
Abstract
The Paleocene-Eocene Thermal Maximum (PETM) was an abrupt global warming event associated with a large injection of carbon into the ocean-atmosphere system, as evidenced by a diagnostic carbon isotope excursion (CIE). Evidence also suggests substantial hydrologic perturbations, but details have been hampered by a lack of appropriate proxies. To address this shortcoming, here we isolate and measure the isotopic composition of hydroxyl groups (OH−) in clay minerals from a highly expanded PETM section in the North Sea Basin, together with their bulk oxygen isotope composition. At this location, we show that hydroxyl O- and H-isotopes are less influenced than bulk values by clay compositional changes due to mixing and/or inherited signals and thus better track hydrologic variability. We find that clay OH− hydrogen-isotope values (δ2HOH) decrease slowly prior to the PETM and then abruptly by ∼8‰ at the CIE onset. Coincident with an increase in relative kaolinite content, this indicates increased rainfall and weathering and implies an enhanced hydrologic cycle response to global warming, particularly during the early stages of the PETM. Subsequently, δ2HOH returns to pre-PETM values well before the end of the CIE, suggesting hydrologic changes in the North Sea were short-lived relative to carbon-cycle perturbations.
Similar content being viewed by others

Introduction
The Paleocene-Eocene Thermal Maximum (PETM) represents an intense global warming event that occurred approximately 55.9 million years ago1,2 (for summary, see ref. 3). Whereas the exact cause of the PETM remains uncertain, several diagnostic markers exist in the geologic record, including a prominent carbon isotope excursion (CIE) of ∼3–4‰ at the PETM onset and widespread dissolution of marine carbonates1,3. The inferred carbon injection led to average global temperature increases of 4–5 °C1,4, oceanographic changes including deoxygenation and acidification leading to mass benthic extinctions5 and perturbations in the hydrologic cycle6.
The global hydrologic cycle is thought to have changed during the PETM relative to today, but large regional variability likely existed (for summary, see ref. 6). Palynological and biomarker isotope studies suggest sustained increases in terrestrial runoff, both in the tropics (e.g. Venezuela7) and in high-latitude regions (e.g. Arctic Spitsbergen8, New Zealand9, North Sea10 and Arctic Ocean11). In other regions, sedimentological, palynological and paleosol data imply either increasing aridity (e.g. southern Rocky Mountains12) or strong seasonality and extreme rainfall events in the subtropics to lower mid-latitudes (e.g. Spanish Pyrenees13, and Normandy14). Despite these general trends, large uncertainty persists in our quantitative understanding of the PETM hydrologic cycle and the timing of these changes relative to the CIE.
Improving constraints on the PETM hydrologic cycle is of particular relevance for ongoing and future climate change. Anthropogenic warming is predicted to exceed 2 °C by the end of this century even under the stabilisation (RCP6.0) greenhouse gas scenario15. Such warming will likely increase hydrologic-cycle intensity, thus exacerbating environmental stress16,17. Current models suggest an enhanced hydrologic cycle with elevated precipitation driven by higher evaporation rates18,19, a pattern which may already be emerging. However, while spatial changes in total precipitation amount can be modelled accurately, climate models are not yet precise enough to predict spatial variations in the frequency, seasonality, intensity or type of precipitation17,20. Testing whether current climate models can simulate the observed hydrologic responses to temperature during the PETM will aid in assessing future hydrological projections in response to global warming.
Large variations in clay mineralogy are recorded in sediments deposited over the PETM in response to changing climate conditions. A major unresolved observation is a conspicuous increase in kaolinite deposition at many mid- to high-latitude sites (e.g. refs. 21,22,23,24,25,26). Kaolinite is a clay mineral that is generally assumed to form predominantly by intense weathering in humid tropical climates27,28,29. The origin and significance of PETM kaolinite deposition is controversial because it could result from either: (i) increased chemical weathering and kaolinite formation under warmer climate and elevated year-round precipitation23,30,31,32, or (ii) enhanced mobilisation, physical transport, and exhumation of previously weathered material (e.g. laterite soils) during strong seasonal rainfall events26,28,33. Whereas the former interpretation of increased chemical weathering is consistent with a direct negative silicate-weathering feedback to elevated temperatures34, the latter of increased physical weathering provides a less direct feedback and may indicate a decoupling between climate and weathering intensity. The two processes are not mutually exclusive and evidence exists that both may have occurred at the time of the PETM in different settings.
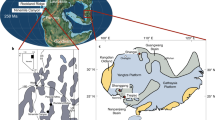
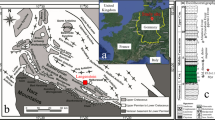
To provide additional constraints on the hydrologic changes across the PETM, we measure the isotopic composition of clay mineral hydroxyl groups from a PETM section in the Sele Formation of the North Sea (well 22/10a-4 (57°44′8.47″N; 1°50′26.59″E; up to ∼500 m paleo-water depth); Fig. 1; refs. 10, 33, 35). During the early Paleogene, the North Sea was a restricted marine basin bounded by Scotland to the north, Greenland to the west and the Fenno-Scandian Shield to the east. The basin contains a highly expanded Paleocene-Eocene transition sequence, uninterrupted but for minor erosion at the base of thin turbidite sandstones10. Terrigenous input was high during the Paleogene and thought to be derived primarily from the Scotland Faeroe-Shetland landmass36, making this an ideal location to study PETM variability in the hydrologic cycle at a mid-paleo-latitude location (57 °N). Furthermore, by isolating and measuring the hydrogen and oxygen isotope composition of clay hydroxyl groups37 (δ2HOH and δ18OOH; Methods) in addition to bulk clay δ18O (δ18Obulk; Methods), our approach aims to avoid potential biases by clay end-member mixing and/or inherited isotopic compositions26. We thus expect this record to robustly reflect PETM variability in the hydrologic cycle.

