
Abstract
Efficient determination of protein ligandability, or the propensity to bind small-molecules, would greatly facilitate drug development for novel targets. Ligandability is currently assessed using computational methods that typically consider the static structural properties of putative binding sites or by experimental fragment screening. Here, we evaluate ligandability of conserved BTB domains from the cancer-relevant proteins LRF, KAISO, and MIZ1. Using fragment screening, we discover that MIZ1 binds multiple ligands. However, no ligands are uncovered for the structurally related KAISO or LRF. To understand the principles governing ligand-binding by BTB domains, we perform comprehensive NMR-based dynamics studies and find that only the MIZ1 BTB domain exhibits backbone µs-ms time scale motions. Interestingly, residues with elevated dynamics correspond to the binding site of fragment hits and recently defined HUWE1 interaction site. Our data argue that examining protein dynamics using NMR can contribute to identification of cryptic binding sites, and may support prediction of the ligandability of novel challenging targets.
Similar content being viewed by others

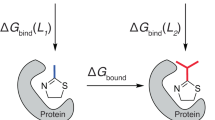

Introduction
Predictions of protein ligandability and/or druggability can tremendously facilitate the development of both chemical probes and novel drug candidates. Ligandability refers to the proclivity of a protein target to bind small molecules with high affinity, whereas druggability reflects the feasibility of developing potent and safe molecules with therapeutic efficacy1. The ligandability, and further, druggability, of a given protein depends on the physicochemical and topological properties of small-molecule binding sites2. Known tractable drug targets, for example, GPCRs, ion channels, and kinases3,4, generally present relatively small binding sites, with Solvent Accessible Surface Areas (SASA) < 1000 A2, and well-defined binding pockets2,5. Conversely, less ligandable targets often present binding sites that are larger (SASAs > 1000 A2)6,7,8, with smaller radii of curvature2. These characteristics reflect larger, “flatter”, and less topologically-defined interfaces for protein–protein interactions (PPIs) and are consequently more challenging targets for drug discovery9,10,11,12.
Various computational approaches have been employed to explore the ligandability of protein targets, typically based on static structural data and topological properties of putative binding sites13,14,15,16,17,18. These methods provide reasonable predictions of ligandability for enzymes and receptors. However, challenging targets necessitate new approaches, particularly when it comes to PPIs. For example, the presence of ligand binding hot spots can be predicted based on static crystal structures employing FTMAP software19. The presence of protein dynamics can be accounted by simultaneous analysis of multiple crystal structures20, inspecting the electron density map alterations21, or by analyzing the conformational heterogeneity from the molecular dynamics simulation data sets22. Additionally, ligandability can be evaluated using experimental methods, and fragment-based screening (FBS) has proven to be a particularly valuable approach for this task. Assessment of protein ligandability by FBS was first developed by Hajduk et al., and is based on both the rate of hits from FBS, and on the physicochemical properties of binding sites14,23. FBS was demonstrated to be highly efficient in predicting ligandability and developing optimized inhibitors, particularly in the case of PPI interfaces24. AstraZeneca proposed a more rigorous approach to predict protein ligandability based on FBS, by combining screen hit rates and affinities of identified ligands25.
BTB domains are common PPI motifs present in transcription factors and epigenetic scaffolding proteins26,27. BTB domain-containing proteins have emerged as pharmacological targets and include BCL6, an oncogenic driver in several subtypes of Diffuse Large B-Cell Lymphomas (DLBCLs)28,29. The BCL6 BTB domain interacts with co-repressors, such as SMRT and BCoR30,31 and various independent academic and industrial groups have undertaken extensive efforts to develop small-molecules that inhibit BCL6 interactions or induce protein degradation32,33. The BCL6 BTB domain has proven to be a highly tractable target and, to date, many BCL6 inhibitors have been reported34,35,36,37,38,39.
The BTB protein family contains numerous other members, several of which are potential targets for inhibitor development. For example, KAISO is a transcriptional repressor that recruits the co-repressor SMRT40. Depletion of KAISO has been reported to attenuate the survival of Triple-Negative Breast Cancer (TNBC) cells, suggesting that KAISO plays a role in TNBC oncogenesis41. LRF is another BTB-containing transcriptional regulator that recruits co-repressor complexes42. LRF regulates B-cell differentiation43, and is also associated with the development of various cancers, including breast cancer44 and prostate45,46. The BTB domain-containing protein MIZ1 functions in both the transcriptional activation and repression of target genes47. MIZ1 BTB domain interacts with HECT-type ubiquitin ligase HUWE1 (also called MULE)48, and recent structural studies revealed an atypical binding mode with the dimeric BTB domain recognizing a single HUWE1 molecule49. MIZ1 also interacts with MYC through a motif outside of its BTB domain and has emerged as an oncogenic co-factor in the MYC-dependent medulloblastomas50.
In this study, we evaluate the ligandability of BTB domains. BCL6, KAISO, LRF, and MIZ1 all possess structurally related and conserved BTB domains, yet small molecule inhibitors have been reported only for BCL6. Therefore, we explore whether the three other members of the BTB family are ligandable. Using fragment-based screening against the BTB domains of KAISO, LRF, and MIZ1, we identify multiple small-molecule ligands that bind to MIZ1. Surprisingly, we find no hits for KAISO and LRF. To rationalize this unexpected finding, and to elucidate the biophysical and structural bases of ligandability of these BTB family members, we investigate their dynamics using solution NMR spectroscopy. Rigorous analysis of spin relaxation data reveals that MIZ1 possesses a distinct dynamics profile compared to KAISO and LRF, featuring specific motions on the µs-ms time scale. Notably, the location of the MIZ1BTB residues with elevated dynamics coincides with the binding site of HUWE149 and the small molecule ligands we discover. We propose that protein dynamics represents a significant mechanism governing the recognition of small-molecule ligands by MIZ1BTB. Our data argue that protein dynamics may be a broadly applicable tool in drug discovery to assess the ligandability of novel and challenging targets.
