
Abstract
Halide perovskites show great optoelectronic performance, but their favorable properties are paired with unusually strong anharmonicity. It was proposed that this combination derives from the ns2 electron configuration of octahedral cations and associated pseudo-Jahn–Teller effect. We show that such cations are not a prerequisite for the strong anharmonicity and low-energy lattice dynamics encountered in these materials. We combine X-ray diffraction, infrared and Raman spectroscopies, and molecular dynamics to contrast the lattice dynamics of CsSrBr3 with those of CsPbBr3, two compounds that are structurally similar but with the former lacking ns2 cations with the propensity to form electron lone pairs. We exploit low-frequency diffusive Raman scattering, nominally symmetry-forbidden in the cubic phase, as a fingerprint of anharmonicity and reveal that low-frequency tilting occurs irrespective of octahedral cation electron configuration. This highlights the role of structure in perovskite lattice dynamics, providing design rules for the emerging class of soft perovskite semiconductors.
Similar content being viewed by others
Introduction
Halide perovskites (HaPs) with formula AMX3 generated enormous research interest because of their outstanding performance in optoelectronic devices, most notably in efficient solar cells1,2,3. These compounds are highly unusual among the established semiconductors because they feature an intriguing combination of properties. Strong anharmonic fluctuations4,5,6 in these soft materials appear together with optoelectronic characteristics that are favorable for technological applications7,8. This confluence raised puzzling questions regarding the microscopic characteristics of the materials and the compositional tuning of their properties alike. On the one hand, the soft anharmonic nature of the HaP structure may be beneficial in self-healing mechanisms of the material9,10,11, allowing for low-energy synthesis routes in their fabrication. On the other hand, pairing of anharmonic fluctuations and optoelectronic processes for key quantities of HaPs, e.g., band gaps12,13,14,15, optical absorption profiles16,17,18, and charge-carrier mobilities8,19,20,21,22,23,24,25, exposed incomplete microscopic rationales for the fundamental physical processes involved in solar-energy conversion. Established materials design rules are now being challenged by these observations, opening a gap in our protocols for making improved compounds.
Significant efforts are now underway to discern the chemical effects giving rise to these remarkable properties of HaPs. Because lattice dynamical and optoelectronic properties appear both to be special and coupled in unusual ways, a common origin in chemical bonding could underlie these phenomena. In this context, an interesting chemical feature is that the octahedral cations in these compounds often bear an ns2 electron configuration (e.g., Pb2+ with configuration [Xe]6s2), which is not present in many other semiconductors26. This particular aspect of HaPs leads to a strong or weak pseudo-Jahn–Teller (PJT) effect27,28,29, depending on the particulars of cation and anion composition and chemical pressure. The weak case influences local structure30,31,32, lattice dynamics33 and ionic dielectric responses26,31,34,35, while the strong case additionally results in the formation of a stereochemically-expressed electron lone pair and impacts average crystal structures33,36,37,38. The weak PJT effect associated with 6s2 Pb2+ coordinated by heavy halides plays a role in optoelectronic properties of these materials: its influence on the dielectric function can modify the Coulomb screening that is relevant for small exciton binding energies, reduced recombination rates and other key properties of HaPs39,40.
Confluences of the propensity for lone-pair formation with structural and lattice-dynamical properties were investigated in previous work exploring the chemical space of HaPs. Gao et al.33 found an inverse relationship between the Goldschmidt tolerance factor, t41, and anharmonic octahedral tilting motions. Similarly, Huang et al. varied the A-site cation to explore interrelations of chemical, structural, and dynamical effects in HaPs35, reporting t-induced modulations of octahedral tiltings and lone-pair stereoactivity. A recent study by several of the present authors found that Cs2AgBiBr6 lacks some expressions of lattice anharmonicity found in other HaP variants42. Because every other octahedral cation (Ag+, 4d10) cannot form a lone pair in this compound, this raised the possibility that changing the electron configuration of the cations may also suppress certain aspects of the lattice dynamics in HaPs. Taken together, previous work assigned a central role of the ns2 electron configuration and associated PJT effect in the anharmonic lattice dynamics of HaPs in addition to their established effect on the electronic structure and dielectric screening. However, exploring the chemical space of HaPs in this way simultaneously changes their structures. Therefore, isolating the convoluted occurrences of cation lone-pair formation propensity and purely structurally-determined changes in the lattice dynamics of HaPs remained challenging, making an assessment of the precise impact of chemical bonding on anharmonicity in these soft semiconductors largely inaccessible.
Here, we address this issue and show that an ns2 cation compatible with lone-pair formation is not required for the strong anharmonicity in the low-energy lattice dynamics of soft HaP semiconductors. We disentangle structural and chemical effects in the lattice dynamics of HaPs by comparing the well-known CsPbBr3 with the far less studied CsSrBr3. Both exhibit almost identical geometrical and structural parameters, but CsSrBr3 exhibits a negligible PJT effect on the octahedral Sr2+ site, owing to substantially weaker vibronic coupling to degenerate excited states than in the Pb2+ case (see Supplementary Note 1), allowing separation of the effects of the ns2 electron configuration and the geometry on the lattice dynamics in a direct manner. Combining electronic structure and molecular dynamics (MD) calculations with X-ray diffraction (XRD), infrared (IR) and Raman spectroscopies, we assess a key fingerprint of vibrational anharmonicity, i.e., the Raman central peak, which is a broad peak towards zero frequency in the Raman spectrum resulting from diffusive inelastic scattering26,33,34,35,43,44,45. While the electronic structure and dielectric properties of CsPbBr3 and CsSrBr3 are very different, their vibrational anharmonicities are found to be remarkably similar. In particular, the crucial dynamic octahedral tiltings giving rise to the Raman central peak are still present even in the absence of ns2 octahedral cations in CsSrBr3. Our results provide microscopic understanding of precisely how the propensity for lone-pair formation influences the anharmonic octahedral tiltings that dynamically break the average cubic symmetry in both compounds, and rule out the weak PJT effect associated with the ns2 main-group cations as the sole reason for the appearance of such anharmonicity in soft HaPs. These findings are important for chemical tuning of HaPs needed for new materials design.
Results
Electronic structure and bonding
We first investigate the electronic structure and bonding of CsPbBr3 and CsSrBr3 using density-functional theory (DFT). Figure 1 shows their band structure, total and projected density of states (DOS), as well as the total and projected crystal-orbital Hamilton population (COHP) of the high-temperature cubic phases of CsPbBr3 and CsSrBr3. The electronic band structure and bonding of CsPbBr3 were extensively investigated before46: the conduction band minimum (CBM) is formed by anti-bonding interactions (positive COHP in Fig. 1c) between Pb-6p and Br-4p/Br-4s orbitals, while the valence band maximum (VBM) is formed by anti-bonding interactions between Br-4p and Pb-6s orbitals.