Results and discussion
We compare all clay isotope results relative to the record of bulk organic matter δ13C from the same section10 and previously reported clay mineralogical data33 (Methods; Fig. 2; Fig. 3). The CIE onset in this section is located between 2614.3 and 2613.5 m10,33, whereas the post-PETM δ13C recovery is not captured in the available δ13C data and likely occurs higher in the section.

The onset of the Carbon Isotope Excursion (CIE) is shown by the light grey bar. a δ13C VPDB (Vienna Pee Dee Belemnite) of total organic carbon (TOC)10; b clay hydroxyl δ2HOH VSMOW (Vienna Standard Mean Ocean Water); c percentage of the dinoflagellate Apectodinium spp. (several species)10; d percentage of low-salinity dinoflagellate cysts excluding Apectodinium10; e clay hydroxyl δ18OOH; and f the bulk clay δ18Obulk. Error bars for b, e and f are ±1 σ analytical uncertainty (Methods).

The onset of the Carbon Isotope Excursion (CIE) is shown by the light grey bar. a kaolinite; b illite-smectite; c illite and d chlorite in the <4 μm size fraction from well 22/10a-433 and e the reconstructed clay mineral (neo)formation source water δ2H (δ2HOH-SW). Also shown in e are end-members assuming 100 % kaolinite (dotted grey line) or 100% illite-smectite (dashed grey line). Reconstructions are calculated using empirical fractionation factors from refs. 39 and 40, respectively (Methods). These end-member values are expected to encompass the range of possible δ2HOH-SW values for a given time point.
Trends in hydroxyl hydrogen and oxygen isotopic composition
Measured δ2HOH values decrease gradually by 13.7 ± 0.6‰ over the 15 m leading up to the CIE onset, reaching −67.8 ± 0.3‰ VSMOW (Vienna Standard Mean Ocean Water) at 2613.5 m (Fig. 2b). The CIE onset at 2613.5 m is followed by an abrupt δ2HOH decrease of 8.1 ± 0.4‰; δ2HOH decreases further throughout the early CIE, culminating in a minimum value of −87.0 ± 0.2‰ VSMOW at 2611.6 m. This δ2HOH excursion occurs only between 2613.5 and 2608.7 m, with δ2HOH values returning to the pre-PETM baseline at 2606.4 m, well before the end of the CIE (Fig. 2b; ref. 10). In total, δ2HOH spans a range of 34.1 ± 0.3‰ (27.6 ± 0.3‰ if the one-point minimum is excluded). Furthermore, the observed δ2HOH excursion is coincident with both an increased abundance of low-salinity-tolerant dinoflagellate cysts excluding Apectodinium, and of Apectodinium itself (a genus abundant during the PETM and possibly associated with enhanced terrestrial runoff9; see ref. 38. for discussion) (Fig. 2b, c, d; ref. 10). The strong correlation between dinoflagellate assemblages and δ2HOH is particularly apparent between 2612.8 and 2608.7 m, which marks the lowest observed δ2HOH values and greatest abundance of low-salinity-tolerant dinoflagellate cysts excluding Apectodinium (and of Apectodinium).
In contrast to δ2HOH, δ18OOH changes appear to be more closely related to changes in clay composition and δ13C (Fig. 2e), but overall correlation with clay oxygen isotope values is weak (e.g. for kaolinite, the strongest correlation, r2 = 0.46; p-value < 0.01; n = 22). At the base of the section, δ18OOH averages 3.2 ± 0.3‰ VSMOW but increases to 10.7 ± 0.2‰ VSMOW (omitting a one-point minimum of 1.38 ± 0.13‰ VSMOW at 2617.4 m, which corresponds to a kaolinite content maximum and illite-smectite (I-S) content minimum; Fig. 3a, b). At the CIE onset, δ18OOH values again decrease by 10.2 ± 0.2‰ to 0.42 ± 0.04‰ VSMOW and remain stable between −0.7 ± 0.2‰ and 3.7 ± 0.2‰ VSMOW for the remainder of the record.
Trends in bulk oxygen isotopic composition
Similar to δ18OOH, bulk clay δ18O values are positively correlated with I-S oxygen content (r2 = 0.30; p-value = 0.01; n = 22). Both δ18OOH and δ18Obulk are also negatively correlated with kaolinite oxygen contribution, with δ18Obulk showing a much stronger correlation (r2 = 0.83; p-value < 0.01; n = 22; Figs. 2f, 3b,c) than δ18OOH. In addition, the magnitude of δ18OOH variability (11.3 ± 0.3 ‰) is much greater than that for δ18Obulk (3.7 ± 0.8‰ VSMOW; Fig. 2e, f). Correlation between δ18Obulk and δ18OOH is weak (r2 = 0.45; p-value < 0.01; n = 22). Prior to the PETM, δ18Obulk averages 18.6 ± 0.5‰ VSMOW at 2634 m and increases to 19.7 ± 0.6‰ VSMOW at 2623.3 m; however, these high values are not maintained until the CIE onset. Rather, δ18Obulk decreases to 17.4 ± 0.7‰ VSMOW between 2621.2 and 2617.4 m, then increases to 18.7 ± 0.3‰ VSMOW at 2616.3 m before decreasing to 16.0 ± 0.6 ‰ VSMOW at the CIE onset at 2612.8 m. Unlike δ18OOH, δ18Obulk begins to return to pre-CIE values after the CIE onset—increasing to 19.6 ± 1.2‰ VSMOW at 2608.7 m—before decreasing to 17.5 ± 0.4‰ VSMOW at the top of the section.