Results
BTB domain of MIZ1 but not KAISO or LRF has a high propensity to bind small molecule ligands
To evaluate the respective ligandabilities of the LRF, KAISO, and MIZ1 BTB domains, we screened each protein against a library of 600 chemically diverse, fragment-like small molecules by protein-observed solution NMR spectroscopy. Specifically, recombinant, uniformly 15N-labeled BTB domains of LRF, KAISO, and MIZ1, referred to herein as LRFBTB, KAISOBTB, and MIZ1BTB, respectively, were screened with mixtures containing 10 compounds via a series of 1H-15N HSQC experiments. First, we screened MIZ1BTB and found that approximately 40 out of 60 screening mixtures yielded detectable chemical shift perturbations. To rank the hits according to their binding potencies, we calculated the sum of chemical shift perturbations of seven selected amide proton resonances (7PA value)34. Out of the 40 mixtures with 7 PA values > 100 Hz, we selected two mixtures yielding the most significant 7 PA values > 300 Hz for further analysis (Supplementary Fig. 1). We deconvoluted these mixtures and identified three compounds, 2GG4, 4CC2, and 5DD7 (Fig. 1a), that yielded the most pronounced chemical shift perturbations upon binding to MIZ1BTB (Fig. 1b). These hits comprise three chemically distinct scaffolds, which suggest that MIZ1BTB can bind structurally diverse compounds. To determine their respective binding affinities, we performed NMR-based titration experiments (Fig. 1b, c), and found that 2GG4 binds MIZ1BTB with the highest affinity (Kd of 68 ± 9 μM, Fig. 1c), which is relatively potent for a small molecule fragment-like compound51. The two remaining compounds presented weaker affinities, with the Kd values of 425 ± 59 and 870 ± 140 μM for 4CC2 and 5DD7, respectively (Fig. 1c). Next, we performed the fragment screen against KAISOBTB and LRFBTB. In contrast to MIZ1BTB, we did not identify any compounds that bind to these proteins. These results emphasize remarkable differences in the propensity of three structurally related BTB domain proteins to bind small-molecule ligands: we found a 7% hit rate for MIZ1BTB (assuming one hit per mixture), whereas no hits were identified for KAISOBTB and LRFBTB.

a The chemical structures of fragments that bind to MIZ1BTB; b The 1H-15N HSQC spectra of MIZ1BTB (red) titrated with the three fragments. The molar ratios of MIZ1BTB-ligands are listed and colors correspond to coloring of the spectra. Selected residues experiencing large chemical shift perturbations are labeled. c Determination of Kd values from NMR titration experiments for the three MIZ1BTB ligands. Averaged binding constants are reported +/− SD and are calculated from fitting the titrations of several amides. Source data for (c) is provided as a Source Data file.
Small molecule ligands bind to a conformationally variable site in MIZ1 BTB domain
The binding of 2GG4, 4CC2, and 5DD7 to MIZ1BTB leads to extensive chemical shift perturbations on the 1H-15N HSQC spectra (Fig. 1b; Supplementary Fig. 1). To characterize the ligand-binding site, we mapped the chemical shift perturbations of methyl groups (Fig. 2a - upper row), as well as the backbone amides (Fig. 2a - bottom row), onto the structure of MIZ1BTB. These perturbations cluster around residues in strands B1, B2, and B4, and helices A2 and A3 (Supplementary Fig. 2 and Fig. 2a, b). Analysis of several independently determined crystal structures of MIZ1BTB, including a structure determined in this study (Supplementary Table 1 and Supplementary Fig. 3), indicates that this region of BTB domain is conformationally variable. Residues 56–64, which comprise the B4 strand in a canonical BTB fold52, namely β-strand, short α-helix, 310-helix, and loop, can adopt a variety of distinct conformations, or can be disordered (Supplementary Fig. 4). The MIZ1BTB domain thus exists in “closed” and multiple “open” conformations resulting from the structural variability around B4. This variable region is consistent with the small molecule binding site mapped by NMR (Fig. 2a, b). Analysis of NMR chemical shifts and through-space HN-HN NOE contacts revealed that residues 56–64 form a β-strand conformation with hydrogen bonds between B2 and B4 (Supplementary Fig. 5). Thus, the BTB domain adopts a predominantly a “closed” conformation in solution, which presents a compact structure. In contrast to the “open” form, the “closed” form lacks the pockets suitable for small molecule binding (Fig. 2a, b). We hypothesize that the compounds we identified bind to a transiently populated “open” conformation that features a pocket that is large enough to accommodate small molecules. Recent studies found that this site in MIZ1BTB is indeed involved in ligand binding and recognition of a peptide fragment from the HUWE1 E3 ligase49. The crystal structure of MIZ1BTB in complex with HUWE1 derived peptide demonstrates that BTB domain adopts an “open” conformation with the B2–B4 interface being involved in PPIs with HUWE1 (Fig. 2c). The MIZ1BTB-HUWE1 interface coincides with the binding site for the fragment compounds we identified (Fig. 2c) indicating that FBS is an unbiased method that can uncover PPI binding sites.

a Mapping of the chemical shift perturbations of methyl groups (upper row) and backbone amides (lower row) resulting from the binding of 2GG4 to MIZ1BTB. The crystal structure is shown in “open” (PDB id: 2Q81:A) and “closed” (PDB id: 2Q81:B) conformations. All methyl-containing residues (ILVAMT) are shown in green with sphere radii corresponding to the magnitude of chemical shift perturbation. Perturbations of backbone amides are in the dark-blue to light-blue colors corresponding to their CSPN/H effect. CSP effects correspond to the MIZ1BTB:2GG4 molar ratio of 1:4. b Mapping of the amide chemical shift perturbations CSPN/H upon binding of the three fragments onto the crystal structure of MIZ1BTB with bound HUWE1 (orange) (PDB id: 7AZX).