DFT-computed electronic band structure of cubic CsPbBr3 a and corresponding total and projected density of states (DOS, b) and crystal-orbital Hamilton population (COHP, c). Panels d–f show the same data for CsSrBr3.
The electronic structure of CsSrBr3 exhibits entirely different characteristics26,47, especially a much larger band gap and weaker covalent interactions. Notably, the magnitude of the COHP is significantly reduced with respect to that of CsPbBr3, indicating much greater ionicity, and the COHP is almost entirely recovered by interactions between Cs and Br. Importantly, all bands derived from antibonding interactions between Sr-5s and Br-4p/Br-4s are empty due to the electron configuration of Sr2+ ([Kr]), and there is no potential for lone pair formation on Sr2+. A manifestation of the lack of ns2 cations in CsSrBr3 is that there is no cross-gap hybridization of the halide valence orbitals. By contrast, Br-4p orbitals hybridize with Pb-6p across the gap of CsPbBr3 (see the pCOHP in Fig. 1c). This leads to large Born effective charges, i.e., large changes in the macroscopic polarization upon ionic displacements48,49,50,51 reported in Table 1, which for CsPbBr3 are more than double the formal charge of Pb (+2) and Br (-1) and much larger than the corresponding values for CsSrBr3. Similarly, there is also a larger electronic contribution to the dielectric response in CsPbBr3 and it features a larger value of the dielectric function at the high-frequency limit (ε∞) compared to CsSrBr3.
Structural properties and phase transitions
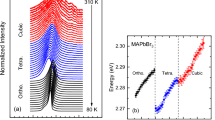
In spite of the markedly different electronic structure and bonding characteristics, CsSrBr3 and CsPbBr3 exhibit the same high-temperature cubic crystal structure (\(Pm\bar{3}m\)) and very similar lattice parameters (see Supplementary Note 3). One can rationalize this through the nearly identical ionic radii of Pb2+ and Sr2+ (119 and 118 pm) and the resulting Goldschmidt factors for the compounds (0.862 and 0.865). Furthermore, both materials exhibit the same sequence of structural phase transitions from the high-temperature cubic to the low-temperature orthorhombic phase (with an intermediate tetragonal phase), as shown by temperature-dependent lattice parameters in Fig. 2 that were determined via XRD. The cubic-to-tetragonal phase transition temperature of CsSrBr3 (~520 K) is noticeably higher than that of CsPbBr3 (~400 K)52,53 and slightly higher (~10 K) than that reported for Eu-doped CsSrBr3:Eu 5%54. The volumetric thermal expansion coefficient (αV) of CsSrBr3 ( ~ 1.32 × 10−4 K−1 at 300 K) is large and similar to that of CsPbBr3 ( ~ 1.29 × 10−4 K−1, see the Supplementary Note 3), in good agreement with the one reported for CsSrBr3:Eu54. Just as for other inorganic HaPs, αV of CsSrBr3 slightly decreases with temperature55,56. The similarity of geometric factors and structural phase transitions suggests that the octahedral tilting dynamics in CsSrBr3 might be similar to those in CsPbBr3, which contrasts with their markedly different electronic structure, and prompts a deeper investigation of the impact of the ns2 cations on structural dynamics.

Temperature-dependent lattice parameters of CsPbBr3 a and CsSrBr3 b determined by XRD throughout the orthorhombic—tetragonal—cubic phases. We show reduced lattice parameters \(\tilde{a}\), \(\tilde{b}\) and \(\tilde{c}\) for better visualization, with the orthorhombic phase expressed in the Pbnm setting. Dashed vertical lines indicate phase-transition temperatures. Error bars from Pawley fitting are smaller than the markers and are omitted.
Lower-temperature lattice dynamics
We conduct IR and Raman spectroscopy at different temperatures as well as DFT-based harmonic-phonon calculations. The measured IR spectra show that the dominant CsSrBr3 features are blue-shifted compared to those of CsPbBr3 (see Fig. 3a). Indeed, our DFT calculations of IR activities find a significant softening of the infrared-active TO modes in CsPbBr3 compared to those in CsSrBr3 (see Fig. 3b): the most prominent IR-active TO mode in CsPbBr3 and CsSrBr3 appears at ~68 and 146 cm−1, respectively, corresponding to the same irreducible representation (B3u) with similar eigenvectors (see Supplementary Fig. 9) in each system. This is in line with the theory of weak PJT effects in general29 and expectations for lone pairs in particular, with significant softening of ungerade modes in CsPbBr3 that would correspond to lone-pair formation in the strong PJT case relative to those in CsSrBr3. Notably, this softening is primarily driven by differences in bonding rather than the difference in the atomic masses (see Supplementary Note 7).

a IR-reflectivity spectra (dashed curves) and fitted imaginary part of the dielectric function (solid curves, see Supplementary Note 4 for details) of CsPbBr3 and CsSrBr3 measured at room temperature. b DFT-calculated IR-absorption spectra within the harmonic approximation for the orthorhombic phases. c Raman spectra of orthorhombic CsPbBr3 and CsSrBr3 measured at 80 K. d DFT-calculated Raman spectra of both compounds within the harmonic approximation for the orthorhombic phases.
Moreover, the LO/TO splitting is enhanced in CsPbBr3 compared to in CsSrBr3 and the LO phonon modes are hardened. Related to this, the CsPbBr3 IR spectrum exhibits a broad feature which is known as the Reststrahlen band as has been reported before for MA-based HaPs57. This particular effect results in near-zero transmission through the material in a frequency range between the TO and LO modes, represented by high IR intensity values, and occurs in polar materials with larger Born-effective charges. Because the TO modes are softened and the LO modes hardened in CsPbBr3 compared to CsSrBr3, and because the latter is less polar (cf. Table 1}, the absence of the ns2 cations leads to a much less pronounced, blue-shifted Reststrahlen band appearing in a smaller frequency window in CsSrBr3 (see Fig. 3a, and Supplementary Note 4).
