well as neighboring heteroatoms. This makes it invaluable for synthesizing various deuterium-labeled compounds, especially those with pharmaceutical significance.
Similar content being viewed by others
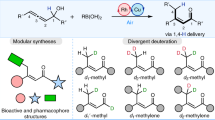

Introduction
Deuterium is a naturally occurring stable isotope of hydrogen containing only an additional neutron. Due to the higher stability of C−D bonds than C−H bonds, drug molecules labeled with deuterium are expected to have longer half-lives, increased efficacy and reduced side effects, making them possibly more desirable than the parent compounds1,2. Recently, deuterium-containing molecules such as Austedo (deutetrabenzine- d6)3 have been approved for use as new drugs, and other drugs like PHA-022121, RT001, and CTP-543 are currently being tested (Fig. 1a)4. Furthermore, deuterium-labeled compounds are valuable for studying kinetic isotope effects during mechanistic investigations and are widely used as internal standards in quantitative mass spectrometry5. Therefore, exploring the chemical space of deuterium-labelled molecules can open new doors to functional-molecule innovation and discovery in several scientific fields. Since that most top-selling commercial drugs contain at least one alkyl moiety, a general synthetic method for achieving isotopically labelled functionalized alkanes would be highly valuable6,7,8. The most popular and efficient synthetic methods, such as hydrogen isotope exchange (HIE)9,10,11,12, reductive deuteration13, and dehalogenative deuteration14,15, have been developed to introduce deuterium atoms directly to target molecules. However, synthesizing functionalized alkanes with precise control over deuterated sites, high efficiency and high deuterium incorporation remains challenging.

a Deuterium-containing drugs. b Deuteration of alkyl (pseudo)halides and carboxylic acids. c Reduction of aryl TT salts to access isotopically labelled arenes. d Metallaphotoredox-catalyzed cross-electrophile coupling of d2-labeled TT salts (This work).
The use of deuterated building blocks in synthetic approaches can lead to the efficient production of deuterium labeled compounds. This technique enables the introduction of both deuterium atoms and functional groups in a single step, resulting in the construction of a target molecule with easily controllable deuterated sites and a high level of deuterium incorporation. Although this method has been utilized in the synthesis of deuterium-containing drugs16,17,18,19,20, its progress has been hindered by the limited availability of readily accessible deuterated reagents. Alkyl halides (I, Br, and Cl) and pseudohalides (OTs, OTf) are widely applied and abundant electrophilic alkylation reagents within the realm of organic synthesis. Due to the low pKa of α C–H bonds and the reactivity of C–X bonds, HIE of alkyl (pseudo)halides to prepare isotopically labeled alkyl electrophiles can be synthetically challenging (Fig. 1b, up). Typically, indirect methods to access deuterated alkyl electrophiles are employed. The state-of-the-art progress involves the utilization of a ternary catalytic system, enabling the efficient α-deuteration of carboxylic acids (Fig. 1b, down)21. However, alkyl carboxylic acids, which possess low electrophilicity, necessitate the formation of redox activated esters to enable decarboxylative substitution. Therefore, chemists need to choose a functional group that possesses a good balance, capable of promoting HIE process while also exhibiting excellent leaving ability.
Sulfonium salts are highly versatile compounds characterized by the presence of positively charged sulfur ions with three organic substituents22,23,24,25,26,27,28,29,30. They serve as immensely useful aryl and alkyl electrophiles in a wide range of chemical reactions. Among these compounds, aryl thianthrenium (TT) salts are particularly renowned for their exceptional versatility as reactive intermediates31,32,33,34,35,36,37,38. Recently, Ritter and coworkers have successfully utilized these compounds for halogenation (F39, Cl, Br, and I40 and hydrogen isotope labeling41 by breaking carbon–sulfur bonds with the assistance of transition metals catalysis (Fig. 1c). Furthermore, alkyl TT salts have been investigated by our group42,43 and other researchers44,45, resulting in a noteworthy expansion of the chemical landscape for alkyl (pseudo)halides.
Here, we introduce a highly adaptable platform utilizing TT salts for the convenient generation of deuterium-labeled alkyl electrophiles. Through a simple pH-dependent hydrogen isotope exchange (HIE) process, a range of d2-labeled alkyl TT salts can be easily synthesized in a modular manner. These compounds possess distinctive benefits in contrast to aryl TT salts, as they can readily engage in electrophilic substitution reactions with a diverse array of inorganic salts, enabling the formation of alkyl halides, cyanides, and azides without the need for catalysts. In the presence of a nickel catalyst and a photocatalyst46,47,48,49, these salts can also undergo cross-electrophile couplings without the need for preformed organometallic reagents50,51,52,53. Such groundbreaking approach involves the formation of d2-labeled alkyl halides in situ, enabling the creation of carbon–carbon bonds with a broad range of aryl, heteroaryl, vinyl, and alkyl halides. Consequently, the precise control over the incorporation of the d2-methylene motif at specific positions in an organic molecule, such as the benzyl or allylic position, as well as within the alkyl chains or neighboring heteroatoms, has revolutionized the synthesis of isotopically labeled compounds.