MIZ1 BTB domain exhibits a dynamic profile distinct from KAISO and LRF
To assess whether ligandability of BTB domains can be predicted using computational methods, we employed FTMAP software, which can determine ligand binding hot spots based on protein structure19. First, we performed analysis of the BCL6BTB domain, which is highly ligandable, and we found multiple fragments mapped to a site encompassed by helices α2, α3, α6 and α1 from the second monomer (Supplementary Fig. 6). Importantly, this region represents a binding site for other known BCL6 inhibitors34,35,36,37,38,39. Then, we performed FTMAP analysis for LRFBTB, KAISOBTB, and MIZ1BTB and found several putative small molecule hot spots for all three BTB domains, including the same ligandable site as in BCL6BTB (Supplementary Fig. 6). Based on the FTMAP results we hypothesized that the propensity of MIZ1BTB to bind small-molecule ligands might result from distinct dynamics when compared to KAISOBTB and LRFBTB. To test this, we performed comprehensive studies of 15N relaxation for KAISOBTB, LRFBTB, and MIZ1BTB using two magnetic fields (Fig. 3; Supplementary Fig. 7). We found that all proteins are dimeric in solution with expected correlation times and size exclusion profiles (Supplementary Fig. 8). The comparison of the raw 15N spin relaxation observables reveals that all three BTB domains are well ordered, except for the flexible N- and C-termini. KAISO has a very rigid BTB domain, with the more flexible, short loop between B3-H4 (residues A64-G65) exhibiting elevated ps-ns motions, as indicated by decreased transverse relaxation rates accompanied by lowered NOE values for those residues (Supplementary Fig. 7). LRFBTB has a similar dynamics profile to KAISOBTB, with a longer, more flexible loop between B3 and H4 (residues S64-Q70) (Supplementary Fig. 7). Importantly, residues in both proteins show flat spectral density function ratio profiles at zero frequency, so-called Residue Consistency, i.e., RCi ratios at 1 (Fig. 3). These flat profiles indicate a lack of significant chemical exchange contribution Rex53. Although several regions in KAISOBTB and LRFBTB present fast ps-ns motions, analysis of RCi values unequivocally indicates no contribution from dynamics on slower µs-ms time scales. In contrast, 15N relaxation parameters determined for MIZ1BTB reveal a distinct dynamics profile, particularly for B2 and B4 strands (Fig. 3). Residues in these regions of MIZ1 have RCi values that are significantly above 1, indicating the presence of elevated dynamics and slow motions on the µs-ms time scale (Fig. 3). Notably, the dynamics profile in MIZ1BTB clearly distinguishes this protein from KAISO and LRF, and correlates with its ability to bind small molecule ligands.

The spectral density analysis plots derived from a comprehensive set of 15N spin relaxation observables, at two magnetic field strengths, for KAISOBTB (a), LRFBTB (b), and MIZ1BTB (c). The RCi ratio errors are propagated from the spin relaxation observables’ uncertainties. Shaded regions indicate the isostructural positions of β4 and β2 strands, that only in MIZ1BTB are experiencing μs-ms time scale dynamics. Source data is provided as a Source Data file.
MIZ1 small molecule ligand binding site shows enhanced μs-ms time scale dynamics
We found that small molecule ligands bind to the MIZ1 B2 and B4 strands (Fig. 2a, b), which show µs-ms time scale dynamics in 15N relaxation studies (Fig. 3; Supplementary Fig. 7). Thus, specific small molecule ligands bind MIZ1BTB at a site with elevated dynamics. To further quantify BTB domain dynamics, we performed a complete analysis of 15N relaxation data from two magnetic fields in the frame of the model-free approach (MFA)54. The MFA allows analysis of fast ps-ns local dynamics by the order parameter S2, which characterizes the amplitude of motion and its internal time - τint. MFA further detects slow µs-ms time scale collective motions, expressed as chemical exchange contribution, Rex. Our MFA analysis confirms that the structurally conserved cores of KAISOBTB and LRFBTB are uniformly well ordered (average S2 = 0.94 ± 0.02), except for the flexible N- and C-termini (Fig. 4a, b). Additionally, the loop linking B3 and B4 in KAISOBTB experiences medium amplitude ps-ns motions (average S2 = 0.72 ± 0.09) (Fig. 4a, b), and is disordered in LRFBTB (average S2 = 0.51 ± 0.07) (Fig. 4a, b). However, in both proteins, B2 and B4 strands are well ordered. In contrast, MIZ1BTB displays elevated local dynamics for two regions encompassing B2 (average S2 = 0.84 ± 0.03) and B4 (average S2 = 0.74 ± 0.08) (Fig. 4a, b). Consistent with the analysis of the spectral density ratios (RCi), the B2 and particularly B4 regions in MIZ1BTB specifically experience a contribution of chemical exchange to transverse relaxation (Rex), indicating µs-ms dynamics around these motifs (Fig. 4c). The MFA analysis demonstrated that the crucial feature that distinguishes MIZ1BTB from KAISOBTB and LRFBTB is the presence of elevated dynamics on the μs-ms time scale for a subset of residues. Data from the fragment screens strongly suggest that elevated dynamics contribute to MIZ1BTB ligandability.

a Order parameters S2 reporting on the local fast dynamics plotted along the MIZ1BTB (orange), LRFBTB (gray), and KAISOBTB (green) primary sequence. Values and corresponding errors are mean +/− SD as error bars obtained from propagated uncertainty after Monte-Carlo simulations. The secondary motifs are marked along the primary sequence and light-blue squares indicate B2 and B4 motifs. Source data is provided as a Source Data file. b The S2 order parameters plotted on the structures of BTB domains scaled yellow to red to indicate increasing fast local dynamics. c Surface representations of the studied BTB domains with mapped residues with identified chemical exchange, Rex contribution, indicating the slow segmental μs-ms dynamics.
Binding to HUWE1 quenches μs-ms dynamics in MIZ1 BTB domain
To evaluate whether μs-ms time scale dynamics in MIZ1BTB is an intrinsic feature of this domain or it is rather related to ligand binding; we determined the dynamics profile of MIZ1BTB in complex with HUWE1 peptide. Binding of HUWE1 breaks the symmetry of BTB domain dimer and results in doubling of the amide resonances of NMR spectra (Supplementary Fig. 9). We then assigned backbone resonances of MIZ1BTB in a complex with HUWE1 and collected spin relaxation observables at two magnetic fields (Supplementary Fig. 10). Overall, binding of HUWE1 is not affecting the fast ps-ns dynamics in MIZ1, and order parameters for MIZ1BTB-HUWE1 complex are in line with apo MIZ1BTB (Supplementary Fig. 11). Interestingly, the RCi profile for MIZ1BTB-HUWE1 complex is flat, particularly for the B4 region, indicating a loss of Rex for the majority of residues at the interface with HUWE1 (Fig. 5a, b, d and Supplementary Fig. 12). Therefore, binding of the HUWE1 ligand suppresses the chemical exchange in MIZ1BTB suggesting that μs-ms dynamics plays a crucial role in ligand binding.

a, b Mapping of residues that show slow μs-ms segmental motions indicated by the chemical exchange Rex (magenta), determined for the MIZ1BTB, onto the structure of the MIZ1BTB dimer (PDB id: 2Q81, panel a) and the structure of MIZ1BTB with bound HUWE1-derived peptide (PDB id: 7AZX; peptide is in orange, panel b); c backbone amide chemical shift perturbations upon 2GG4 binding mapped onto the structure of the MIZ1BTB dimer (PDB id: 2Q81); d the chemical exchange Rex (magenta) determined for MIZ1BTB bound to HUWE1 peptide mapped on the structure of MIZ1BTB-HUWE1 complex (PDB id: 7AZX).