Figure 3c shows the 80 K Raman spectra of CsPbBr3 and CsSrBr3, which are in good agreement with the Raman activities calculated for harmonic phonons (Fig. 3d). Specifically, the experimental spectrum of CsPbBr3 in Fig. 3b finds three broader features at frequencies below and one weaker-intensity feature at frequencies above 100 cm−1. Conversely, CsSrBr3 exhibits a structured feature around 50 cm−1, a pronounced signal close to 100 cm−1, and then a series of weaker intensities between 100 and 150 cm−1. While the DFT-computed Raman activities calculated in the harmonic approximation are in broad agreement with these findings (see Fig. 3d), we note a slightly larger deviation of approximately 20 cm−1 for the higher-frequency peak in CsPbBr3. These findings lead us to conclude that unlike in IR, the Raman spectrum of CsSrBr3 exhibits no substantial energy shifts with respect to CsPbBr3. Computing the phonon DOS for the orthorhombic phase of both compounds with DFT (see Supplementary Fig. 8), we find that they exhibit similar phonon DOS below 100 cm−1, i.e., in the region of most of the Raman-active modes. The similar phonon DOS and the contributions of the M-site at low frequencies explain the limited shift of the CsSrBr3 Raman spectrum, which might be surprising at first sight given the different atomic masses of Sr and Pb. Above this range, CsPbBr3 exhibits few vibrational states while CsSrBr3 shows its most pronounced phonon DOS peaks, which correspond well with the strongest IR mode calculated from the harmonic approximation.
High-temperature lattice dynamics
A key signature of vibrational anharmonicity in HaPs at higher temperatures is the Raman central peak26,33,34,35,43,44,45. We use this feature that is nominally symmetry-forbidden in the cubic phase as a fingerprint to directly investigate how the propensity for cation lone-pair formation or lack thereof determines anharmonicity in these materials, using Raman spectroscopy and DFT-based MD simulations. Interestingly, a central peak also appears in the high-temperature Raman spectrum of CsSrBr3 (see Fig. 4 and Supplementary Note 5 for full temperature range). We note that differences in Raman intensity imply that the scattering cross-section of CsSrBr3 is notably weaker than that of CsPbBr3, which is due to its significantly higher bandgap and weaker dielectric response at the Raman excitation wavelength (785 nm) and because a powder sample of CsSrBr3 has been used for which scattering of light in the back-scattering direction is considerably lower. The presence of a central peak in CsSrBr3 shows that local fluctuations associated with a cation lone-pair are not required for the low-frequency diffusive Raman scattering and anharmonicity to occur. This result, together with the identical phase-transition sequences of both materials (see Fig. 2), led us to investigate the role of tilting instabilities in CsSrBr3 and CsPbBr3.

Raman spectra of CsPbBr3 a and CsSrBr3 b in the high-temperature cubic phase measured experimentally and calculated using DFT-MD. The central peak appears for both compounds in the experiments and computations despite significant differences in bonding: [PbBr6]4− is proximate to lone-pair formation (i.e., exhibits a weak PJT effect)29, while PJT effects associated with [SrBr6]4− are negligible.
We first calculate the Raman spectrum for both compounds using MD calculations (see Fig. 4 and Methods section). Remarkably, a central peak appears also in the MD-computed high-temperature Raman spectrum of CsPbBr3 and CsSrBr3. We find good agreement between experiment and theory, both showing a feature between 50 and 100 cm−1 in the Raman spectra of the two materials in addition to the central peak.
Next, we compute harmonic phonon dispersions of both compounds (see Fig. 5) and find these to be remarkably similar for cubic CsSrBr3 and CsPbBr3 in the low frequency region, in line with the aforementioned similarities in the phonon DOS of the orthorhombic phase. Specifically, both compounds exhibit the same dynamic tilting instabilities at the edge of the Brillouin zone (BZ), governed by in-phase (M point) and three degenerate out-of-phase (R point) rotations. These rotation modes are not only active in the phase transitions, but they also have been discussed to drive the dynamic disorder of halide perovskites4,14,58,59,60,61.

a Harmonic phonon dispersion of cubic CsPbBr3 and CsSrBr3 showing the dynamic instabilities in the high-temperature, cubic phase of both compounds. The imaginary modes at the M and R points are the in-phase and out-of-phase tilting depicted in b, c, respectively. The tilting modes are almost identical for CsSrBr3 and CsPbBr3.
Finally, using the MD trajectories of CsPbBr3 and CsSrBr3 in the cubic phase, we calculate the frequency-resolved dynamic changes of octahedral rotation angles, Φα(ω) (see Fig. 6 and Eq. (1) in the Methods Section). Figure 6b shows Φα(ω) for CsPbBr3 and CsSrBr3 and indicates strong low-frequency tilting components in both CsPbBr3 and CsSrBr3. Recently, a phenomenological model for the description of the temperature-dependent Raman spectra of cubic HaPs proposed the inclusion of a low-frequency anharmonic feature, which was associated with transitions between minima of a double-well potential energy surfaces45 that correspond to different octahedral tiltings60,62,63,64. Our results confirm that substantial octahedral dynamics correspond to low-frequency features dynamically breaking the cubic symmetry in CsPbBr3 and CsSrBr34,14,44,65,66. Interestingly, this low-frequency component appears irrespective of the presence of ns2 cations and induces the formation of relatively long-lived (tens of ps) structural distortions (see Supplementary Fig. 11), which strongly deviate from the average cubic symmetry. This suggests that the dynamic deviations from the long-range, crystallographic structure enable the low-frequency Raman response without violating the selection rules.

a Schematic representation of the MBr6 octahedron aligned along the z Cartesian axis. The octahedral rotation angle around z, ϕz, is defined as the average of the angles formed by the x/y Cartesian axis and the vector connecting two in-plane Br atoms at opposing edges of the octahedron (\({\phi }_{z}^{(x)}\) in red and \({\phi }_{z}^{(y)}\) in blue). Note that a clockwise rotation is defined as positive and counter-clockwise as negative. Fourier transform of the octahedral rotation angle, Φα(ω), and cross-correlation between rotation angle and M-site displacement, Cαβ(ω), calculated using DFT-MD trajectories of cubic CsPbBr3 (b, c, respectively, T = 525 K) and CsSrBr3 (d, e, respectively, T = 570 K).
We investigate the impact of the M-site chemistry on octahedral tilting tendencies33 by computing the Fourier-transform of cross-correlations between rotation angles and M-site displacements, Cαβ(ω) (see Eq. (2) in the Methods section). Larger values of Cαβ generally indicate stronger coupling between octahedral rotations and Pb displacements. Absence of the propensity for lone-pair formation becomes evident in the low intensity of Cαβ(ω) for CsSrBr3 (Fig. 6c), which is less than half of that of CsPbBr3, especially at low-frequencies relevant for the slow, anharmonic, symmetry-breaking rotational features. This suggests that the presence of the ns2 cations in CsPbBr3 enhances the low-frequency octahedral tilting, in line with the literature33. M-site displacements and octahedral rotations are correlated because the latter is accompanied by changes of the Br-Pb-Br resonant network17 affecting the charge density in the vicinity of the M-site. While this effect is very weak in CsSrBr3 (see Supplementary Fig. 12), the non-zero Cαβ for this case shows that the presence of ns2 cations is not necessary to couple octahedral rotations and M-site displacements because the ions are still interacting through other types of interactions, e.g., electrostatically or due to Pauli repulsion. In CsPbF3, the interaction of tilting and M-site displacements is strong enough to drive the adoption of an unusual tilt pattern37. We speculate that the lone-pair-enhanced tilting could contribute to the fact that CsPbBr3 has a lower tetragonal-to-cubic phase transition temperature compared to that of CsSrBr3.