Results
Reaction design
The unique reactivity of alkyl TT salts can be attributed to the positive charge located at the sulfur ring, which also contributes to the acidity of the α-hydrogen of TT salts. Computational studies have investigated the characteristics of an olefin-containing alkyl TT salt 1a, representing a typical exampleg (see Supplementary Table 2 and 3). These studies have revealed that the pKa value of the α-hydrogen in 1a, as well as the bond dissociation energy (BDE), are comparatively lower than those of conventional alkyl (pseudo)halides, including Cl (I), Br (II), I (III), OTs (IV), and OTf (VI) (Fig. 2a). These findings suggest that TT salts offer reactivity advantages for pH-dependent HIE of alkyl groups in equilibrium with D2O, as well as serving as attractive electrophiles. Based on these theoretical findings, we investigated the HIE reaction conditions using TT salt 1a as a model substrate (Fig. 2b). It was determined that the HIE reaction of 1a is most effective when conducted in MeCN solvent at room temperature (r.t.) for 6 h, using D2O as a readily available source of deuterium and 2.0 equivalents of K2CO3 as a base. The desired d2-TT salt 2a was obtained in 93% yield and 96% deuterium incorporation, while the formation of alcohol byproduct 2a’ was efficiently suppressed. Interestingly, we also discovered a switchable process that promotes the generation of alcohol 2a’ by simply changing the solvent to DMSO. As shown in Fig. 2c, we conducted further investigations to explore the reaction’s applicability. Remarkably, under the simple reaction conditions employed, we achieved highly efficient H/D exchange of alkyl TT salts encompassing diverse functional groups, including aryl (2b), heteroaryl (2c–e), phthalimid (2f), ether (2g–h), and trifluoroethyl (2i). Notably, the chlorine leaving group was effectively preserved in the resulting product 2j, showcasing its potential for subsequent nucleophilic substitution reactions. Moreover, we examined alkene-substituted TT salts (2k–l) and an internally alkyne-substituted TT salt 2m in detail. In these cases, the selective deuteration predominantly occurred at α position of the TT group. Notably, for the substrate 2n, featuring a terminal alkyne, exchange took place not only at the position adjacent to the TT moiety but also at the acetylenic C–H bond. Furthermore, we successfully synthesized d2-alkyl compounds (2o–q) with impressive efficiency from alkyl TT salts (1o–q) derived from natural sources such as lithocholic acid, estrone, and cholesterol. Due to their excellent leaving ability, these TT salts have been found to exhibit high reactivity in halogenation reactions, leading to the formation of isotopically labeled alkyl halides (Fig. 2d). For instance, considering the complex molecule 2o as an example, the reaction readily undergoes iodination (3oa), bromination (3ob), chlorination (3oc), and even fluorination (3od) with the related halide salts, resulting in good yields and deuterium incorporation. Moreover, other transformations such as trifluoromethylthiolation (3oe), cyanation (3of), and azidation (3og) can be also accomplished adeptly utilizing the related inorganic salts.

a Absolute pKa values of α C–H bond in water and the corresponding bond dissociation energy (BDE) of the C–X bond using the theory of M062X/6-311+g(2d,p), SMD solvation calculation (for details, see Supplementary Date). b HIE of TT salt 1a with D2O to 2a and substitution reaction to alcohol 2a’. c Facile construction of a library of d2-alkyl TT salts. d, Reaction of TT salt 2o with various inorganic salts for the production of isotopically labelled compounds. Deuterium incorporation was determined by 1H NMR spectroscopy and/or HRMS. *The yield determined by 1H NMR spectrum with CH2Br2 as a standard.