Discussion
Prediction of protein ligandability or druggability is of paramount importance for discovery of small-molecule inhibitors of new protein targets. However, despite significant progress in the development of computational and experimental methods, inhibitor development remains challenging for many targets. Here, we uncovered striking differences in the ligandability of structurally related members of the BTB domain protein family. Our rigorous analysis indicates that ligandability of BTB domains correlates with the presence of μs-ms time scale dynamics.
BTBs are conserved domains, abundant in transcriptional regulators, and involved in the formation of protein–protein complexes. Since these proteins are associated with a range of diseases27, BTB domains represent attractive targets for inhibitor development. To date, BTB domain inhibitors have been reported only for BCL633, raising the question of whether other BTB family members are ligandable. In this study, we performed fragment screens against three previously unexplored BTB domains of KAISO, LRF, and MIZ1. Surprisingly, we discovered that only the screen against MIZ1BTB yielded small molecule ligand hits, in stark contrast with the screens against KAISOBTB and LRFBTB. We, therefore, examined why domains with conserved three-dimensional structures have such distinct propensities to bind small-molecule ligands, and tested what defines the ligandability of BTB domains. Computational prediction of ligand binding hot-spots solely based on the three-dimensional structures did not show any preference for ligandability among analyzed BTB domains, including the highly ligandable BCL6 (Supplementary Fig. 6). Thus, we used NMR spectroscopy to compare protein dynamics in solution. On the bases of experimentally determined order parameters S2, we demonstrate that the cores of the three BTB domains are relatively rigid (<S2 > = 0.93), except for disordered N- and C-termini, and the flexible internal loop in LRF encompassing residues S64-Q70. We found that MIZ1BTB has a uniquely elevated dynamics profile on the slow µs-ms time scale in B2 and, particularly, in B4 region, which is indicated by significant contributions from Rex (Fig. 5a, b). The same B4 region experiences elevated fast local dynamics as evidenced by the average S2 ranging from 0.74 to 0.84 (Fig. 4a, b). Corresponding regions in KAISO and LRF do not show such µs-ms dynamics, and moreover, show local rigidity, with an average S2 of 0.93 and 0.92 for KAISO and LRF, respectively. The KAISO and LRF BTB domains lack significant contribution from Rex (Fig. 4). Importantly, regions with elevated dynamics and Rex in MIZ1BTB correspond to the binding site for small-molecule ligands identified through FBS (Fig. 5c). We also found that binding of HUWE1 ligand does not affect fast local ps-ns motions in MIZ1BTB (Supplementary Fig. 11), in contrast, it quenches slow µs-ms motions (Fig. 5d). We determined that in solution, the interface of the free BTB domain involved in HUWE1 binding is different and adopts predominantly “closed” conformation, not accessible to ligand binding. However, µs-ms dynamics in concert with conformational heterogeneity observed in MIZ1BTB crystal structures (Supplementary Fig. 4) may reflect the presence of cryptic pockets suitable for ligand binding, and suggest that binding of HUWE1 might selects one of the conformations already present in MIZ1BTB. Overall, this example highlights the important role of µs-ms dynamics, which could be explored for ligand design (Supplementary Fig. 13).
BCL6 is an example of a highly tractable member of the BTB family, as demonstrated by multiple fragment hits identified in FBS campaigns34,35, and by several reports of potent inhibitors36,37,38,39. We excluded BCL6BTB from the studies reported here, as wild-type BCL6BTB shows low solubility and requires several point mutations to obtain recombinant protein amenable to biophysical and structural characterization by NMR34,55. We anticipated that such mutations might perturb the protein’s native dynamics pattern. Nevertheless, independent studies by our group34 and by others56 have found that 1H-15N HSQC spectra of BCL6BTB feature peak broadening and lacking resonances, suggesting the presence of slow segmental μs-ms dynamics. Increased dynamics in the BCL6BTB at the interface with co-repressor peptide has additionally been observed in MD simulations57. Therefore, it is likely that BCL6BTB undergoes elevated dynamics, comparable to the dynamics measured here for MIZ1, which facilitates ligand binding.
BCL6 recruits binding partners in the lateral groove, formed at the interface of the BTB domain dimer30,31. The lateral groove on BCL6BTB also constitutes the binding site for all reported fragment-like compounds and small molecule BCL6 inhibitors34,35,36,37,39. Comparison of the crystal structure of BCL6BTB-SMRT co-repressor complex, with the crystal structures of KAISOBTB, LRFBTB, and MIZ1BTB, confirmed that the putative lateral groove is present in all these BTB domains, and have been identified via FTMAP as potential small molecule binding sites (Supplementary Fig. 6). Our fragment screening campaigns failed to identify any small molecule ligands that bind to the lateral grooves on KAISO, LRF, and MIZ1, but did identify compounds binding to MIZ1BTB at a distinct site centered around B2, B4 strands. The recently reported crystal structure of MIZ1BTB in complex with a bound peptide fragment of HUWE1 revealed an unreported PPI binding site on MIZ1BTB49, which involves the residues in B2 and B4 strands and coincides with the binding site we identified through FBS. This again emphasizes that FBS is a robust approach to assess protein ligandability and identify functional binding sites in an unbiased manner. Here, we propose that the presence of slow dynamics in such sites may support the selection of proteins for the development of small molecule inhibitors.
Previous work established that small-molecule inhibitor of HUWE1 E3 ligase stabilizes MIZ1 and inhibits MYC-dependent transactivation in colorectal cancer cells58. Thus, disrupting MIZ1-HUWE1 interaction may prove to be viable strategy to develop new anti-cancer agents. To date, no inhibitors of MIZ1 have been reported and it is likely that the small molecule fragments we identified can be further developed into potent MIZ1BTB PPI interaction inhibitors.
In summary, we performed a comprehensive analysis that relates ligandability to the backbone dynamics of structurally related BTB domains. Based on these studies, we found that protein dynamics, in addition to structural and topological features, governs the ability of BTB domains to bind small-molecule ligands. We anticipate that elevated μs-ms dynamics may represent a general feature of druggable proteins, and comprehensive studies of protein dynamics employing NMR relaxation may reveal novel, highly tractable drug discovery targets.