Discussion
We directly disentangled structural and chemical effects in HaPs by comparing CsPbBr3 and CsSrBr3, two compounds with similar ionic radii and structural properties but entirely different orbital interactions that imbue CsPbBr3 with the weak PJT effect common to technologically-relevant Pb perovskites and CsSrBr3 with negligible PJT effects. While the ns2 configuration of the octahedral cations is paramount for the optoelectronic and dielectric properties of these materials, using the Raman central peak at higher temperatures as a fingerprint to detect anharmonicity we found it to appear also for CsSrBr3 with 5s0 cations and to correlate with slow, anharmonic rotations of the octahedra. Notably, the anharmonicity of the tilting motions is different from that of intra-octahedral distortions associated with the PJT effect29. Altogether, these findings demonstrate that the perovskite structure allows for anharmonic vibrational dynamics to occur, irrespective of the presence of ns2 cations with the propensity to form lone pairs, which establishes this somewhat unusual behavior as a generic effect in this material class. We note that recent work by some of the present authors has investigated the commonalities and differences between oxide perovskites and HaPs in this context45.
Since octahedral dynamics impact the optoelectronic characteristics of these systems, our results have implications for synthesis of new HaPs with improved properties for technological applications. For instance, Pb-Sr alloying has been proposed as a method to tune the band gap of HaPs for light emission and absorption applications47. Our work implies that such Sr alloying for tuning electronic and dielectric properties preserves the strongly anharmonic lattice dynamics. Furthermore, investigating related compounds with distinct electronic configurations on the octahedral cation, such as CsEuBr3, may provide further insight about chemical trends in tuning of the HaP properties.
The relevance of these findings for material design strategies of HaP compounds is additionally affirmed when putting our results in the context of previous work discussing anharmonic effects in this class of materials. Specifically, cubic CsPbBr3, CsSnBr3, CsGeBr3, (CH3NH3)0.13(CH3CH2NH3)0.87PbBr3, CH(NH2)2PbBr3, and, here, CsSrBr3 are all reported to exhibit dynamic hopping between low symmetry minima on the potential energy surface33,34,35,67. By contrast, the high symmetry phase of Cs2AgBiBr6 is anharmonically stabilized and exhibits well-defined normal modes and a soft-mode transition on cooling42. Cs2SnBr6, on the other hand, lacks any phase transitions and similarly exhibits well-defined normal modes68. Where previously the strength of the PJT effect associated with ns2 cations or the density of such cations appeared to be a plausible predictor of broad, nominally symmetry-forbidden Raman scattering resulting in a central peak, our work suggests that instead the differing symmetry in both the structure and the chemical bonding of metal halide perovskites and double-perovskites may be a controlling factor. Notably, CsGeBr3, which exhibits no octahedral tilting transitions69 and a broad Raman central peak in the cubic phase with a mode reflecting persistent pyramidal [GeBr3]− anions33, corresponds to the strong PJT29 case: Stereochemically expressed cation lone pairs are evident in the low temperature average structure69 and in the local fluctuations of the cubic phase33. Dynamic symmetry-breaking giving rise to a broad Raman central peak is thus observed for three distinct bonding regimes with regard to pseudo-Jahn–Teller effects: strong PJT (CsGeBr3)33, weak PJT (CsPbBr3 and others)34, and negligible PJT (CsSrBr3).
In conclusion, the ns2 electron configuration in HaPs that can result in the formation of lone-pairs is crucial to several favorable electronic features26,39,46 and gives rise to the elevated ionic dielectric response via enhancement of Born effective charges39,48. However, we found that presence of a strong or weak PJT effect associated with ns2 cations is not necessary to produce dynamic symmetry-breaking of the sort that gives rise to broad, intense Raman scattering in the high temperature phases of HaPs and that has been associated with the unique optoelectronic properties in these compounds such as long charge-carrier lifetimes and photoinstabilities. Instead, such dynamic symmetry breaking is common to all cubic bromide and iodide (single-)perovskites thus far studied to the best of our knowledge. These results highlight the key role of structural chemistry in the anharmonic dynamics of halide perovskites, providing an additional criterion for the design of soft optoelectronic semiconductors.
Methods
Electronic structure calculations
DFT calculations were performed with Vienna ab-initio simulation package (VASP) code70 using the projector augmented wave (PAW) method71. We employed the Perdew-Burke-Ernzerhof (PBE) exchange-correlation functional72 and the Tkatchenko-Scheffler (TS) scheme73—using an iterative Hirshfeld partitioning of the charge density74,75 – to account for dispersive interactions. This setup has been shown to accurately describe the structure of HaPs, also in regard to the omission of SOC which impacts electronic-structure properties but does not result in significant changes of quantities related to the total energy76,77. All static calculations used an energy convergence threshold of 10−6 eV, a plane-wave cutoff of 500 eV, and a Γ-centered k-grid of 6 × 6 × 6 (6 × 4 × 6) for the \(Pm\bar{3}m\) (Pnma) structures. Lattice parameters were optimized by a fitting procedure using the Birch-Murnaghan equation of state78,79 The final structures used in all subsequent calculations were obtained by relaxing the ionic degrees of freedom until the maximum residual force was below 10−4 eV Å−1. The total and projected electronic DOS and COHP, were calculated by partitioning the DFT-calculated band structure into bonding and antibonding contributions using the LOBSTER code80,81. For this task, the DFT-computed electronic wave functions were projected onto Slater-type orbitals (basis set name: pbeVaspFit2015)80 including Cs 6s, 5p and 5s, Pb 6s and 6p, and Br 4p and 4s states. The maximum charge spilling in this procedure was 1.3%. Spin-orbit coupling was not included in our calculations, since it is currently not supported by the LOBSTER code. We emphasize that our focus is on the orbital contributions to the (anti) bonding interactions, rather than on a quantitative descriptions of the energy.
Phonon calculations
Phonon dispersions and DOSs were obtained via the finite displacements method implemented in the phonopy package82. For these calculations, we used 2 × 2 × 2 supercells with 40 (160) atoms of the \(Pm\bar{3}m\) (Pnma) CsMBr3 structures reducing k-space sampling accordingly. IR and Raman spectra were computed with the phonopy-spectroscopy package83, using zone-center phonon modes, Born-effective charges and polarizabilities, calculated with density functional perturbation theory (DFPT)84.