Based on the above results, we then focused on the cross-electrophile coupling of TT salt 2a (96% D) with aryl bromide 4a. A crucial step in this process involved the in situ formation of isotopically labelled alkyl halides (Fig. 3a). We conducted a systematic screening of reaction conditions and successfully obtained the desired coupling product 5aa with a remarkable 73% isolated yield and excellent deuterium incorporation (96% D). To achieve these results, we employed a catalytic system comprising a mixture of NiBr2· dtbpy (5.0 mol%) and 4CzIPN (1.0 mol%) as the photocatalyst. The reductant used was (TMS)3SiH, and we utilized 2.0 equivalents of Cs2CO3 as the base along with 2.0 equivalents of LiBr as a key additive. The reaction was carried out in a MeOAc/DMSO solvent mixture and exposed to blue LED light (440 nm) irradiation at room temperature for 12 h (entry 1). Control experiments were performed to assess the individual contributions of each component in the catalytic system. The absence of the photocatalyst (entry 2), nickel complex (entry 3), LiBr (entry 4), TMS3SiH (entry 5), Cs2CO3 (entry 6), or visible light (entry 7) resulted in poor outcomes, confirming the vital role played by each component. Notably, when we substituted the 4CzIPN photocatalyst with an iridium counterpart, product 5aa was still obtained, albeit with a slightly lower yield of 65% (entry 8). Ultimately, this sequential reaction can be scaled up to the gram level with a significant decrease in yield (entry 9). Furthermore, the resulting TT reagent shows nearly perfect recovery, almost reaching quantitative retrieval. Having optimized the reaction conditions, we conducted a comprehensive examination of the substrate scope for the cross-electrophile coupling reaction. The results are summarized in Fig. 3b. To evaluate the reactivity of different d2-alkyl TT salts, we selected methyl 4-bromobenzoate (4a) as the substrate. Remarkably, the reaction conditions exhibited excellent tolerance towards a wide range of functional groups. Various groups, including phenyl (5ba), thiophene (5ca), benzofuran (5da), carbazole (5ea), phthalimide (5fa), methoxy (5ga), benzyloxy (5ha), trifluoromethyl (5ia), and chloride (5ja), were easily accommodated. Furthermore, d2-alkyl TT salts carrying carbon-carbon double bonds (5ka, 5la) and triple bonds (5ma, 5na) could also be efficiently converted into the desired coupling products. Notably, when complex d2-alkyl TT salts derived from natural compounds were employed, the corresponding deuterated products (5oa–qa) were obtained without any significant impact on C–C bond formation and deuterium incorporation. These results underscore the practicality and versatility of this method.

a Optimizations of the reaction of aryl bromide 4a using TT salt 2a. b The scope of d2-TT salts coupled with aryl bromide 4a. Standard reaction conditions: aryl bromide 1c (0.2 mmol), TT salt (0.4 mmol), 4CzIPN (1.0 mol%), NiBr2•dtbpy (5.0 mol%), TMS3SiH (0.22 mmol), Cs2CO3 (0.4 mmol), LiBr (0.4 mmol), MeOAc (1.6 mL)/DMSO (0.4 mL), Blue LEDs, r.t., 24 h, isolated yields, deuterium incorporation was determined by 1H NMR spectroscopy and/or HRMS.
Encouraged by these results, we conducted further investigations into the reactivity of aryl bromides with d2-TT salt 2a (Fig. 4). Both electron-deficient (4b) and electron-rich (4c) aryl bromides proved to be suitable substrates, yielding the desired coupling products 5ab and 5ac in good yields with high deuterium incorporation. Even the steric hindrance of ortho-substituted aryl bromides did not impede the smooth progress of the reaction (5ad). Additionally, a range of heteroaryl bromides, such as pyridinyl (4e), quinolinyl (4f), isoquinolinyl (4g), indazolyl (4h), and pyrimidinyl (4i) bromides, displayed compatibility with this reaction, affording the desired d2-alkyl substituted heteroarenes 5ae-ai in good yields. The impressive functional group compatibility and excellent deuterium incorporation observed in these molecules prompted us to evaluate the reaction using commercially available pharmaceuticals and biologically active compounds. Substrates bearing amide or ester derivatives of natural products, such as nortropinone (4j), d-phenylalanine (4k), and d-glucose (4l), which possess carbonyl, ester, and amide functional groups susceptible to sensitivity, proved to be amenable substrates, yielding the corresponding products 5aj–al in good yields. Furthermore, drug molecule derivatives, such as Ezetimibe (3m), Indomethacin (4n), Sulfadimethoxine (4o), Thalidomide (4p), and the Celecoxib derivative 4q featuring an electron-neutral aryl bromide unit, all successfully underwent the reaction with d2-TT salt 2a, resulting in the formation of products 5am–aq with excellent deuterium incorporation. Expanding the substrate scope to derivatives of drugs containing heteroaryl bromide units, such as Linagliptin (4r) and Theophylline (4s), proved to be compatible with this transformation. Remarkably, even vinyl bromides showed reactivity as coupling partners. Both (E)-5-(2-bromovinyl)-1,2,3-trimethoxybenzene (4t) and styryl-containing complex molecules 4u–v participated effectively in the coupling reaction with 2a. Furthermore, treatment of brominated pirfenidone (4w) and Galactose (4x) under these conditions led to the formation of products with exceptional deuterium incorporation. Lastly, enol triflates derived from Pirfenidone (4y) and Indometacin (4z) also exhibited successive cross-coupling reactions.

Standard reaction conditions: aryl bromide (0.2 mmol), TT salt (0.4 mmol), 4CzIPN (1.0 mol%), NiBr2•dtbpy (5.0 mol%), TMS3SiH (0.22 mmol), Cs2CO3 (0.4 mmol), LiBr (0.4 mmol), MeOAc (1.6 mL)/DMSO (0.4 mL), Blue LEDs, r.t., 24 h, isolated yields, deuterium incorporation was determined by 1H NMR spectroscopy and/or HRMS.