Estimation of clay hydroxyl source water
Finally, we estimate the hydrogen-isotope composition of clay mineral (neo)formation source water (δ2HOH-SW) using mineral-specific 2H fractionation factors39,40 combined with measured δ2HOH and clay mineral content trends (Methods; Fig. 3e). Resulting δ2HOH-SW closely tracks the measured δ2HOH value (r2 = 0.90; p-value < 0.01; n = 22). Absolute values are less negative (ranging from −2.5 to −50.9‰ VSMOW) and exhibit greater variability (48.5‰) relative to δ2HOH (34.1 ± 0.3‰), as the most enriched δ2HOH values correspond to depths with higher I-S concentrations, while the most depleted δ2HOH values correspond to depths with higher kaolinite concentrations, despite kaolinite having a larger fractionation factor.
Implications of bulk and hydroxyl isotopes on clay origin
The origin of the pervasive increase in PETM kaolinite deposition is debated. Early studies interpreted this trend as reflecting contemporaneous PETM kaolinite formation, indicating increased pedogenesis and weathering under a warm, humid climate with an intensified hydrologic cycle23,30,31,32. More recent studies considered the time required for regolith kaolinitization—with estimates on the order of one million years28—and suggested instead that this increase in kaolinite represents increased erosion and exhumation of previously deposited kaolinite that formed well before the PETM26,33,41. However, such long timescales refer to the formation of kaolinite from unweathered bedrock but do not necessarily apply to the kaolinization of pre-existing clays in soil profiles. The soil transformation of 2:1-type phyllosilicates (e.g. smectite and illite) into 1:1 types (e.g. kaolinite) has been found to occur on the order of 100 days under laboratory conditions at 150 °C42, and significant kaolinization of smectites can occur over a 5–10 ka period under tropical weathering conditions43.
Clay oxygen isotopes provide information about possible mineral sources and their genesis. As such, bulk oxygen isotope measurements of PETM-age clays have been analysed previously; for example, from the Bass River section on the New Jersey margin, which similarly contains a mineral assemblage of smectite, illite, kaolinite and quartz26. In that study, a strong correlation was observed between smectite and δ18Obulk (r2 = 0.96) and between kaolinite and δ18Obulk (r2 = 0.88). This suggested that δ18Obulk variability resulted from compositional changes in the proportion of kaolinite and smectite—each with constant δ18Obulk values—rather than changes to the δ18Obulk of the individual clay minerals across the PETM. Applying a similar analysis to our δ18Obulk measurements, we find that only kaolinite shows a similarly strong correlation (r2 = 0.83) with δ18Obulk (Table 1). This implies that kaolinite across the section has a fairly constant δ18Obulk value, suggesting that kaolinite δ18Obulk does not necessarily capture isotopic changes across the PETM; this could be the result of kaolinite erosion from a pre-existing source or from kaolinization of existing soil profiles (where δ18Obulk will only be partially affected by the addition of hydroxyl groups).
By contrast, the relative amounts of oxygen contributed by smectite, illite, quartz and chlorite components do not strongly correlate with δ18Obulk, δ18OOH or δ2HOH, with r2 values never exceeding 0.52 (Table 1). Kaolinite content—which contributes up to 75% of the oxygen and hydrogen for the hydroxyl measurements—also shows poor correlation to δ18OOH and δ2HOH, suggesting that the variation in the hydroxyl isotopes over the section is not driven by changes in the clay-sized mineral proportions.
We thus do not observe strong clay compositional effects in the section studied here. In fact, the δ2HOH-SW variability increases when measured isotopic values are corrected for clay composition (i.e. by mineral-specific fractionation factors), suggesting a greater change in source water isotopic composition than is actually measured. Furthermore, the measured δ2HOH-SW decrease (48.5‰) is consistent with n-alkane δ2H data from the same region (the Normandy Vasterival section), where a decrease of 60‰ was measured across the early PETM14. The observed changes in δ2HOH are thus best explained by the formation and alteration of clays during the PETM, rather than by the erosion of pre-existing deposits.
In order for kaolinite formation to capture changing source water isotopic composition across the PETM, it must have formed via a faster mechanism than regolith kaolinization (as regolith kaolinization is estimated to take longer than the duration of the PETM28). We therefore suggest that kaolinite was formed by the transformation of 2:1-type phyllosilicate clays (e.g. smectite and illite) into 1:1-type clays (e.g. kaolinite) within soil profiles. This kaolinization process of smectitic precursors leads to the addition of new hydroxyl groups—which will isotopically reflect meteoric water—and has been shown to occur under warm, humid conditions at rates sufficiently fast to capture changes in rainfall isotope composition across the PETM44,45.
If, as argued above, clay formation and alteration during the PETM is the major contributor to δ2HOH in this section, then the effect of temperature on δ2HOH must also be considered, as temperatures in the North Sea region increased over the PETM. Current models for clay hydroxyl group H-isotope fractionation factors (2HαOH) predict values less than one39,40, meaning that clay hydroxyl group H-isotopes are depleted relative to the hydroxyl group source water. Less isotopic fractionation occurs at higher temperatures, so we would therefore expect temperature increases to result in higher δ2HOH values over the CIE in contrast to the observed lower values.
The δ2HOH signal may be somewhat biased towards marine values by post-depositional isotopic exchange; however, in contrast to exposed terrestrial clays, there is little evidence for hydrogen isotopic exchange in deep-sea buried clays over ∼3 Myr outside of the <0.1 μm fraction46, so significant hydroxyl exchange is unlikely. Contamination by organic matter may also affect the accuracy of δ2HOH measurements; however, we consider this effect to be relatively minor to the overall δ2HOH trend (see the Supplementary Information for a more complete discussion).
Paleohydrologic implications of δ2HOH
Taking into account the weak or insignificant correlations between measured isotope values and clay mineral proportions, and the negative relationship between temperature and 2HαOH, we interpret the hydroxyl isotope record as capturing changes in the isotopic composition of the source water. Although the decrease in δ2HOH at the PETM onset is the largest observed change, both the measured δ2HOH and the calculated δ2HOH-SW show a clear trend towards more negative values in the period leading up to the PETM onset. This trend towards more negative δ2HOH values precedes the CIE, suggesting that hydrologic changes during the PETM were not solely a result of changes in the carbon cycle. The pre-PETM decrease in δ2HOH may instead reflect increased terrestrial input to the North Sea, as sea level in the basin was lowered by ∼100 m during this time47. However, there is additional palynological evidence from a close-by North Sea well, where significant changes to vegetation were documented prior to the CIE, implying the pre-PETM decrease in δ2HOH does reflect hydrologic changes which predate the CIE48.