Methods
Expression and purification of BTB domains (LRFBTB, MIZ1BTB, KAISOBTB)
Synthetic genes encoding Human BTB domains KAISO (residues 1–122), LRF (residues 1–130), and MIZ1 (residues 1–115) were ordered from Life Technologies and sub-cloned using BamHI and HindIII restriction sites into pet32a expression plasmid containing N-terminal Thioredoxin (Trx) and hexa-histidine (his6) tags with an N-terminal PreScission-cleavage site. Recombinant pet32a-BTB domain plasmids were transformed into E. coli strain BL21 (DE3) cells and transformations were grown in Luria Broth (LB) or 15N-labeled M9 media with Ampicillin selection. Protein was expressed by induction with 0.25 mM IPTG for 16 h at 18 °C. Cells were lysed in buffer containing 50 mM Tris (pH 7.5), 300 mM NaCl, 30 mM imidazole, 1 mM TCEP, and 0.5 mM PMSF, and the clarified lysate was applied to Ni-NTA resin. Protein was eluted using lysis buffer with 300 mM imidazole, and Trx-his6-tags were proteolytically cleaved with PreScission enzyme. Eluate was re-applied to Ni-NTA resin to extract Trx-his6-tags, and KAISOBTB and MIZ1BTB were dialyzed extensively against 50 mM Tris (pH 7.5), 150 mM NaCl, and 1 mM TCEP at 4 °C for protein-observed solution-NMR experiments. For LRFBTB, protein was dialyzed against 50 mM HEPES (pH 7.5), 150 mM NaCl, and 1 mM TCEP. For MIZ1BTB crystallization experiments, protein was dialyzed against 20 mM Tris (pH 7.5), 300 mM NaCl, and 1 mM TCEP. For the NMR resonance assignment, titrations with ligands and 15N spin relaxation studies the uniformly labeled U-15N and U-13C,15N BTB domain samples were obtained from M9 media by using 1 g/L of 15NH4Cl as a sole source of nitrogen and 2.5 g/L 13C-glucose as a sole source of carbon. In order to obtain the perdeuterated U-2H,13C,15N KAISOBTB sample, the M9 media was prepared in 99.9% D2O (CIL) with 2.5 g/L of 2H,13C-glucose and 1 g/L of 15NH4Cl. The synthetic HUWE13870–3897 peptide was ordered from GeneScript with >99% purity, TFA removal service and dialyzed to the buffer containing 50 mM HEPES (pH 7.5), 150 mM NaCl, 1 mM TCEP before the further use.
Fragment screening by protein-observed solution-NMR spectroscopy
150 μM 15N-labeled KAISOBTB and MIZ1BTB in buffer comprised of 50 mM Tris (pH 7.5), 150 mM NaCl, 1 mM TCEP, and 7.5% D2O was prepared for screening. LRFBTB was prepared in 50 mM HEPES (pH 7.5), but protein concentration and buffer conditions were otherwise maintained for KAISOBTB and MIZ1BTB screening. The library used for screening was comprised of commercially available fragment-like small molecules, and in-house synthesized compounds. Protein was incubated with fragment-like small molecules in mixtures of 10 compounds per sample, at 500 μM per compound, and 5% DMSO (v/v). 1H-15N HSQC spectra were acquired at 30 °C on 600 MHz Bruker Avance III Spectrometer equipped with cryogenic probe running Topspin version 3.5. Processing and visualization of HSQC spectra were performed in NMRPipe ver. 11.0 and Sparky ver. 3.314. Fragment hits were identified by chemical shift perturbations on 1H-15N HSQC spectra. The chemical shift perturbation of 1H/15N resonances was determined with the equation (1): CSPi = [(ΔδH,i)2 + 0.1·(ΔδN,i)2]0.5, where ΔδH,i and ΔδN,i are detected chemical shift changes of proton and nitrogen, respectively.
K D determination by protein-observed solution-NMR spectroscopy
1H-15N HSQC spectra were obtained using samples with 250 μM 13C,15N-labeled MIZ1BTB in buffer composed of 50 mM Tris (pH 7.5), 150 mM NaCl, 1 mM TCEP, 7.5% D2O, and 5% DMSO. NMR-titration experiments were performed using increasing concentrations of compound 2GG4 (125, 250, 500, and 1000 μM), and of compounds 4CC2 and 5DD7 (250, 500, 1000, and 2000 μM). Dissociation constants were derived by least-squares fitting of chemical shift perturbations as function of ligand concentration (2):
with a = (Ka/δb) × [Pt], b = 1 + Ka([Lti] + [Pt]), and c = δb × Ka × [Lti], where δi is the absolute change in chemical shift for each titration point, [Lti] is the total ligand concentration at each titration point, [Pt] is the total protein concentration, Ka = 1/Kd is the binding constant, and δb is the chemical shift of the resonance in question in the complex. Kd and δb were used as fitting parameters in the analysis.
Determination of MIZ1BTB crystal structures
Screening by the hanging-drop vapor-diffusion technique was used to first obtain crystals of MIZ1BTB. Crystals were then optimized using the sitting-drop technique, via incubation of equal volumes (1.5 μL) of protein [6 mg/mL in 20 mM Tris (pH 7.5), 300 mM NaCl, and 1 mM TCEP] and crystallant solution (30% PEG-1500 [w/v]). Crystals formed over one week at 4 °C. Prior to freezing, crystals were transferred to cryoprotectant solution comprised of crystallant solution with 25% glycerol.
Crystallographic data collection and structure determination
Diffraction data for MIZ1BTB was collected at 21-ID-D beamlines at the Life Sciences Collaborative Access Team at the Advanced Photon Source. Data were integrated and scaled using HKL-2000, and structures were solved by molecular replacement using known native MIZ1BTB structure as the search model. The model was refined using REFMAC, COOT, CCP4 program suite, and PHENIX program suite. Structure validation was performed using MOLPROBITY and ADIT servers. Data collection and structure refinement statistics are reported in Supplementary Table 1.