Methods for the coupling of two alkyl centers are still relatively uncommon in the field of cross-electrophile coupling54. Our specific aim was to synthesize functionalized alkanes with a deuterium label located internally by utilizing alkyl bromides and d2-alkyl TT salts (Fig. 5a. For the optimization conditions, see details in Table S2 of SI). Our approach proved to be remarkably effective in coupling heterocyclic bromides, such as piperidines (6a–b) and azetidines (6c), which are commonly found in medicinal chemistry. These reactions resulted in the formation of the desired deuterated adducts 7aa–ac, displaying moderate yields but with high deuterium incorporation. Regarding acyclic systems, we discovered that both secondary (6d–f) and primary (6g–h) alkyl bromides were amenable to coupling with d2-TT salt 2a, allowing for the synthesis of the desired alkanes 7ad–ah. Notably, these reactions exhibited over 96% deuterium incorporation at specific positions. Additionally, other d2-alkyl TT salts, including 2b, 2h–j, were employed for reaction with alkyl bromide 6a, leading to the formation of desired products with high yields, alongside 96% deuterium incorporation. These findings underscore the remarkable potential and versatility of our metallaphotoredox-catalyzed cross-electrophile coupling reaction in the synthesis of functionalized alkanes bearing deuterium labels at internal positions. Based on the above results, we further investigated whether cross-electrophile coupling could occur between two alkyl TT salts. As shown in Fig. 5b, we conducted a coupling between alkyl TT salt 1a and 1f under optimized conditions. As a result, we obtained the desired coupling product 8 in a modest yield, simultaneously by-products of hydrogenation and self-coupling were formed. To achieve controlled distinct deuterated sites and high deuterium incorporation, we employed d2-TT salt 2a or 2f as reagent. This allowed us to obtain two isomers, 9 and 10. Additionally, when compound 2a and 2f was used as the substrates, we obtained the neighbouring d4-alkane 11 with high deuterium incorporation. Such finding opens up new possibilities for synthetic methodologies and the development of deuterated compounds with specific isotopic labelling patterns.

a Substrate scope of coupling alkyl bromides with d2-alkyl TT salts. b Cross-electrophile couplings between two alkyl TT salts. Reaction conditions: [6 (0.2 mmol), 2 (0.8 mmol)] or [1a/2a (0.2 mmol), 1f/2f (0.8 mmol)], 4CzIPN (3.0 mol%), NiBr2•dtbpy (15.0 mol%), (TMS)3SiOH (0.3 mmol), Cs2CO3 (0.4 mmol), Bu4NBr (0.8 mmol), MeOAc (1.6 mL)/DMF (0.4 mL), Blue LEDs, r.t., 16 h, isolated yields, deuterium incorporation was determined by 1H NMR spectroscopy and/or HRMS. a Using NiBr2•DME (15.0 mol%) and 1,3-Bis(4,5-dihydro-2-oxazolyl)benzene (15.0 mol%) instead of NiBr2•dtbpy (15.0 mol%).
Synthetic applications
The approval of deuterium-modified drugs such as Austedo and Donafenib has generated interest in the synthesis of drugs incorporating deuterium. To demonstrate the practicality of the metallaphotoredox process, we conducted a thorough investigation of the cross-electrophile coupling protocol by synthesizing deuterium-containing drugs using commercially available starting materials (Fig. 6). We successfully synthesized deuterated Prothionamide 1555, an orally administered antitubercular drug, by coupling d2-alkyl salt 12 with pyridinyl bromide 13, followed by sulfidation of the cyano group (Fig. 6a). Likewise, we achieved efficient synthesis of deuterated Metoprolol (18)56, a drug used for lung cancer treatment. In these syntheses, the d2-TT salt 2g was coupled with aryl bromide 16 to form the central C–C bond, which subsequently reacted with propan-2-amine to yield the desired molecule (Fig. 6b). Additionally, we utilized the developed method to access deuterated Bezafibrate (19)57, a medication used to regulate lipid levels in the blood. Treatment of d2-TT salt 2f with aryl bromide 4b resulted in the efficient formation of the 5fb product. Subsequent deprotection and benzoylation afforded the final product with satisfactory yield (Fig. 6c). The successful synthesis of these drugs with high deuterium incorporation underscores the compatibility and practicality of this method in terms of functional group transformations.

a Synthesis of d2-Prothionamide from TT salt 11. b Synthesis of d2-Metoprolol from TT salt 2g. c Synthesis of d2-Bezafibrate from TT salt 2f.