First-principles molecular dynamics
DFT-based MD calculations were performed for 2 × 2 × 2 supercells of the \(Pm\bar{3}m\) structures using a Nosé-Hoover thermostat within the canonical ensemble (NVT), as implemented in VASP85. The simulation temperature was set to T=525 and 570 K for CsPbBr3 and CsSrBr3, respectively. An 8 fs timestep, reduced k-grid of 3 × 3 × 3, and energy convergence threshold of 10−5 eV were used for the 10 ps equilibration and 115 ps production runs.
Raman spectra from molecular dynamics
DFT-based MD calculations were used to compute the high-temperature Raman spectra of CsPbBr3 and CsSrBr3. We calculated Raman intensities from the autocorrelation function of the polarisability, as detailed elsewhere86. The polarizabilities were calculated with DFPT84 on 400 evenly-spaced snapshots every 0.11 ps for a total of 44.8 ps. The k-grid employed for the DFPT calculations was set to 4 × 4 × 4 after testing convergence of the polarisability tensor for several snapshots.
Octahedral rotation dynamics and cross-correlations
We quantified the octahedral dynamics using the rotation angles, ϕα, around a given Cartesian axis α (see Fig. 6a). The frequency-resolved rotational dynamics were calculated as the Fourier transform of ϕα:
where Nsteps is the number of snapshots. To compute the angles we selected 1000 equally spaced snapshots. We calculated the frequency-resolved cross-correlation between octahedral rotation angles (around a Cartesian direction α) and the displacements (along a Cartesian direction β) of the corresponding M-site, \({d}_{\beta }^{{{{{{{{\rm{M}}}}}}}}}(t)\), as:
Polycrystalline sample preparation
CsBr (Alfa Aesar, 99.9%), anhydrous SrBr2 (Alfa Aesar, 99%), Cs2CO3, PbO, and concentrated aqueous HBr were purchased and used as received. Guided by the reported pseudo-binary phase diagram87, polycrystalline CsSrBr3 for X-ray powder diffraction and Raman spectroscopy was prepared by a solid-state reaction at 600 °C. CsBr (5 mmol, 1064 mg) and SrBr2 (5 mmol, 1237 mg) were ground and pressed into a 5 mm diameter pellet, placed in an alumina crucible, and flame-sealed under ~ 1/3 atmosphere of argon in a fused silica ampoule. The reaction yields a porous, colorless pellet which is easily separated from the crucible and ground in inert atmosphere. Polycrystalline CsPbBr3 for X-ray powder diffraction was prepared in ambient atmosphere by precipitation from aqueous hydrobromic acid. PbO (2 mmol, 446.4 mg) was dissolved in 2 mL hot concentrated HBr under stirring. Cs2CO3 (1 mmol, 325.8 mg) was added slowly resulting in an immediate bright orange precipitate. 13 mL additional HBr was added and the mixture left to stir. After an hour, stirring was stopped and the mixture allowed to cool to room temperature. Excess solution was decanted, and the remaining mixture was evaporated to dryness on a hotplate and ground. Phase purity of all prepared compounds was established by powder XRD.
Single crystal preparation
Single crystals of CsSrBr3 were grown by the Bridgman method from a stoichiometric mixture of the binary metal bromides in a 10 mm diameter quartz ampoule. CsSrBr3 was pulled at 0.5 mm h−1 through an 800 °C hot zone, yielding a multi-crystalline rod from which several-mm single crystal regions could be cleaved. CsSrBr3 is hygroscopic and all preparation and handling was performed in an inert atmosphere.
The vertical Bridgman method was used to grow large, high-quality single crystals of CsPbBr3. After synthesis and purification (see Supplementary Note 2 for details), the ampoule was reset to the hot zone for the Bridgman Growth. The zone 1 temperature was set to 650 °C with a 150 °C h−1 ramp rate, and held for 12 h to ensure a full melt before sample motion occurred. The zone 2 and 3 temperatures were set to 375 °C. These temperatures were held for 350 h while the ampoule was moved through the furnace at a rate of 0.9 mm h−1 under 0.3 rpm rotation. After the motion had ceased, the zone 1 temperature ramped to 375 °C to make the temperature profile in the furnace uniform. The cooling program was set to slow during the phase transitions occurring near 120 and 90 °C, with a 10 °C h−1 cooling rate from 375 °C to 175 °C, a 2.5 °C h−1 slow cooling rate from 175 °C to 75 °C, and a 10 °C h−1 rate to 30 °C. The resulting CsPbBr3 ingot was orange-red and had large (≥5 mm) transparent single-crystalline domains, though the edges of some portions exhibited twinning.
X-ray diffraction
Polycrystalline samples were ground with silicon powder (as an internal standard and diluent) and packed in borosilicate glass capillaries. Powder XRD patterns were measured in STOE geometry using a STOE Stadi P diffractometer (Mo Kα1 radiation, Ge-(111) monochromator, Mythen 1K Detector) equipped with a furnace. Data were analyzed by sequential Pawley refinement using GSAS-II88.
Infrared reflectivity measurements
IR-reflection spectra in the THz range were measured as a combination of THz spectroscopy (TDS) for the low-frequency end and bolometer detection for the higher frequencies. Bolometer spectra were measured using a Bruker 80v Fourier-transform IR spectrometer with a globar source and a bolometer detector cooled to liquid He temperatures. The crystals were mounted for reflection measurements and the instrument was sealed in vacuum. A gold mirror was used as reflection reference. TDS was performed using a Spectra Physics Mai Tai-Empower-Spitfire Pro Ti:Sapphire regenerative amplifier. The amplifier generates 35 fs pulses centered at 800 nm at a repetition rate of 5 kHz. THz pulses were generated by a spintronic emitter, which was composed of 1.8 nm of Co40Fe40B20 sandwiched between 2 nm of tungsten and 2 nm of platinum, all supported by a quartz substrate. The THz pulses were detected using electro-optic sampling in a (100)-ZnTe crystal. A gold mirror was used as reflection reference. The sample crystals, THz emitter and THz detector were held under vacuum during the measurements.
TDS offers better signal at low frequency, while bolometer measurements have an advantage over TDS at higher frequencies. Therefore, the spectra were combined and merged at 100 cm−1. Owing to scattering losses, the absolute intensity of reflected light can not be taken quantitatively. Therefore, the spectra were scaled to the signal level at 100 cm−1 before merging the data. The final reflectivity spectra are given in arbitrary units. The phonon frequencies and overall spectral shape allows for fitting to the dielectric function.