Sequence-specific resonance assignment of globular BTB domains
The backbone 1HN, 15N, Cα, Cβ and C chemical shifts of LRFBTB, MIZ1BTB, KAISOBTB were assigned based on a set of TROSY-type 3D triple resonance experiments, i.e., HNCA, HNcoCA, HNcoCACB, HNCACB, CBCAcoNH, HNCO, HNcaCO and HNCB supported by 3D 15N-edited NOESY-HMQC experiment (mixing times 100 ms for U-13C,15N-labeled samples and 200 ms for perdeuterated U-2H,13C,15N samples)59,60. In contrast to LRFBTB and MIZ1BTB, where a decent quality of 3D triple resonance spectra was obtained from uniformly double labeled U-13C,15N-samples at concentrations of 90–160 μM (per dimer), the KAISOBTB domain and MIZ1BTB:HUWE1 complex required perdeuteration. Analogical 3D triple resonance TROSY-type experiments were recorded utilizing 2H-decoupling on 100 μM (per dimer) samples of U-2H,13C,15N KAISOBTB and U-2H,13C,15N MIZ1BTB with unlabeled HUWE13870–3897 peptide (1:3 molar ratio). The following buffer conditions were used for all samples: 50 mM HEPES (pH 7.5), 150 mM NaCl, 1 mM TCEP, and 1% (v/v) D2O. All spectra were recorded at 30 °C on 700 and 950 MHz Bruker Avance NEO spectrometers equipped with 5 mm TCI cryogenic probes. Spectra were processed in TOPSPIN 4.0.7 and analyzed in CARA ver. 1.8 software. The secondary structure motif propensities for structures in solution were determined with Talos-N software from backbone 1HN, 15N, 13Cα, 13Cβ, and 13C chemical shifts61.
Complete methyl group assignment of MIZ1BTB and titrations with ligands
The complete set of 1H/13C resonances from the methyl moiety containing amino acids, i.e., alanine, threonine, leucine, isoleucine, valine and methionines, was achieved for uniformly labeled 13C,15N-MIZ1BTB sample of 200 µM (per dimer). The side-chain methyl assignment was started from the backbone resonance assignments and a combination of high-resolution through-bond correlations in 3D spectra recorded on 950 MHz instrument: 3D (H)CCH-TOCSY (mixing times of 5.6 ms giving nearly COSY-type correlation mainly through three bonds, and 16.4 ms for total through multiple C–C bonds correlations). Moreover, the through-space correlations from 3D 15N-edited NOESY-HMQC (mixing time 100 ms) and 3D 13C-edited NOESY-HSQC (mixing time 100 and 200 ms) centered on the aliphatic region with 1JC–H set to 125 Hz were used together with available static X-ray structures from PDB to verify the assignments. The hydrogens were added in Coot ver. 0.8.9.2 software. The methionine methyl groups missing the TOCSY-type correlations were identified based on the negative sign on 13.3 ms constant time 2D 1H-13C CT-HSQC spectra and their methyls correlated to their amide via NOE cross peaks present on 13C- and 15N-edited NOESY spectra. Two 2D 1H-13C HSQC constant time (CT time of 13.3 with Methionine -S-CH3 and -CH2- groups negative and 26.6 ms with all correlations positive) at high-resolution allowed to unequivocally resolve methyl signals. The NMR titrations of 13C,15N MIZ1BTB with selected ligands were performed as described above and monitored with 2D 1H-13C CT-HSQC spectra centered on the methyl group region. Spectra were processed in TOPSPIN 4.0.7 and analyzed in CARA software.
Relaxation measurements and BTB domain dynamics analysis
The 15N backbone amide spin relaxation experiments were collected at 700 and 950 MHz (1H frequency) instruments on uniformly labeled U-15N samples of all three BTB domains and the MIZ1BTB:HUWE1 complex at an identical concentration of 100 μM each with 50 mM HEPES (pH 7.5), 150 mM NaCl, 1 mM TCEP, and 1% (v/v) D2O. The NMR experimental temperature was carefully calibrated to 30 °C with standard ethylene glycol (80%) in DMSO-d6 sample at two magnetic fields operating at 1H frequencies of 700 and 950 MHz. The temperature was monitored before and after each experiment and found to be stable within ±0.3 °C. The upgraded TROSY-versions of 15N-R1, 15N-R2, and {1H}-15N heteronuclear NOE experiments optimized for larger molecular weight proteins were utilized62,63. The recycle delay of 4 s for 15N-R1 and 15N-R2 and 10 s for {1H}-15N NOE (10 s of the 1H-saturation period) were used. The sixteen delays from 0 to 3520 ms were used for 15N-R1 and nineteen from 0 to 250.16 ms for 15N-R2, for both cases in a randomized order. The relaxation rates were fit from the intensities to exponential decay curves with two parameters in a non-linear least-square procedure, and standard errors were derived from variance-covariance matrix analysis using QtiPlot ver. 0.9. The {1H}-15N heteronuclear NOE values were obtained from the ratio of intensities and errors from the propagation of the signal-to-noise values of two signals corresponding to an individual residue. Residues with the complete set of 15N-R1, 15N-R2, and {1H}-15N NOE parameters from two fields were subjected to model-free analysis (MFA). The dynamics interpretation based on the collected 15N spin relaxation data was performed according to our previous approaches54,64,65. In short: a fully anisotropic model of motion was applied for all proteins, with the assumption of C2 symmetry for each homo-dimer resulting in four global parameters, D1, D2, D3 and γ Euler angle (while α = β = 0o). Local parameters of order parameter S2 reporting on the amplitude of motion [0,1] and local τint time characterizing the ps-ns fast motions, and Rex term sensitive to slow μs-ms time scale motions, being the addition to the transverse 15N-R2 relaxation rates. The coordinates were taken from the corresponding PDB files (KAISOBTB 3M4T; MIZ1BTB 2Q81:B symmetrized to reflect the major form in solution; LRFBTB 2NN2; MIZ1BTB:HUWE1 7AZX; the missing heavy and H-atoms were added, and energy-minimized in Coot ver. 0.8.9.2 software). Errors were obtained after Monte-Carlo procedure after 200 minimizations.
Size exclusion chromatography with multi-angle static light scattering (SEC-MALS)
The SEC-MALS measurements were performed with MIZ1BTB, KAISOBTB, LRFBTB domains at 100 μM passed through the Superdex 200 30/300 column (GE Healthcare) using Agilent HPLC following DAWN® MALS detector. The running buffer contained 50 mM HEPES (pH 7.5), 150 mM NaCl, and 1 mM TCEP. Data were analyzed with the ASTRA® software provided by the company (Wyatt Technologies). The presented results are mean values with standard error from the three sample replicates.
Data availability
The experimental crystallographic data and structural coordinates of the MIZ1 BTB domain determined in this study were deposited in the Protein Data Bank (PDB) under the accession code: PDB id 7T58. The following PDB data sets were used in the study: 2Q81, 1R29, 2NN2, 3M8V, 7AZX, 3M4T. Source data are provided with this paper.
References
Brown, K. K. et al. Approaches to target tractability assessment—a practical perspective. Medchemcomm 9, 606–613 (2018).