The 8‰ VSMOW decrease in δ2HOH at the onset of the PETM is interpreted to reflect a decrease in the δ2HOH of rainfall. We propose that the mechanism causing this δ2HOH decrease is the amount effect49—an observed negative correlation between monthly mean rainwater isotope composition and the total amount of precipitation, typically seen in the tropics at low elevation due to increased precipitation intensity in those regions. Increased PETM precipitation amount is further evidenced in this section by the concurrency of the δ2HOH excursion and the increased abundances of the low-salinity-tolerant dinoflagellate cysts (and Apectodinium), implying that δ2HOH accurately reflects the δ2H of rainfall during the PETM.
The measured δ2HOH decrease during the PETM may reflect high intensity precipitation events where rainfall is depleted in 18O and 2H; for example, those resulting from tropical cyclone activity50. TEX86-derived sea-surface temperatures for the North Sea suggest a temperature increase of at least ∼10 °C across the CIE onset with temperatures exceeding 30 °C51. Such high temperatures exceed the tropical convective threshold52, and may have contributed to the high rainfall intensity. Our δ2HOH results are consistent with previous interpretations that global warming during the PETM caused an intensified hydrologic cycle and northward migration of storm tracks10,11,41.
The δ2HOH values return to their pre-PETM baseline before the CIE termination, suggesting that increased rainfall intensity around the North Sea was short-lived; this too is consistent with prior δ2H measurements from the region, where short-lived increases in seasonality have been inferred from n-alkane δ2H in the Normandy Vasterival section14.
Similar hydrological behaviour during the CIE is suggested by mineralogical data from the Fur section in Denmark, where the total clay fraction, clay mineral proportions and the clay chemical index of alteration return to pre-PETM values before the end of the CIE53. Lithium isotopic composition measurements from the Fur section similarly show an excursion at the PETM onset before beginning to return to pre-PETM isotopic values during the CIE54. However, relative changes in proxies around the CIE onset differ in the Fur section compared to ours—in particular, only the total clay fraction seems to increase before the CIE onset, and kaolinite in this section is mostly present after the CIE onset. The Fur section is less expanded over the start of the PETM (i.e. the Stolleklint clay layer), suggesting that early hydrological changes may not have been fully captured.
Also of note is that the 8 ‰ VSMOW decrease in δ2HOH at the PETM onset appears to precede the CIE in this section. This may offer further evidence that hydrologic changes during the PETM were not a result of carbon excursion. However, the CIE onset recorded in this section is complex and potentially influenced by multiple confounding factors, including variable mixing of different carbon sources at time of deposition55 (where carbon sources could be either marine or terrestrial, and may or may not reflect newly sequestered carbon), and possible variability caused by a pre-onset carbon isotope excursion, which has been recorded in other sections56 (although is not present in this section’s measured record).
Future Direction
This study uses the recently developed differential thermal isotope analysis (DTIA) method to report clay δ2HOH across the PETM37. Clay hydroxyl isotopic composition is not a well-developed area of research; future work will benefit from better constraints on hydrogen isotopic fractionation factors for clay hydroxyl groups (2HαOH). There currently exist a limited number of 2HαOH values in the literature39,40, and reported values assume that fractionation is not influenced by variations in chemical composition (i.e. the interlayer metal ions present) or mineralogical composition (i.e. the ratio of illite to smectite within illite-smectite). New 2HαOH estimates for a suite of clay minerals under environmentally relevant conditions are needed to more accurately correct for changing clay composition and estimates of paleo-precipitation δ2H variability. Methodological improvements for the removal of organic matter without alteration of the clay hydroxyl isotope composition, or better constraints for the influence of organic matter on DTIA δ2HOH measurements, will also improve the certainty of this proxy. Similar clay isotope measurements should be made in other PETM sections to determine whether the δ2HOH values reported here indicate pervasive hydroclimate changes at the PETM onset or were driven by local effects in the North Sea Basin. We emphasise the importance in future studies of carefully evaluating the effects of changing clay composition on the δ18Obulk, δ18OOH and δ2HOH before an environmental interpretation is proffered.
In conclusion: we have applied a recently developed method for measuring δ2H and δ18O of the hydroxyl group in clay minerals to reconstruct past hydrological changes during the PETM. We observe a large decrease in δ2HOH coinciding with the CIE, which we interpret to reflect increased precipitation—possibly resulting from increased tropical cyclone activity—in response to global warming during the PETM. Our interpretation is supported by concomitant increases in the abundances of low-salinity dinoflagellates10, indicating increased runoff to the North Sea Basin. Hydrologic changes are short-lived relative to the CIE, as evidenced by δ2HOH values and low-salinity-tolerant dinoflagellate abundances returning to pre-PETM values before recovery of the carbon cycle. Taken together, these results imply increased kaolinization, silicate-weathering intensity, and hence humidity, in response to warming events in a greenhouse world. Comparing these and future PETM clay δ2HOH measurements to predictions from isotopically enabled models offers the promise of quantitative reconstructions of paleohydrologic conditions that can enhance our understanding of hydrology and pedology in a warming world. Moreover, clay hydroxyl group isotopes are a largely untapped proxy, which can greatly improve our understanding of clay provenance and paleoclimate in the past, provided the compositional changes in clay mineralogy are fully characterised.