Raman spectroscopy
All the measurements were taken in a home-built back scattering Raman system. For all measurements, the laser was focused with a 50× objective (Zeiss, USA), and the Rayleigh scattering was then filtered with a notch filter (Ondax Inc., USA). The beam was focused into a spectrometer 1 m long (FHR 1000, Horiba) and then on a CCD detector. To get the unpolarized Raman spectrum for the single crystals (CsSrBr3 and CsPbBr3), two orthogonal angles were measured in parallel and cross configurations (four measurements overall). The unpolarized spectrum is a summation of all four spectra. The samples were cooled below room temperature by a Janis cryostat ST-500 controlled by Lakeshore model 335 and were heated above room temperature by a closed heating system (Linkam Scientific). Due to the sensitivity of CsSrBr3 to ambient moisture, CsSrBr3 powder was flame-sealed in a small quartz capillary for the high-temperature measurements, and a single crystal was loaded into a closed cell under an Ar environment for the low temperatures measurements. CsSrBr3 low temperature measurements were taken with a 2.5 eV CW diode laser (Toptica Inc.). CsSrBr3 high-temperature measurement and all the CsPbBr3 measurements were taken with a 1.57 eV CW diode laser (Toptica Inc.). We note that while Raman spectra on quartz show a contribution towards zero frequency89, it is narrower in frequency than what we observe. Results from control experiments (see Supplementary Fig. 7) show that the main signals from quartz do not contribute to the measured Raman spectra of CsSrBr3.
References
Snaith, H. J. Perovskites: the emergence of a new era for low-cost, high-efficiency solar cells. J. Phys. Chem. Lett. 4, 3623–3630 (2013).
Correa-Baena, J.-P. et al. Promises and challenges of perovskite solar cells. Science 358, 739–744 (2017).
Nayak, P. K., Mahesh, S., Snaith, H. J. & Cahen, D. Photovoltaic solar cell technologies: analysing the state of the art. Nat. Rev. Mater. 4, 269–285 (2019).
Beecher, A. N. et al. Direct observation of dynamic symmetry breaking above room temperature in methylammonium lead iodide perovskite. ACS Energy Lett. 1, 880–887 (2016).
Whalley, L. D., Skelton, J. M., Frost, J. M. & Walsh, A. Phonon anharmonicity, lifetimes, and thermal transport in CH3NH3PbI3 from many-body perturbation theory. Phys. Rev. B 94, 1–5 (2016).
Carignano, M. A., Aravindh, S. A., Roqan, I. S., Even, J. & Katan, C. Critical fluctuations and anharmonicity in lead iodide perovskites from molecular dynamics supercell simulations. J. Mater. Chem. C. 121, 20729–20738 (2017).
Brenner, T. M., Egger, D. A., Kronik, L., Hodes, G. & Cahen, D. Hybrid organic—inorganic perovskites: low-cost semiconductors with intriguing charge-transport properties. Nat. Rev. Mater. 1, 15007 (2016).
Egger, D. A. et al. What remains unexplained about the properties of halide perovskites? Adv. Mater. 30, 1800691 (2018).
Ceratti, D. R. et al. Self-healing inside APbBr3 halide perovskite crystals. Adv. Mater. 30, 1706273 (2018).
Cahen, D., Kronik, L. & Hodes, G. Are defects in lead-halide perovskites healed, tolerated, or both? ACS Energy Lett. 6, 4108–4114 (2021).
Parida, S. et al. Self-healing and -repair of nanomechanical damages in lead halide perovskites. Adv. Funct. Mater. 33, 2304278 (2023).
Patrick, C. E., Jacobsen, K. W. & Thygesen, K. S. Anharmonic stabilization and band gap renormalization in the perovskite CsSnI3. Phys. Rev. B 92, 201205 (2015).
Wiktor, J., Rothlisberger, U. & Pasquarello, A. Predictive determination of band gaps of inorganic halide perovskites. J. Phys. Chem. Lett. 8, 5507–5512 (2017).
Lanigan-Atkins, T. et al. Two-dimensional overdamped fluctuations of the soft perovskite lattice in CsPbBr3. Nat. Mater. 20, 977–983 (2021).
Seidl, S. A. et al. Anharmonic fluctuations govern the band gap of halide perovskites. Phys. Rev. Mater. 7, L092401 (2023).
Gehrmann, C. & Egger, D. A. Dynamic shortening of disorder potentials in anharmonic halide perovskites. Nat. Commun. 10, 3141 (2019).
Gehrmann, C., Caicedo-Dávila, S., Zhu, X. & Egger, D. A. Transversal halide motion intensifies band-to-band transitions in halide perovskites. Adv. Sci. 9, 2200706 (2022).
Wu, B. et al. Indirect tail states formation by thermal-induced polar fluctuations in halide perovskites. Nat. Commun. 10, 1–10 (2019).
Mayers, M. Z., Tan, L. Z., Egger, D. A., Rappe, A. M. & Reichman, D. R. How lattice and charge fluctuations control carrier dynamics in halide perovskites. Nano Lett. 18, 8041–8046 (2018).
Lacroix, A., De Laissardière, G. T., Quémerais, P., Julien, J. P. & Mayou, D. Modeling of electronic mobilities in halide perovskites: adiabatic quantum localization scenario. Phys. Rev. Lett. 124, 1–6 (2020).
Iaru, C. M. et al. Fröhlich interaction dominated by a single phonon mode in CsPbBr3. Nat. Commun. 12, 5844 (2021).
Schilcher, M. J. et al. The significance of polarons and dynamic disorder in halide perovskites. ACS Energy Lett. 6, 2162–2173 (2021).
Lai, R. et al. Transient suppression of carrier mobility due to hot optical phonons in lead bromide perovskites. J. Phys. Chem. Lett. 13, 5488–5494 (2022).
Zhang, K.-C., Shen, C., Zhang, H.-B., Li, Y.-F. & Liu, Y. Effect of quartic anharmonicity on the carrier transport of cubic halide perovskites CsSnI3 and CsPbI3. Phys. Rev. B 106, 235202 (2022).
Schilcher, M. J. et al. Correlated anharmonicity and dynamic disorder control carrier transport in halide perovskites. Phys. Rev. Mater. 7, L081601 (2023).
Fabini, D. H., Seshadri, R. & Kanatzidis, M. G. The underappreciated lone pair in halide perovskites underpins their unusual properties. MRS Bull. 45, 467–477 (2020).
Van der Vorst, C. & Maaskant, W. Stereochemically active (5s)2 lone pairs in the structures of α-Incl and β-Incl. J. Solid State Chem. 34, 301–313 (1980).
Maaskant, W. J. A. & Bersuker, I. B. A combined Jahn-Teller and pseudo-Jahn-Teller effect: an exactly solvable model. J. Phys. Condens. Matter 3, 37–47 (1991).