Cheng, A. C. et al. Structure-based maximal affinity model predicts small-molecule druggability. Nat. Biotechnol. 25, 71–75 (2007).
Hopkins, A. L. & Groom, C. R. The druggable genome. Nat. Rev. Drug Discov. 1, 727–730 (2002).
Makley, L. N. & Gestwicki, J. E. Expanding the number of ‘druggable’ targets: Non-enzymes and protein–protein interactions. Chem. Biol. Drug Des. 81, 22–32 (2013).
Smith, R. D. et al. Exploring protein-ligand recognition with Binding MOAD. J. Mol. Graph Model 24, 414–425 (2006).
Jones, S. & Thornton, J. M. Principles of protein–protein interactions. Proc. Natl Acad. Sci. USA 93, 13–20 (1996).
Lo Conte, L., Chothia, C. & Janin, J. The atomic structure of protein–protein recognition sites. J. Mol. Biol. 285, 2177–2198 (1999).
Hwang, H., Vreven, T., Janin, J. & Weng, Z. Protein–protein docking benchmark version 4.0. Proteins 78, 3111–3114 (2010).
Guo, W., Wisniewski, J. A. & Ji, H. Hot spot-based design of small-molecule inhibitors for protein–protein interactions. Bioorg. Med. Chem. Lett. 24, 2546–2554 (2014).
Wells, J. A. & McClendon, C. L. Reaching for high-hanging fruit in drug discovery at protein–protein interfaces. Nature 450, 1001–1009 (2007).
Arkin, M. R., Tang, Y. & Wells, J. A. Small-molecule inhibitors of protein–protein interactions: Progressing toward the reality. Chem. Biol. 21, 1102–1114 (2014).
Linhares, B. M., Grembecka, J. & Cierpicki, T. Targeting epigenetic protein–protein interactions with small-molecule inhibitors. Future Med. Chem. 12, 1305–1326 (2020).
Volkamer, A. & Rarey, M. Exploiting structural information for drug-target assessment. Future Med. Chem. 6, 319–331 (2014).
Hajduk, P. J., Huth, J. R. & Tse, C. Predicting protein druggability. Drug Discov. Today 10, 1675–1682 (2005).
Nisius, B., Sha, F. & Gohlke, H. Structure-based computational analysis of protein binding sites for function and druggability prediction. J. Biotechnol. 159, 123–134 (2012).
Fauman, E. B., Rai, B. K. & Huang, E. S. Structure-based druggability assessment–identifying suitable targets for small molecule therapeutics. Curr. Opin. Chem. Biol. 15, 463–468 (2011).
Fuller, J. C., Burgoyne, N. J. & Jackson, R. M. Predicting druggable binding sites at the protein–protein interface. Drug Disco. Today 14, 155–161 (2009).
Vukovic, S. & Huggins, D. J. Quantitative metrics for drug-target ligandability. Drug Discov. Today 23, 1258–1266 (2018).
Kozakov, D. et al. The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat. Protoc. 10, 733–755 (2015).
Egbert, M., Jones, G., Collins, M. R., Kozakov, D. & Vajda, S. FTMove: A web server for detection and analysis of cryptic and allosteric binding sites by mapping multiple protein structures. J. Mol. Biol. 434, 167587 (2022).
Beglov, D. et al. Exploring the structural origins of cryptic sites on proteins. Proc. Natl Acad. Sci. USA 115, E3416–E3425 (2018).
Evans, D. J. et al. Finding druggable sites in proteins using TACTICS. J. Chem. Inf. Model 61, 2897–2910 (2021).
Hajduk, P. J., Huth, J. R. & Fesik, S. W. Druggability indices for protein targets derived from NMR-based screening data. J. Med. Chem. 48, 2518–2525 (2005).
Valkov, E., Sharpe, T., Marsh, M., Greive, S. & Hyvonen, M. Targeting protein–protein interactions and fragment-based drug discovery. Top. Curr. Chem. 317, 145–179 (2012).
Edfeldt, F. N., Folmer, R. H. & Breeze, A. L. Fragment screening to predict druggability (ligandability) and lead discovery success. Drug Discov. Today 16, 284–287 (2011).
Perez-Torrado, R., Yamada, D. & Defossez, P. A. Born to bind: The BTB protein–protein interaction domain. Bioessays 28, 1194–1202 (2006).
Chaharbakhshi, E. & Jemc, J. C. Broad-complex, tramtrack, and bric-a-brac (BTB) proteins: Critical regulators of development. Genesis 54, 505–518 (2016).
Parekh, S., Prive, G. & Melnick, A. Therapeutic targeting of the BCL6 oncogene for diffuse large B-cell lymphomas. Leuk. Lymphoma 49, 874–882 (2008).
Hatzi, K. & Melnick, A. Breaking bad in the germinal center: How deregulation of BCL6 contributes to lymphomagenesis. Trends Mol. Med. 20, 343–352 (2014).
Ahmad, K. F. et al. Mechanism of SMRT corepressor recruitment by the BCL6 BTB domain. Mol. Cell 12, 1551–1564 (2003).
Ghetu, A. F. et al. Structure of a BCOR corepressor peptide in complex with the BCL6 BTB domain dimer. Mol. Cell 29, 384–391 (2008).
Cardenas, M. G. et al. The expanding role of the BCL6 oncoprotein as a cancer therapeutic target. Clin. Cancer Res. 23, 885–893 (2017).
Leeman-Neill, R. J. & Bhagat, G. BCL6 as a therapeutic target for lymphoma. Expert Opin. Ther. Targets 22, 143–152 (2018).
Cheng, H. et al. Identification of thiourea-based inhibitors of the B-cell lymphoma 6 BTB domain via NMR-based fragment screening and computer-aided drug design. J. Med. Chem. 61, 7573–7588 (2018).
McCoull, W. et al. Discovery of pyrazolo[1,5-a]pyrimidine B-cell lymphoma 6 (BCL6) binders and optimization to high affinity macrocyclic inhibitors. J. Med. Chem. 60, 4386–4402 (2017).
Yasui, T. et al. Discovery of a novel B-cell lymphoma 6 (BCL6)-corepressor interaction inhibitor by utilizing structure-based drug design. Bioorg. Med. Chem. 25, 4876–4886 (2017).
Sameshima, T. et al. Discovery of an irreversible and cell-active BCL6 inhibitor selectively targeting Cys53 located at the protein–protein interaction interface. Biochemistry 57, 1369–1379 (2018).