Discussion
Mechanistic investigation
Several experiments were then conducted to offer insights into the metallaphotocatalytic cross-coupling (Fig. 7). We first conducted an analysis to determine which species in the solution can absorb blue LED light at around 450 nm. By obtaining UV/vis absorption spectra of the components and their combinations, we identified the light-absorbing properties (Fig. 7a). The data clearly shows that a solution containing 4CzIPN can absorb light at 450 nm, while the nickel complex solution cannot. Furthermore, the TT salt 2b exhibited no discernible absorption within the spectrum of visible light, and the introduction of LiBr induced a blue shift, elucidating the emergence of a new compound. To further substantiate the assertion, a reaction profile between the reactions of TT salt 2b and aryl bromide 4a under standard conditions was conducted (Fig. 7b). As the product 5ba emerged, substrate 4a gradually diminished, concomitant with the rapid generation of a significant quantity of alkyl bromide 20 right from the outset58. A competition reaction between TT salt 2a and alkyl bromide 21 resulted in the formation of a product mixture containing 5aa and 22 in a ratio of 1.2/1 (Fig. 7c). This observation further suggests that the bromination process is rapid, exerting no influence on the overall rate of the reaction. When TEMPO was used as a radical scavenger in the reaction, the TEMPO-captured product 23 was detected by HRMS, while the coupling product 5aa was not observed (Fig. 7d). Furthermore, when a well-designed TT salt 24, containing a styrene unit, was used as a substrate, a trace amount of direct coupling product 25 was observed. Additionally, compound 26, resulting from the cascade of a 5-exo cyclization followed by coupling with aryl bromide 4a, was isolated in 31% yield. These observations strongly support the formation of alkyl radicals during the reaction.

a UV/vis study of the reaction components. b Tracing the reaction between 2b and 4a. c Competition reaction between alkyl TT salt 2a and alkyl bromide 21. d Radical trapping experiments.
In order to delve deeper into the conversion process of TT salts into alkyl bromides, we first conducted density functional theory (DFT) calculations pertaining in into the bromination of TT salt 1b (Fig. 8a). TBAB attacks 1n through the transition state TS1A in an SN2 manner, with an energy barrier of 17.1 kcal mol−1, which is much lower compared to the intramolecular SN2 nucleophilic substitution via transition state TS2B (17.1 vs 35.2 kcal mol−1). The mechanism involving SN1-typed nucleophilic attack is well excluded, as the C–S bond cleavage requires a high activation free energy of 36.9 kcal mol−1 (see Supplementary Fig. 10 for details). Based on the above results and previous studies59, a proposed mechanism for the reaction is presented in Fig. 8b. The photoredox catalyst 4CzlPN absorbs photons and gets excited to its photoexcited state (4CzlPN*). This species further undergoes single-electron oxidation of a bromine anion, generating a bromine radical (Br•) and the reduced photocomplex 4CzlPN•−. The electrophilic bromine radical (Br•) abstracts a hydrogen atom from TMS3SiH, resulting in the formation of a stabilized silyl radical (TMS3Si•). Meanwhile, bromination of alkyl TT salt 2 leads to the formation of the alkyl bromide I. This compound then undergoes bromine atom abstraction by the silyl radical (TMS3Si•), producing the corresponding nucleophilic alkyl radical species II. Simultaneously, oxidative addition of a Ni0 complex A to the aryl bromide 4 yields an intermediate B. This species undergoes radical addition with II, giving rise to the intermediate C. Importantly, an excess amount of alkyl source and silane is utilized during this step to enhance the selectivity for the desired cross-coupling product60. Further reductive elimination of C generates the desired product 5, and nickel complex D. Single-electron reduction of D by the reduced organic photocatalyst (4CzlPN•−) completes both the photocatalytic cycle and the nickel catalytic cycle, leading to the restoration of the starting Ni0 catalyst A and photocatalysis.

a DFT-calculated energy profile for the bromination of alkyl TT salt 1b with TBAB. b Full elucidated mechanism of the nickel cycle (left) and photoredox cycle (right).
In summary, a robust method has been developed for the synthesis of versatile alkanes containing two deuterium atoms. A library of novel deuteroalkyl reagents was generated by subjecting alkyl-substituted TT salts to a pH-dependent HIE process using D2O as the source of deuterium. These readily available d2-TT salts can then undergo a metallaphotoredox-catalyzed cross-electrophile coupling with a range of electrophiles, including aryl, heteroaryl, alkenyl, and alkyl bromides, as well as other alkyl TT salts. Our approach possesses several notable strengths. Firstly, it employs commercially available catalysts, rendering it practical and accessible for wide-ranging applications. Secondly, it provides precise control over the placement of deuterium atoms, ensuring cross selectivity and regioselectivity. Importantly, the reaction conditions do not necessitate the use of metal reductants, thereby simplifying the process and circumventing the need for additional steps. Additionally, our strategy accomplishes high levels of deuterium incorporation, enabling accurate positioning of deuterium atoms within the synthesized compounds. We anticipate that this powerful platform will facilitates research in medicine, biology, and chemistry.