Bersuker, I. B. Pseudo-Jahn–Teller Effect—A two-state paradigm in formation, deformation, and transformation of molecular systems and solids. Chem. Rev. 113, 1351–1390 (2013).
Worhatch, R. J., Kim, H., Swainson, I. P., Yonkeu, A. L. & Billinge, S. J. L. Study of local structure in selected organic-inorganic perovskites in the \(Pm\bar{3}m\) phase. Chem. Mater. 20, 1272–1277 (2008).
Fabini, D. H. et al. Dynamic stereochemical activity of the Sn2+ lone pair in perovskite CsSnBr3. J. Am. Chem. Soc. 138, 11820–11832 (2016).
Laurita, G., Fabini, D. H., Stoumpos, C. C., Kanatzidis, M. G. & Seshadri, R. Chemical tuning of dynamic cation off-centering in the cubic phases of hybrid tin and lead halide perovskites. Chem. Sci. 8, 5628–5635 (2017).
Gao, L. et al. Metal cation s lone-pairs increase octahedral tilting instabilities in halide perovskites. Mater. Adv. 2, 4610–4616 (2021).
Yaffe, O. et al. Local polar fluctuations in lead halide perovskite crystals. Phys. Rev. Lett. 118, 1–6 (2017).
Huang, X. et al. Understanding electron–phonon interactions in 3D lead halide perovskites from the stereochemical expression of 6s2 lone pairs. J. Am. Chem. Soc. 144, 12247–12260 (2022).
Walsh, A., Payne, D. J., Egdell, R. G. & Watson, G. W. Stereochemistry of post-transition metal oxides: revision of the classical lone pair model. Chem. Soc. Rev. 40, 4455 (2011).
Smith, E. H., Benedek, N. A. & Fennie, C. J. Interplay of octahedral rotations and lone pair ferroelectricity in CsPbF3. Inorg. Chem. 54, 8536–8543 (2015).
Radha, S. K., Bhandari, C. & Lambrecht, W. R. Distortion modes in halide perovskites: to twist or to stretch, a matter of tolerance and lone pairs. Phys. Rev. Mater. 2, 1–11 (2018).
Du, M. H. Efficient carrier transport in halide perovskites: theoretical perspectives. J. Mater. Chem. A 2, 9091–9098 (2014).
Herz, L. M. How lattice dynamics moderate the electronic properties of metal-halide perovskites. J. Phys. Chem. Lett. 9, 6853–6863 (2018).
Goldschmidt, V. M. Die Gesetze der Krystallochemie. Naturwissenschaften 14, 477–485 (1926).
Cohen, A. et al. Diverging expressions of anharmonicity in halide perovskites. Adv. Mater. 34, 2107932 (2022).
Ferreira, A. C. et al. Direct evidence of weakly dispersed and strongly anharmonic optical phonons in hybrid perovskites. Commun. Phys. 3, 48 (2020).
Sharma, R. et al. Elucidating the atomistic origin of anharmonicity in tetragonal CH3NH3PbI3 with Raman scattering. Phys. Rev. Mater. 4, 092401 (2020).
Menahem, M. et al. Disorder origin of Raman scattering in perovskite single crystals. Phys. Rev. Mater. 7, 044602 (2023).
Goesten, M. G. & Hoffmann, R. Mirrors of bonding in metal halide perovskites. J. Am. Chem. Soc. 140, 12996–13010 (2018).
Straus, D. B. & Cava, R. J. Tuning the band gap in the halide perovskite CsPbBr3 through Sr substitution. ACS Appl. Mater. Interfaces 30, 34884–34890 (2022).
Du, M.-H. & Singh, D. J. Enhanced Born charge and proximity to ferroelectricity in thallium halides. Phys. Rev. B 81, 144114 (2010).
Sun, J. & Singh, D. J. Electronic properties, screening, and efficient carrier transport in NaSbS2. Phys. Rev. Appl. 7, 024015 (2017).
Ran, Z. et al. Bismuth and antimony-based oxyhalides and chalcohalides as potential optoelectronic materials. npj Comput Mater. 4, 14 (2018).
Kang, B. & Biswas, K. Exploring polaronic, excitonic structures and luminescence in Cs4PbBr6/CsPbBr3. J. Phys. Chem. Lett. 9, 830–836 (2018).
Rodová, M., Brožek, J. & Nitsch, K. Phase transitions in ternary caesium lead bromide. J. Therm. Anal. 71, 667–673 (2003).
Stoumpos, C. C. et al. Crystal growth of the perovskite semiconductor CsPbBr3: a new material for high-energy radiation detection. Cryst. Growth Des. 13, 2722–2727 (2013).
Loyd, M. et al. Crystal structure and thermal expansion of CsCaI3:Eu and CsSrBr3:Eu scintillators. J. Cryst. Growth 481, 35–39 (2018).
Fabini, D. H. et al. Reentrant structural and optical properties and large positive thermal expansion in perovskite formamidinium lead iodide. Angew. Chem. Int. Ed. 55, 15392–15396 (2016).
Schueller, E. C. et al. Crystal structure evolution and notable thermal expansion in hybrid perovskites formamidinium tin iodide and formamidinium lead bromide. Inorg. Chem. 57, 695–701 (2018).
Sendner, M. et al. Optical phonons in methylammonium lead halide perovskites and implications for charge transport. Mater. Horiz. 3, 613–620 (2016).
Armstrong, R. L. Displacive order—disorder crossover in perovskite and antifluorite crystals undergoing rotational phase transitions. Prog. Nucl. Magn. Reson. Spectrosc. 21, 151–173 (1989).
Woodward, P. M. Octahedral tilting in perovskites. I. Geometrical considerations. Acta Crystallogr B 53, 32–43 (1997).
Yang, R. X., Skelton, J. M., Da Silva, E. L., Frost, J. M. & Walsh, A. Spontaneous octahedral tilting in the cubic inorganic cesium halide perovskites CsSnX3 and CsPbX3 (X = F, Cl, Br, I). J. Phys. Chem. Lett. 8, 4720–4726 (2017).
Yang, R. X., Skelton, J. M., da Silva, E. L., Frost, J. M. & Walsh, A. Assessment of dynamic structural instabilities across 24 cubic inorganic halide perovskites. J. Chem. Phys. 152, 024703 (2020).
Klarbring, J. Low-energy paths for octahedral tilting in inorganic halide perovskites. Phys. Rev. B 99, 1–7 (2019).
Bechtel, J. S., Thomas, J. C. & Van Der Ven, A. Finite-temperature simulation of anharmonicity and octahedral tilting transitions in halide perovskites. Phys. Rev. Mater. 3, 113605 (2019).