Methods
Isotope notation
All isotope ratios are reported in traditional “delta” notation; for example, written here for 2H/1H as
where 2R is the 2H/1H ratio and “standard” refers to Vienna Standard Mean Ocean Water (VSMOW) for \({\delta }^{2}{{\mbox{H}}}\) and \({\delta }^{18}{{\mbox{O}}}\) of hydration water and clay bulk oxygen or to Vienna Pee Dee Belemnite (VPDB) for \({\delta }^{13}{{\mbox{C}}}\) for calcite. All results are reported in units of “per mil” (‰).
Samples
We measured 22 samples in this study consisting of separated <4 μm size fractions of sediment samples across the PETM from the Sele Formation, taken from well 22/10a-4 (57°44′8.47″N; 1°50′26.59″E; up to ∼500 m water depth) located in the central North Sea.
Bulk organic matter δ13C has been reported elsewhere for the same section10. In the late Paleocene prior to the PETM, δ13C remains relatively constant at −25 ± 1‰ VPDB (Fig. 2a). CIE onset in this section is located between 2614.3 and 2613.5 m10,33, with a large δ13C decrease to −30 ± 1‰ VPDB at 2612.0 m. Carbon isotope values remain low (−30.7 ± 0.3‰) until at least 2605.0 m; post-PETM δ13C recovery has not been captured with the available data and likely occurs higher than measured in this section.
Samples of the same <4 μm size fractions were used previously for a clay mineralogical study33. The clay mineral assemblage in the section comprises a mixture of kaolinite, illite, illite-smectite (I-S) and chlorite. Illite (13–23 wt.%) and I-S (44–54 wt.%) dominate near the base of the section (2634.0–2620.0 m), with chlorite content also at its highest in this interval (7–10 wt.%; Fig. 3). In contrast, kaolinite concentrations are low during this period (1–4 wt.%), but begin to increase at 2620.0 m and rise irregularly to a maximum of 44 wt.% just after the CIE onset at 2613.5 m; this is mirrored by a decrease in I-S concentration to 19 wt.% (Fig. 3a, b). Following this peak, between 2612.0 and 2608.0 m kaolinite, illite and chlorite concentrations decline to 6–32 wt.%, 9–20 wt.% and 1–3 wt.%, respectively, whereas I-S returns to 27–52 wt.% (mirroring the pattern in kaolinite concentrations). In the upper-most section between 2608.0 and 2596.0 m, I-S again dominates (53–68 wt.%), but illite (10–16 wt.%) and chlorite (1–3 wt.%) are less abundant than in the base of the section (Fig. 3c, d), whereas kaolinite content remains higher (12–24 wt.%) than before the PETM onset.
Sample preparation and measurement—hydroxyl isotopes
Details of the initial preparation and isolation of the <4 µm samples are given in ref. 33. Before beginning isotope measurements, a test sample was heated using a heating ramp of 5 °C per minute to 1030 °C to identify the temperature range of dehydroxylation (Fig. 4).

These dehydration profiles were obtained by using a heating rate of 5 °C per minute between 30 and 1030 °C. No chlorite dehydroxylation peak is seen in our PETM samples, suggesting that the chlorite is sedimentary and undergoes dehydroxylation at lower temperatures61. The pure kaolinite sample was provided by IMERYS from Blackpool Pit, St. Austell pluton, Cornwall, UK25. The rectorite standard was purchased from the Clay Minerals Society, originating from Garland County, Arkansas, USA. The chlorite sample is a metamorphic chlorite of unknown origin.
Before making the hydroxyl isotope measurements, we first treated the <4 µm samples with 2 ml of 2 M sodium acetate/acetic acid buffer solution to remove any traces of carbonate. Following acid treatment, the samples were dried overnight at 40 °C and then measured with the Differential Thermal Isotope Analysis (DTIA) system37, using a heating programme optimised for maximum separation of the different forms of water contained in the clay minerals (Fig. 5). All heating ramps used were at a 40 °C / min rate to give sharp water peaks. Sample sizes varied between 16 and 31 mg and were adjusted to give a hydroxyl peak height of 11,000–14,000 ppm water in the optical cavity of the Picarro 2130 analyzer. The δ18O and δ2H were calculated by integrating the H2O, δ18O and δ2H traces for the hydroxyl peak and correcting them for background and calibration of the Picarro cavity ringdown laser spectrometer.

The dehydration profiles shown are from depths a 2616.34 m, b 2613.48 m and c 2611.59 m. The first and second peaks, which are measured between 30–250 and 250–390 °C, respectively, likely reflect exchangeable interlayer water. At 250 °C and 390 °C, 10 min isothermal steps were used to increase peak separation. The second peak varies in size across the samples, being between 12 and 51% of the size of the third peak, and thus is too small for accurate isotopic measurements. Including the second peak as part of the hydroxyl peak in isotope analysis leads to only small changes in reported values, and does not change the overall trends in δ18OOH and δ2HOH.
Three to four DTIA measurements were taken at each depth (at 2610.82 m, only two measurements could be taken, due to having limited sample). Errors (1 σ) were calculated from the standard deviation of the measurements at each depth.
Sample analysis—hydroxyl isotopes
To calculate the fractionation factor corrected δ2H values from the hydroxyl group δ2H, which should represent the approximate source water hydrogen isotopic composition, a number of assumptions are required. Not all clays have hydroxyl fractionation factors reported in the literature, so we must assume that the hydroxyl water was derived from a mixture of illite-smectite and kaolinite only. We then calculate the proportion of hydroxyl water that each clay mineral would contribute to the total hydroxyl water, by comparing the proportions of each clay in the assemblage to the relative weight percentage of hydroxyl in the clay chemical formulae, assuming a typical clay elemental composition. For illite-smectite, we assume that changes to the percentage composition of illite, K0.65Al2.65Si3.35O10(OH)2, and montmorillonite, Na0.1Ca0.23Al2Si4O10(OH)2(H2O)11.66 (assuming the montmorillonite is 36.1 % water by weight), do not affect the fractionation factor, but do change the proportion of hydroxyl H-isotopes contributed. The source water values were then estimated by applying the fractionation factors weighted by the water amounts contributed by each clay:
where:
α2Hmineral is the water-mineral hydrogen-isotope fractionation factor, and δ2HSW is the estimated source water value for δ2H. Hydrogen-isotope fractionation factors at 30 °C are taken from refs. 39, 40. for kaolinite and illite-smectite respectively (Table 2). We also estimate δ2HSW assuming compositions of 100% kaolinite and 100% illite-smectite, to account for the spread of possible values for δ2HSW in order to estimate the errors for the calculations (where the largest error is caused by not having fractionation factors for an average of ∼25% of the hydroxyl isotope contribution).