Methods
General procedures for metallaphotoredox-catalyzed Csp2-Csp3 coupling
In a nitrogen-filled glove box, alkyl TT salt (0.4 mmol, 2.0 equiv.), Cs2CO3 (130.3 mg, 0.4 mmol, 2.0 equiv.), LiBr (34.7 mg, 0.4 mmol, 2.0 equiv.), 4CzIPN (1.8 mg, 1 mol%), and NiBr2•dtbpy (4.9 mg, 5 mol%) were added to an 8 mL oven-dried vial equipped with a stir bar. Then, anhydrous MeOAc (1.6 mL) and anhydrous DMSO (0.4 mL) were added using a syringe, followed by the addition of (hetero)aryl bromide (0.2 mmol, 1.0 equiv.) and tris(trimethylsilyl)silane (68 µL, 0.22 mmol, 1.1 equiv.). The vial was sealed and removed from the glovebox. Subsequently, the reaction mixture was stirred and irradiated with a 40 W blue LED lamp for 12 h. The final reaction mixture was diluted with EtOAc (60 mL) and saturated aqueous LiCl solution (20 mL). The organic layer was washed with brine (2 × 20 mL) and concentrated. The aryl alkylation product was purified by flash column chromatography on silica gel.
General procedures for metallaphotoredox-catalyzed Csp3-Csp3 coupling
In a nitrogen-filled glove box, alkyl TT salts (0.8 mmol, 4.0 equiv), Cs2CO3 (130.3 mg, 0.4 mmol, 2.0 equiv), TBAB (257.9 mg, 0.8 mmol, 4.0 equiv), 4CzIPN (4.8 mg, 3 mol%), and NiBr2•dtbpy (14.6 mg, 15 mol%) were added to an 8 mL oven-dried vial equipped with a stir bar. Anhydrous MeOAc (1.6 mL) and anhydrous DMF (0.4 mL) were then added via a syringe, followed by the addition of an alkyl bromide (0.2 mmol, 1.0 equiv) and tris(trimethylsilyl)silanol (95 µL, 0.3 mmol, 1.5 equiv). The vial was sealed and removed from the glovebox. Subsequently, the reaction mixture was stirred and irradiated with a 40 W blue LED lamp for 16 h. The final reaction mixture was diluted with EtOAc (60 mL) and saturated aqueous LiCl solution (20 mL). The organic layer was washed with brine (2 × 20 mL) and concentrated. The alkyl alkylation product was purified by flash column chromatography on silica gel.
Data availability
The data supporting the findings of this study are available within the paper and its Supplementary Information files. Coordinates of the optimized structures are provided in a source data file. Raw data are available from the corresponding author on request. Source data are provided with this paper.
References
Mullard, A. Deuterated drugs draw heavier backing. Nat. Rev. Drug Discov. 15, 219–221 (2016).
Pirali, T., Serafini, M., Cargnin, S. & Genazzani, A. A. Applications of deuterium in medicinal chemistry. J. Med. Chem. 62, 5276–5297 (2019).
Schmidt, C. First deuterated drug approved. Nat. Biotechnol. 35, 493–494 (2017).
Mullard, A. First de novo deuterated drug poised for approval. Nat. Rev. Drug Discov. 21, 623–625 (2022).
Wiberg, K. B. The deuterium isotope effect. Chem. Rev. 55, 713–734 (1955).
Suna, Q. & Soulé, J.-F. Broadening of horizons in the synthesis of CD3-labeled molecules. Chem. Soc. Rev. 50, 10806–10835 (2021).
Li, N. et al. Radical deuteration. Chem. Soc. Rev. 51, 6291–6306 (2022).
Kopf, S. et al. Recent developments for the deuterium and tritium labeling of organic molecules. Chem. Rev. 122, 6634–6718 (2022).
Atzrodt, J., Derdau, V., Fey, T. & Zimmermann, J. The renaissance of H/D exchange. Angew. Chem. Int. Ed. 46, 7744–7765 (2007).
Prakash, G. et al. C–H deuteration of organic compounds and potential drug candidates. Chem. Soc. Rev. 51, 3123–3163 (2022).
Li, H. et al. One-pot sequential hydrogen isotope exchange/reductive deuteration for the preparation of α,β-deuterated alcohols using deuterium oxide. Org. Lett. 24, 5319–5323 (2022).
Peng, M. et al. Pentafluorophenyl group as activating group: Synthesis of α-deuterio carboxylic acid derivatives via Et3N catalyzed H/D exchange. Adv. Synth. Catal. 364, 2184–2189 (2022).
Nishimura, S. Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis (Wiley-VCH, New York, 2001).
Li, Y. et al. Organophotocatalytic selective deuterodehalogenation of aryl or alkyl chlorides. Nat. Commun. 12, 2894 (2021).