Zhu, X., Caicedo-Dávila, S., Gehrmann, C. & Egger, D. A. Probing the disorder inside the cubic unit cell of halide perovskites from first-principles. ACS Appl. Mater. Interfaces 14, 22973–22981 (2022).
Wiktor, J., Fransson, E., Kubicki, D. & Erhart, P. Quantifying Dynamic Tilting in Halide Perovskites: Chemical Trends and Local Correlations. Chem. Mater. 35, 6737–6744 (2023).
Baldwin, W. J. et al. Dynamic local structure in caesium lead iodide: spatial correlation and transient domains. Small 20, 2303565 (2023).
Reuveni, G. et al. Static and dynamic disorder in formamidinium lead bromide single crystals. J. Phys. Chem. Lett. 14, 1288–1293 (2023).
Kaltzoglou, A. et al. Optical-vibrational properties of the Cs2SnX6 (X = Cl, Br, I) defect perovskites and hole-transport efficiency in dye-sensitized solar cells. J. Phys. Chem. C 120, 11777–11785 (2016).
Thiele, G., Rotter, H. W. & Schmidt, K. D. Kristallstrukturen und Phasentransformationen von Caesiumtrihalogenogermanaten (II) CsGeX3 (X= Cl, Br, I). Z. Anorg. Allg. Chem. 545, 148–156 (1987).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Tkatchenko, A. & Scheffler, M. Accurate molecular Van Der Waals interactions from ground-state electron density and free-atom reference data. Phys. Rev. Lett. 102, 073005 (2009).
Bučko, T., Lebègue, S., Hafner, J. & Ángyán, J. G. Improved density dependent correction for the description of London dispersion forces. J. Chem. Theory Comput. 9, 4293–4299 (2013).
Bučko, T., Lebègue, S., Ángyán, J. G. & Hafner, J. Extending the applicability of the Tkatchenko-Scheffler dispersion correction via iterative Hirshfeld partitioning. J. Chem. Phys. 141, 034114 (2014).
Egger, D. A. & Kronik, L. Role of dispersive interactions in determining structural properties of organic-inorganic halide perovskites: Insights from first-principles calculations. J. Phys. Chem. Lett. 5, 2728–2733 (2014).
Beck, H., Gehrmann, C. & Egger, D. A. Structure and binding in halide perovskites: Analysis of static and dynamic effects from dispersion-corrected density functional theory. APL Mater. 7, 021108 (2019).
Birch, F. Finite elastic strain of cubic crystals. Phys. Rev. 71, 809–824 (1947).
Murnaghan, F. D. The Compressibility of Media under Extreme Pressures. Proc. Natl Acad. Sci. USA 30, 244–247 (1944).
Maintz, S., Deringer, V. L., Tchougréeff, A. L. & Dronskowski, R. LOBSTER: A tool to extract chemical bonding from plane-wave based DFT. J. Comput. Chem. 37, 1030–1035 (2016).
Nelson, R. et al. LOBSTER : Local orbital projections, atomic charges, and chemical-bonding analysis from projector-augmented-wave-based density-functional theory. J. Comput. Chem. 41, 1931–1940 (2020).
Togo, A. & Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 108, 1–5 (2015).
Skelton, J. M. et al. Lattice dynamics of the tin sulphides SnS2, SnS and Sn2S3: vibrational spectra and thermal transport. Phys. Chem. Chem. Phys. 19, 12452–12465 (2017).
Gajdoš, M., Hummer, K., Kresse, G., Furthmüller, J. & Bechstedt, F. Linear optical properties in the projector-augmented wave methodology. Phys. Rev. B 73, 1–9 (2006).
Kresse, G. & Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metalamorphous- semiconductor transition in germanium. Phys. Rev. B 49, 14251–14269 (1994).
Thomas, M., Brehm, M., Fligg, R., Vöhringer, P. & Kirchner, B. Computing vibrational spectra from ab initio molecular dynamics. Phys. Chem. Chem. Phys. 15, 6608–6622 (2013).
Riccardi, R., Sinistri, C., Campari, G. Y. & Magistris, A. Binary systems formed by alkali bromides with barium or strontium bromide. Z. Naturforsch. A 25, 781–785 (1970).
Toby, B. H. & Dreele, R. B. V. GSAS-II: the genesis of a modern open-source all purpose crystallography software package. J. Appl. Crystallogr. 46, 544–549 (2013).
Bates, J. B. & Quist, A. S. Polarized Raman spectra of β-Quartz. J. Chem. Phys. 56, 1528–1533 (1972).
Caicedo-Dávila, S. et al. Disentangling the effects of structure and lone-pair electrons in the lattice dynamics of halide perovskites. https://doi.org/10.5281/zenodo.10975217 (2024).
Acknowledgements
Funding provided by the Alexander von Humboldt-Foundation in the framework of the Sofja Kovalevskaja Award, endowed by the German Federal Ministry of Education and Research, by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) via Germany’s Excellence Strategy - EXC 2089/1-390776260, and by TU Munich - IAS, funded by the German Excellence Initiative and the European Union Seventh Framework Programme under Grant Agreement No. 291763, are gratefully acknowledged. Funding was provided by the Engineering and Physical Sciences Research Council (EPSRC), UK. The Gauss Centre for Supercomputing eV is acknowledged for providing computing time through the John von Neumann Institute for Computing on the GCS Supercomputer JUWELS at Jülich Supercomputing Centre. D.H.F. gratefully acknowledges financial support from the Alexander von Humboldt Foundation and the Max Planck Society. D.H.F. thanks Maximilian A. Plass for assistance with flame-sealing the Raman capillaries. K.M.M. and M.V.K. acknowledge financial support by ETH Zürich through the ETH+ Project SynMatLab Laboratory for Multiscale Materials Synthesis.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
S.C.-D. performed the theoretical calculations, analyzed the data and wrote the initial manuscript under the supervision of D.A.E and with additional support by M.G. A.C. performed Raman measurements and analyzed data under the supervision of O.Y. S.M. performed IR measurements and analyzed data under the supervision of L.M.H. D.H.F. prepared polycrystalline samples, performed XRD measurements, analyzed data, and modeled the PJT effect. M.I. prepared the CsSrBr3 single crystals. K.M.M. prepared the CsPbBr3 single crystals under the supervision of M.V.K. D.H.F. and D.A.E. conceived and supervised the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Yongping Fu, Daniel B. Straus and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Caicedo-Dávila, S., Cohen, A., Motti, S.G. et al. Disentangling the effects of structure and lone-pair electrons in the lattice dynamics of halide perovskites. Nat Commun 15, 4184 (2024). https://doi.org/10.1038/s41467-024-48581-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-48581-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