No reliable values for hydroxyl oxygen isotope fractionations in clays exist in the literature. As such, it was not possible to carry out this analysis for hydroxyl oxygen. Fractionation factors for bulk oxygen isotope compositions exist; however, our samples contain considerable amounts of quartz of unknown provenance (up to 31%). For such an analysis to be useful, the quartz would have to be separated from the clay fraction, or the oxygen contribution of the quartz otherwise constrained.
Sample preparation and measurement—bulk oxygen isotopes
Samples were first treated with 1 M hydrochloric acid (HCl) at a greater than 40:1 liquid to solid ratio for four hours to remove carbonates. This is unsuitable for hydroxyl δD measurements as it leads to hydrogen-isotope exchange, but it does not cause problems for bulk oxygen isotope measurements. Samples were then treated with 30% hydrogen peroxide (H2O2) for 48 h at room temperature to remove organic matter, again at a greater than 40:1 liquid to solid ratio, and were subsequently centrifuged, decanted and rinsed three times with 18.1 MΩ Milli-Q water to remove residual H2O2. HCl and H2O2 treatments are shown to negligibly impact bulk δ18O values for a kaolinite standard material, supporting the use of these methods (raw δ18O = 21.6 ± 0.6‰, n = 2; HCl treated δ18O = 22.5 ± 0.5‰, n = 2; H2O2 treated δ18O = 20.5 ± 0.7‰, n = 9).
Following this, samples were dried overnight at 65 °C in a vacuum oven, loaded into a 21-well laser fluorination tray and dried for a further 72 h at 65 °C in a vacuum oven. This tray was then inserted into a laser fluorination chamber, heated to ∼65 °C under vacuum for ∼6 h, and allowed to cool to room temperature before being reacted with 40 Torr fluorine gas (F2) for 15 h to remove any remaining hydration water (“pre-fluorination”). Pre-fluorination product O2 was then removed under vacuum. To analyse each sample, the chamber was again charged with 40 Torr F2 gas and same material was reacted by use of a 50 W CO2 laser (Teledyne) following ref. 57; laser power was initially low to prevent sputtering and was gradually increased to ∼10% until no visible sample residue remained. Fluorinated compounds were then removed by liquid nitrogen, residual F2 gas was passivated by KBr salt held at 200 °C, and O2 gas was purified by passage through a 3 m long gas chromatography column packed with 5 Å molecular sieve as described in ref. 58.
Analyte O2 gas was measured on a ThermoFischer Scientific MAT 253 isotope ratio mass spectrometer (IRMS) operated in dual inlet mode; for each sample, gas was analysed in 4-acquisition blocks of 10 cycles each and averaged. Resulting δ18O values were corrected to the 2-point calibration SMOW/SLAP scale following ref. 59. by directly fluorinating and analysing VSMOW-2 and SLAP-2 standard material using a CoF3 reactor as described in ref. 60. Uncertainty was taken as the difference between replicate sample aliquots.
Data availability
The clay isotope data generated in this study, as well as source data for all figures, are provided in the Supplementary Data.
Code availability
A Mathematica script which can be used in the analysis of raw Picarro L2130i cavity ringdown spectrometer data is included in the Supplementary Software.
References
Thomas, D. J., Zachos, J. C., Bralower, T. J., Thomas, E. & Bohaty, S. Warming the fuel for the fire: Evidence for the thermal dissociation of methane hydrate during the Paleocene-Eocene thermal maximum. Geology 30, 1067 (2002).
Westerhold, T. et al. Astronomical calibration of the Ypresian timescale: implications for seafloor spreading rates and the chaotic behavior of the solar system. Clim. Past 13, 1129–1152 (2017).
McInerney, F. A. & Wing, S. L. The Paleocene-Eocene Thermal Maximum: a perturbation of carbon cycle, climate, and biosphere with implications for the future. Annu. Rev. Earth Planet Sci. 39, 489–516 (2011).
Frieling, J. et al. Extreme warmth and heat-stressed plankton in the tropics during the Paleocene-Eocene Thermal Maximum. Sci. Adv. 3, e1600891 (2017).
Schmidt, D. N., Thomas, E., Authier, E., Saunders, D. & Ridgewell, A. Strategies in times of crisis—insights into the benthic foraminiferal record of the Palaeocene–Eocene Thermal Maximum. Philos. Trans. R. Soc. A 376, 20170328 (2018).
Carmichael, M. J. et al. Hydrological and associated biogeochemical consequences of rapid global warming during the Paleocene-Eocene Thermal Maximum. Glob. Planet. Change 157, 114–138 (2017).
Jaramillo, C. et al. Effects of rapid global warming at the paleocene-eocene boundary on neotropical vegetation. Science 330, 957–961 (2010).
Harding, I. C. et al. Sea-level and salinity fluctuations during the Paleocene–Eocene thermal maximum in Arctic Spitsbergen. Earth Planet. Sci. Lett. 303, 97–107 (2011).
Crouch, E. M. et al. The Apectodinium acme and terrestrial discharge during the Paleocene–Eocene thermal maximum: new palynological, geochemical and calcareous nanoplankton observations at Tawanui, New Zealand. Palaeogeogr. Palaeoclimatol. Palaeoecol. 194, 387–403 (2003).