Li, P. et al. Facile and general electrochemical deuteration of unactivated alkyl halides. Nat. Commun. 13, 3774 (2022).
Hesk, D. et al. Synthesis of 3H, 2H4, and 14C-MK 3814 (Preladenant). J. Label. Compd. Radiopharm. 60, 194–199 (2017).
Dietz, C. et al. Convergent synthesis of a deuterium-labeled serine dipeptide lipid for analysis of biological samples. J. Label. Compd. Radiopharm. 60, 274–285 (2017).
Pedras, M. S. C. & To, Q. H. Synthesis of stable isotope-labeled nasturlexins and potential precursors to probe biosynthetic pathways of cruciferous phytoalexins. J. Label. Compd. Radiopharm. 61, 94–106 (2018).
Mosaferi, S., Jelley, R. E., Fedrizzi, B. & Barker, D. Scalable synthesis of the aroma compounds d6-β-ionone and d6-β-cyclocitral for use as internal standards in stable isotope dilution assays. Tetrahedron Lett. 61, 152642 (2020).
Bergare, J. et al. The synthesis of one H-2 labeled and two H-3 labeled leukotriene C4 synthase inhibitors. J. Label. Compd. Radiopharm. 63, 434–441 (2020).
Tanaka, T. et al. Ternary catalytic α-deuteration of carboxylic acids. Nat. Synth. 1, 824–830 (2022).
Kaiser, D. et al. Bond-forming and -breaking reactions at sulfur(IV): sulfoxides, sulfonium salts, sulfur ylides, and sulfinate salts. Chem. Rev. 119, 8701–8780 (2019).
Lou, J. et al. Transition-metal mediated carbon–sulfur bond activation and transformations: an update. Chem. Soc. Rev. 49, 4307–4359 (2020).
Peter, A., Perry, G. J. P. & Procter, D. J. Radical C–C bond formation using sulfonium salts and light. Adv. Synth. Catal. 362, 2135–2142 (2020).
Fan, R. et al. A leap forward in sulfonium salt and sulfur ylide chemistry. Chin. Chem. Lett. 32, 299–312 (2021).
Yorimitsu, H. Catalytic transformations of sulfonium salts via C-S bond activation. Chem. Rec. 21, 3356–3336 (2021).
Aukland, M. H. et al. Metal-free photoredox-catalysed formal C–H/C–H coupling of arenes enabled by interrupted pummerer activation. Nat. Catal. 3, 163–169 (2020).
Wang, D. J., Targos, K. & Wickens, Z. K. Electrochemical synthesis of allylic amines from terminal alkenes and secondary amines. J. Am. Chem. Soc. 143, 21503–21510 (2021).
Liu, M.-S., Du, H.-W. & Shu, W. Metal-free allylic C–H nitrogenation, oxygenation, and carbonation of alkenes by thianthrenation. Chem. Sci. 13, 1003–1008 (2022).
Liu, M.-S., Du, H.-W., Cui, J.-F. & Shu, W. Intermolecular metal-free cyclopropanation and aziridination of alkenes with XH2 (X = N, C) by thianthrenation. Angew. Chem. Int. Ed. 61, e202209929 (2022).
Meng, H., Liu, M.-S. & Shu, W. Organothianthrenium salts: synthesis and utilization. Chem. Sci. 13, 13690–13707 (2022).
Engl, P. S. et al. C–N cross-couplings for site-selective late-stage diversification via aryl sulfonium salts. J. Am. Chem. Soc. 141, 13346–13351 (2019).
Sang, R. et al. Site-selective C−H oxygenation via aryl sulfonium salts. Angew. Chem. Int. Ed. 58, 16161–16166 (2019).
Xu, P. et al. Site-selective late-stage aromatic [18F]fluorination via aryl sulfonium salts. Angew. Chem. Int. Ed. 59, 1956–1960 (2020).
Cai, Y. & Ritter, T. Meerwein-type bromoarylation with arylTT salts. Angew. Chem. Int. Ed. 61, e202209882 (2022).
Berger, F. et al. Site-selective and versatile aromatic C−H functionalization by thianthrenation. Nature 567, 223–228 (2019).
Juliá, F. et al. High site selectivity in electrophilic aromatic substitutions: mechanism of C–H thianthrenation. J. Am. Chem. Soc. 143, 16041–16054 (2021).
Lansbergen, B., Granatino, P. & Ritter, T. Site-selective C–H alkylation of complex arenes by a two-step aryl thianthrenation-reductive alkylation sequence. J. Am. Chem. Soc. 143, 7909–7914 (2021).
Li, J. et al. Photoredox catalysis with aryl sulfonium salts enables site-selective late-stage fluorination. Nat. Chem. 12, 56–62 (2020).