Kerres, N. et al. Chemically induced degradation of the oncogenic transcription factor BCL6. Cell Rep. 20, 2860–2875 (2017).
McCoull, W. et al. Development of a novel B-cell lymphoma 6 (BCL6) PROTAC to provide insight into small molecule targeting of BCL6. ACS Chem. Biol. 13, 3131–3141 (2018).
Prokhortchouk, A. et al. The p120 catenin partner Kaiso is a DNA methylation-dependent transcriptional repressor. Genes Dev. 15, 1613–1618 (2001).
Bassey-Archibong, B. I. et al. Kaiso depletion attenuates the growth and survival of triple negative breast cancer cells. Cell Death Dis. 8, e2689 (2017).
Lunardi, A., Guarnerio, J., Wang, G., Maeda, T. & Pandolfi, P. P. Role of LRF/Pokemon in lineage fate decisions. Blood 121, 2845–2853 (2013).
Sakurai, N. et al. The LRF transcription factor regulates mature B cell development and the germinal center response in mice. J. Clin. Invest. 121, 2583–2598 (2011).
Aggarwal, A. et al. Expression of leukemia/lymphoma-related factor (LRF/POKEMON) in human breast carcinoma and other cancers. Exp. Mol. Pathol. 89, 140–148 (2010).
Aggarwal, H., Aggarwal, A. & Agrawal, D. K. Epidermal growth factor increases LRF/Pokemon expression in human prostate cancer cells. Exp. Mol. Pathol. 91, 496–501 (2011).
Aggarwal, H. et al. Expression of leukemia/lymphoma related factor (LRF/Pokemon) in human benign prostate hyperplasia and prostate cancer. Exp. Mol. Pathol. 90, 226–230 (2011).
Moroy, T., Saba, I. & Kosan, C. The role of the transcription factor Miz-1 in lymphocyte development and lymphomagenesis-Binding Myc makes the difference. Semin. Immunol. 23, 379–387 (2011).
Adhikary, S. et al. The ubiquitin ligase HectH9 regulates transcriptional activation by Myc and is essential for tumor cell proliferation. Cell 123, 409–421 (2005).
Orth, B. et al. Identification of an atypical interaction site in the BTB domain of the MYC-interacting zinc-finger protein 1. Structure 29, 1230–1240 e5 (2021).
Vo, B. T. et al. The interaction of Myc with Miz1 defines medulloblastoma subgroup identity. Cancer Cell 29, 5–16 (2016).
Lamoree, B. & Hubbard, R. E. Current perspectives in fragment-based lead discovery (FBLD). Essays Biochem. 61, 453–464 (2017).
Stogios, P. J., Downs, G. S., Jauhal, J. J., Nandra, S. K. & Prive, G. G. Sequence and structural analysis of BTB domain proteins. Genome Biol. 6, R82 (2005).
Morin, S. & S, M. G. Simple tests for the validation of multiple field spin relaxation data. J. Biomol. NMR 45, 361–372 (2009).
Jaremko, L., Jaremko, M., Nowakowski, M. & Ejchart, A. The quest for simplicity: Remarks on the free-approach models. J. Phys. Chem. B 119, 11978–11987 (2015).
Lin, L. Y. et al. Backbone resonance assignment of the BCL6-BTB/POZ domain. Biomol. NMR Assign. 12, 47–50 (2018).
Cerchietti, L. C. et al. A small-molecule inhibitor of BCL6 kills DLBCL cells in vitro and in vivo. Cancer Cell 17, 400–411 (2010).
Guvench, O. & MacKerell, A. D. Jr Computational fragment-based binding site identification by ligand competitive saturation. PLoS Comput. Biol. 5, e1000435 (2009).
Peter, S. et al. Tumor cell-specific inhibition of MYC function using small molecule inhibitors of the HUWE1 ubiquitin ligase. EMBO Mol. Med. 6, 1525–1541 (2014).
Salzmann, M., Pervushin, K., Wider, G., Senn, H. & Wuthrich, K. TROSY in triple-resonance experiments: New perspectives for sequential NMR assignment of large proteins. Proc. Natl Acad. Sci. USA 95, 13585–13590 (1998).
Frueh, D. P. Practical aspects of NMR signal assignment in larger and challenging proteins. Prog. Nucl. Magn. Reson Spectrosc. 78, 47–75 (2014).
Shen, Y. & Bax, A. Protein structural information derived from NMR chemical shift with the neural network program TALOS-N. Methods Mol. Biol. 1260, 17–32 (2015).
Lakomek, N. A., Ying, J. & Bax, A. Measurement of (1)(5)N relaxation rates in perdeuterated proteins by TROSY-based methods. J. Biomol. NMR 53, 209–221 (2012).
Jaremko, M. et al. High-resolution NMR determination of the dynamic structure of membrane proteins. Angew. Chem. Int. Ed. Engl. 55, 10518–10521 (2016).
Kharchenko, V., Nowakowski, M., Jaremko, M., Ejchart, A. & Jaremko, L. Dynamic (15)N{(1)H} NOE measurements: A tool for studying protein dynamics. J. Biomol. NMR 74, 707–716 (2020).
Jaremko, L., Jaremko, M., Ejchart, A. & Nowakowski, M. Fast evaluation of protein dynamics from deficient (15)N relaxation data. J. Biomol. NMR 70, 219–228 (2018).
Acknowledgements
This publication is based upon work supported by the King Abdullah University of Science and Technology (KAUST) Office of Sponsored Research (OSR) under Award No. OSR-CRG2018-3792 and Award No. OSR-CRG2019-4088 (L.J.). This work was funded by the National Institute of Health (NIH) R01 grant CA207272 (T.C.) and T.C. is a Rogel Scholar. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817).
Author information
Authors and Affiliations
Contributions
L.J. and T.C. were responsible for initiating and supervising the project and writing the manuscript. V.K., B.L., M.B., and I.Cz. prepared the proteins and performed biophysical characterization. V.K., M.J., J.G., L.J., and T.C. performed the structural biology, the NMR studies and dynamics studies. All authors contributed to data analysis and writing of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
T.C. and J.G. received prior research support from Kura Oncology Inc. for an unrelated project, served as consultants for Kura Oncology, and have equity ownership in the company. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Gilbert Privé, Ben Davis and Fabien Ferrage for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kharchenko, V., Linhares, B.M., Borregard, M. et al. Increased slow dynamics defines ligandability of BTB domains. Nat Commun 13, 6989 (2022). https://doi.org/10.1038/s41467-022-34599-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-34599-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