Kender, S. et al. Marine and terrestrial environmental changes in NW Europe preceding carbon release at the Paleocene–Eocene transition. Earth Planet. Sci. Lett. 353-354, 108–120 (2012).
Pagani, M. et al. Arctic hydrology during global warming at the Palaeocene/Eocene thermal maximum. Nature 442, 671–675 (2006).
Bowen, G. J. & Bowen, B. B. Mechanisms of PETM global change constrained by a new record from central Utah. Geology 36, 379 (2008).
Schmitz, B. & Pujalte, V. Abrupt increase in seasonal extreme precipitation at the Paleocene-Eocene boundary. Geology 35, 215 (2007).
Garel, S. et al. Paleohydrological and paleoenvironmental changes recorded in terrestrial sediments of the Paleocene–Eocene boundary (Normandy, France). Palaeogeogr. Palaeoclimatol. Palaeoecol. 376, 184–199 (2013).
Stocker, T. et al. Technical summary. in Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change (Cambridge University Press, Cambridge, 2013).
Trenberth, K. E., Dai, A., Rasmussen, R. M. & Parsons, D. B. The changing character of precipitation. Bull. Am. Meteorol. Soc. 84, 1205–1218 (2003).
Dai, A. Precipitation characteristics in eighteen coupled climate models. J. Clim. 19, 4605–4630 (2006).
Houghton, J. T. et al. Climate Change 2001: The Scientific Basis. Contribution of Working Group I to the Third Assessment Report of the IPCC (Cambridge University Press, 2001).
Seager, R., Naik, N. & Vecchi, G. A. Thermodynamic and dynamic mechanisms for large-scale changes in the hydrological cycle in response to global warming. J. Clim. 23, 4651–4668 (2010).
Sun, Y., Solomon, S., Dai, A. & Portmann, R. W. How often does it rain. J. Clim. 19, 916–934 (2006).
Gibson, T. G., Bybell, L. M. & Owens, J. P. Latest Paleocene lithologic and biotic events in neritic deposits of southwestern New Jersey. Paleoceanography 8, 495–514 (1993).
Gibson, T. G., Bybell, L. M. & Mason, D. B. Stratigraphic and climatic implications of clay mineral changes around the Paleocene/Eocene boundary of the northeastern US margin. Sediment. Geol. 134, 65–92 (2000).
Robert, C. & Kennett, J. P. Antarctic subtropical humid episode at the Paleocene-Eocene boundary: clay-mineral evidence. Geology 22, 211 (1994).
Bolle, M.-P. et al. Climatic evolution on the southern and northern margins of the Tethys from the Paleocene to the early Eocene. GFF 122, 31–32 (2000).
Kelly, D. C., Zachos, J. C., Bralower, T. J. & Schellenberg, S. A. Enhanced terrestrial weathering/runoff and surface ocean carbonate production during the recovery stages of the Paleocene-Eocene thermal maximum. Paleoceanography 20, PA4023 (2005).
John, C. M. et al. Clay assemblage and oxygen isotopic constraints on the weathering response to the Paleocene-Eocene thermal maximum, east coast of North America. Geology 40, 591–594 (2012).
White A. F., & Brantley S. L. Chemical weathering rates of silicate minerals; an overview (Mineralogical Society of America, 1995).
Thiry, M. Palaeoclimatic interpretation of clay minerals in marine deposits: an outlook from the continental origin. Earth Sci. Rev. 49, 201–221 (2000).
Egger, H., Homayoun, M. & Schnabel, W. Tectonic and climatic control of Paleogene sedimentation in the Rhenodanubian Flysch basin (Eastern Alps, Austria). Sediment. Geol. 152, 247–262 (2002).
Gawenda, P., Winkler, W., Schmitz, B. & Adatte, T. Climate and bioproductivity control on carbonate turbidite sedimentation (Paleocene to earliest Eocene, Gulf of Biscay, Zumaia, Spain. J. Sediment. Res. 69, 1253–1261 (1999).
Bolle, M.-P. & Adatte, T. Palaeocene-early Eocene climatic evolution in the Tethyan realm: clay mineral evidence. Clay Min. 36, 249–261 (2001).
Clechenko, E. R., Kelly, D. C., Harrington, G. J. & Stiles, C. A. Terrestrial records of a regional weathering profile at the Paleocene-Eocene boundary in the Williston Basin of North Dakota. Geol. Soc. Am. Bull. 119, 428–442 (2007).
Kemp, S. J., Ellis, M. A., Mounteney, I. & Kender, S. Palaeoclimatic implications of high-resolution clay mineral assemblages preceding and across the onset of the Palaeocene–Eocene Thermal Maximum, North Sea Basin. Clay Min. 51, 793–813 (2016).
Walker, J. C. G., Hays, P. B. & Kasting, J. F. A negative feedback mechanism for the long-term stabilization of Earth’s surface temperature. J. Geophys. Res. Oceans 86, 9776–9782 (1981).
Kender, S. et al. Paleocene/Eocene carbon feedbacks triggered by volcanic activity. Nat. Commun. 12, 5186 (2021).
Knox, R.W.O’. B. in Late Paleocene–Early Eocene climatic and biotic events in the marine and terrestrial records (eds. Aubrey, M. P., Lucas, S. G., and Berggren, W. A.) (Columbia University Press, 1998).
Bauska, T. K., Walters, G., Gázquez, F. & Hodell, D. A. Online differential thermal isotope analysis of hydration water in minerals by cavity ringdown laser spectroscopy. Anal. Chem. 90, 752–759 (2017).
Frieling, J. & Sluijs, A. Towards quantitative environmental reconstructions from ancient non-analogue microfossil assemblages: ecological preferences of Paleocene – Eocene dinoflagellates. Earth Sci. Rev. 185, 956–973 (2018).
Gilg, H. A. & Sheppard, S. M. F. Hydrogen isotope fractionation between kaolinite and water revisited. Geochim. Cosmochim. Acta 60, 529–533 (1996).