Nie, S. et al. Nickel meets aryl thianthrenium salts: Ni(I)-catalyzed halogenation of arenes. J. Am. Chem. Soc. 145, 9988−9993 (2023).
Zhao, D., Petzold, R., Yan, J., Muri, D. & Ritter, T. Tritiation of aryl TT salts with a molecular palladium catalyst. Nature 600, 444–449 (2021).
Chen, C. et al. Enabling the use of alkyl thianthrenium salts in cross-coupling reactions by copper catalysis. Angew. Chem. Int. Ed. 60, 21756–21760 (2021).
Chen, C. et al. Generation of non-stabilized alkyl radicals from thianthrenium salts for C-B and C-C bond formation. Nat. Commun. 12, 4526 (2021).
Holst, D. E., Wang, D. J., Kim, M. J., Guzei, I. A. & Wickens, Z. K. Aziridine synthesis by coupling amines and alkenes via an electrogenerated dication. Nature 596, 74–79 (2021).
Alvarez, E. M. et al. O-, N- and C-bicyclopentylation using thianthrenium reagents. Nat. Synth. 2, 548–556 (2023).
Twilton, J. et al. The merger of transition metal and photocatalysis. Nat. Rev. Chem. 1, 0052 (2017).
Tellis, J. C., Primer, D. N. & Molander, G. A. Single-electron transmetalation in organoboron cross-coupling by photoredox/nickel dual catalysis. Science 345, 433–436 (2014).
Zuo, Z. W. et al. Merging photoredox with nickel catalysis: coupling of α-carboxyl sp3-carbons with aryl halides. Science 345, 437–440 (2014).
Pipal, R. W. et al. Metallaphotoredox aryl and alkyl radiomethylation for PET ligand discovery. Nature 589, 542–547 (2021).
Weix, D. J. Methods and mechanisms for cross-electrophile coupling of Csp2 halides with alkyl electrophiles. Acc. Chem. Res. 48, 1767–1775 (2015).
Poremba, K. E., Dibrell, S. E. & Reisman, S. E. Nickel-catalyzed enantioselective reductive cross-coupling reactions. ACS Catal. 10, 8237−8246 (2020).
Liu, J., Ye, Y., Sessler, J. L. & Gong, H. Cross-electrophile couplings of activated and sterically hindered halides and alcohol derivatives. Acc. Chem. Res. 53, 1833–1845 (2020).
Yi, L. et al. Nickel-catalyzed reductive cross-couplings: new opportunities for carbon–carbon bond formations through photochemistry and electrochemistry. CCS Chem. 4, 9–30 (2022).
Jana, R., Pathak, T. P. & Sigman, M. S. Advances in transition metal (Pd,Ni,Fe)-catalyzed cross-coupling reactions using alkyl-organometallics as reaction partners. Chem. Rev. 111, 1417–1492 (2011).
Wang, F. et al. Mechanism of thioamide drug action against tuberculosis and leprosy. J. Exp. Med. 204, 73–78 (2007).
Grassi, G. Metoprolol in the treatment of cardiovascular disease: a critical reappraisal. Curr. Med. Res. Opin. 34, 1635–1643 (2018).
Witte, E. C. et al. 4-(Benzamidoalkyl)phenoxyalkanoic acids and esters—with hypolipaemic and hypocholesterolaemic activity; German Patent DE 2149070; April 5, 1973.
Li, Z. et al. Electrochemically enabled, nickel-catalyzed dehydroxylative cross-coupling of alcohols with aryl halides. J. Am. Chem. Soc. 143, 3536–3543 (2021).
Shang, T.-Y. et al. Recent advances of 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (4CzIPN) in photocatalytic transformations. Chem. Commun. 55, 5408–5419 (2019).
Ben-Tal, Y. & Lloyd-Jones, G. C. Kinetics of a Ni/Ir-photocatalyzed coupling of ArBr with RBr: intermediacy of ArNiII(L)Br and rate/selectivity. Factors J. Am. Chem. Soc. 144, 15372–15382 (2022).
Acknowledgements
We would like to thank the Financial support from National Key R&D Program of China (2022YFA1503200), National Natural Science Foundation of China (Grants 22025104, 22171134, 21972064, 22071100, 22271148 and 21901111), the Fundamental Research Funds for the Central Universities (Grant 020514380254) for their financial support. We are also grateful to the High-Performance Computing Center of Nanjing University for performing the numerical calculations in this paper on its blade cluster system.
Author information
Authors and Affiliations
Contributions
M.J. & J.Z. performed and analyzed experiments. M.W. performed the DFT calculation. L.H. & Z.S. supervised the project and wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Volker Derdau and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jiao, M., Zhang, J., Wang, M. et al. Metallaphotoredox deuteroalkylation utilizing thianthrenium salts. Nat Commun 15, 5067 (2024). https://doi.org/10.1038/s41467-024-48590-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-48590-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