Hyeong, K. & Capuano, R. M. Hydrogen isotope fractionation factor for mixed-layer illite/smectite at 60° to 150 °C: New data from the northeast Texas Gulf Coast. Geochim. Cosmochim. Acta 68, 1529–1543 (2004).
Carmichael, M. J., Pancost, R. D. & Lunt, D. J. Changes in the occurrence of extreme precipitation events at the Paleocene–Eocene thermal maximum. Earth Planet. Sci. Lett. 501, 24–36 (2018).
Ryan, P. C. & Huertas, F. J. Reaction pathways of clay minerals in tropical soils: insights from Kaolinite-Smectite synthesis experiments. Clays Clay Min. 61, 303–318 (2013).
Ryan, P. C. & Huertas, F. J. The temporal evolution of pedogenic Fe-smectite to Fe-kaolin via interstratified kaolin-smectite in a moist tropical soil chronosequence. Geoderma 151, 1–15 (2009).
Dudek, T., Cuadros, J. & Fiore, S. Interstratified kaolinite-smectite: nature of the layers and mechanism of smectite kaolinization. Am. Min. 91, 159–170 (2006).
Cuadros, J., Nieto, F. & Wing-Dudek, T. Crystal-chemical changes of mixed-layer kaolinite-smectite with progressive kaolinization, as investigated by TEM-AEM and HRTEM. Clays Clay Min. 57, 742–750 (2009).
Yeh, H.-W. & Epstein, S. Hydrogen isotope exchange between clay minerals and sea water. Geochim. Cosmochim. Acta 42, 140–143 (1978).
Knox, R. W. O. ’B. Correlation of the early Paleogene in northwest Europe: an overview. Geo. Soc. Spec. Publ. 101, 1–11 (1996).
Eldrett, J. S., Greenwood, D. R., Polling, M., Brinkhuis, H. & Sluijs, A. A seasonality trigger for carbon injection at the Paleocene–Eocene Thermal Maximum. Clim 10, 759–769 (2014).
Dansgaard, W. Stable isotopes in precipitation. Tellus 16, 436–468 (1964).
Müller, T. et al. Stable isotope composition of Cyclone Mekunu rainfall, Southern Oman. Water Resour. Res. 56, e2020WR027644 (2020).
Stokke, E. W., Jones, M. T., Tierney, J. E., Svensen, H. H. & Whiteside, J. H. Temperature changes across the Paleocene-Eocene Thermal Maximum – a new high-resolution TEX86 temperature record from the Eastern North Sea Basin. Earth Planet. Sci. Lett. 544, 116388 (2020).
Folkins, I. & Braun, C. Tropical rainfall and boundary layer moist entropy. J. Clim. 16, 1807–1820 (2003).
Stokke, E. W. et al. Rapid and sustained environmental responses to global warming: the Paleocene–Eocene Thermal Maximum in the eastern North Sea. Clim 17, 1989–2013 (2021).
Pogge von Strandmann, P. A. E. et al. Lithium isotope evidence for enhanced weathering and erosion during the Paleocene-Eocene Thermal Maximum. Sci. Adv. 7, eabh4224 (2021).
Lyons, L. L. et al. Palaeocene–Eocene Thermal Maximum prolonged by fossil carbon oxidation. Nat. Geosci. 12, 54–60 (2019).
Babila, T. L. et al. Surface ocean warming and acidification driven by rapid carbon release precedes Paleocene-Eocene Thermal Maximum. Sci. Adv. 8, eabg1025 (2022).
Sharp, Z. D. In situ laser microprobe techniques for stable isotope analysis. Chem. Geol. 101, 3–19 (1992).
Cowie, B. R. & Johnston, D. T. High-precision measurement and standard calibration of triple oxygen isotopic compositions (δ18O, Δ′17O) of sulfate by F2 laser fluorination. Chem. Geol. 440, 50–59 (2016).
Schoenemann, S. W., Schauer, A. J. & Steig, E. J. Measurement of SLAP2 and GISP δ17O and proposed VSMOW-SLAP normalization for δ17O and 17Oexcess. Rapid Commun. Mass Spectrom. 27, 582–590 (2013).
Barkan, E. & Luz, B. High precision measurements of 17O/16O and 18O/16O ratios in H2O. Rapid Commun. Mass Spectrom. 19, 3737–3742 (2005).
Földvári, M. Handbook of Thermogravimetric System of Minerals and Its Use in Geological Practice. (Geological Institute of Hungary, 2011).
Acknowledgements
This research was supported by the ERC WIHM Project (#339694) to D.A.H., and the NSF Early-Concept Grants for Exploratory Research (Grant EAR1839341) and American Chemical Society Petroleum Research Fund (Grant 59455-ND2) to both J.D.H. and D.T.J. S.J.K. publishes with the permission of the Director of the British Geological Survey, part of United Kingdom Research and Innovation (UKRI). We are grateful to B.G. Group for initial access to the 22/10a-4 core samples. We thank James Rolfe for technical assistance in the Godwin Laboratory, and Simon Redfern, Thomas Bauska and Tao Liu for discussions and guidance.
Author information
Authors and Affiliations
Contributions
D.A.H. conceived the study and G.L.W. performed DTIA experiments and the analysis of experimental results, S.J.K. provided samples and performed sample preparation, J.D.H. performed bulk oxygen measurements, D.T.J. supervised bulk oxygen measurements and D.A.H. supervised DTIA experiments and analysis. G.L.W. wrote the text with input from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Joost Frieling, Morgan Jones and Torsten Vennemann for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Walters, G.L., Kemp, S.J., Hemingway, J.D. et al. Clay hydroxyl isotopes show an enhanced hydrologic cycle during the Paleocene-Eocene Thermal Maximum. Nat Commun 13, 7885 (2022). https://doi.org/10.1038/s41467-022-35545-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-35545-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
