
Abstract
Treating hazardous waste Ni from the electroplating industry is mandated world-wide, is exceptionally expensive, and carries a very high CO2 footprint. Rather than regarding Ni as a disposable waste, the chemicals and petrochemicals industries could instead consider it a huge resource. In the work described herein, we present a strategy for upcycling waste Ni from electroplating wastewater into a photothermal catalyst for converting CO2 to CO. Specifically, magnetic nanoparticles encapsulated in amine functionalized porous SiO2, is demonstrated to efficiently scavenge Ni from electroplating wastewater for utilization in photothermal CO2 catalysis. The core-shell catalyst architecture produces CO at a rate of 1.9 mol·gNi−1·h−1 (44.1 mmol·gcat−1·h−1), a selectivity close to 100%, and notable long-term stability. This strategy of upcycling metal waste into functional, catalytic materials offers a multi-pronged approach for clean and renewable energy technologies.
Similar content being viewed by others

Introduction
The annual discharge of heavy metal wastewater produced by the electroplating industry has exceeded 4 billion tons just in China1. Ni electroplating is particularly widely practiced and allowed to enter natural water systems, causing serious lung, kidney, stomach, intestine, and skin diseases in humans2. While Ni is less costly than precious metals, it has more health and safety regulatory standards by authorities like the EPA3,4,5. The discharged Ni has been commonly regarded as waste or hazard that needs to be treated via remediation methods, like chemical precipitation and electrodeposition6,7. They require preconcentration, post-treatment of sludge, and electricity input, which are often accompanied by high energy costs and a large CO2 footprint8,9. In the meantime, other industries like chemicals and petrochemicals, as well as electroplating itself that requires an excessive amount of Ni are stimulating mining of more Ni from minerals and paying the added price of reagent-grade Ni chemicals, leaving the waste Ni from electroplating buried underground. This bleak situation is worrisome in the climate-challenged world, where the greenhouse effect is looming, and the transition towards sustainable energy and a green economy is of great urgency10,11,12,13,14,15,16. Fortunately, emerging wastewater techniques, including adsorption5,17,18,19 and ion exchange20, many coupled with magnetic separation21,22,23,24, provide energy-saving solutions to scavenge and recycle Ni from the wastewater, but the big question is: what happens next to the adsorbents and exchangers? Landfilling them causes further environmental and health issues, whereas regenerating them and retrieving Ni costs more reagents and energy.
Richard Buckminster-Fuller in Life Magazine said, “Pollution is nothing but resources we’re not harvesting”. Today we are not unfamiliar with the concept of “recycling” waste, but industries’ bottom lines often demand that the resulting product of recycling must be better or even higher value than the original item. This paradigm of so-called “upcycling” has become a keyword in the new world of technologies being eco-friendly25,26. In this context, an economically and environmentally viable strategy should be to directly transform the scavenged Ni from electroplating wastewater to a valuable product that avoids energy- and CO2-intensive mining and rather stores energy in chemicals and fuels made from CO2. Although nickel has been widely accepted as an excellent catalyst for CO2 reduction27,28,29,30, this vision is challenging because electroplating wastewater has a complex composition containing Ni salts with multiple anions, including sulfate, chloride, and amino-sulphonate, which may hinder the recovery of pure Ni chemical feedstocks, and even if an adsorbent is used to retrieve Ni regardless of the anion mix, the adsorbing components can be redundant or even detrimental to the new product. For example, Mikhail et al.’s work suggests that the presence of Na and K impurities in Ni/CeZrOx catalysts resulted in decreased CO2 conversions, lower selectivity to CH4, and increased power consumption in DBD plasma-catalytic CO2 methanation31. In contrast, the impurities can also function as promoters rather than inhibitors. In many cases, different from Maria’s case, the introduction of alkali metals (e.g., Na and K) could instead improve the catalytic performance32,33,34. Nevertheless, the existence of impurities in wastewater still brings complications and uncertainties, so the judicious design of the upcycling process, materials, and catalysts are critical and must be validated.
Targeting this challenge, we demonstrate a two-fold direct recycling and upcycling strategy to scavenge the majority of the Ni from zero-cost and abundant electroplating wastewater and utilize it as a heterogeneous photothermal catalyst for converting CO2 to chemicals and fuels. A carefully designed adsorbent comprised of a sphere-shaped, multifunctional ternary heterostructure Fe3O4@SiO2@mSiO2-NH2 was utilized to capture high dispersions of Ni in real electroplating tailings, employing the chemical tethering property of –NH2 enhanced by the large surface area of mesopores in SiO2. The ternary nanocomposite Fe3O4@SiO2@mSiO2-NH2@Ni is comprised of earth-abundant elements. It displays a remarkable 100% selectivity and high stability towards the reverse water-gas shift reaction CO2 + H2 → CO + H2O, qualities not enjoyed by traditional Ni-sourced heterogeneous catalysts. The Fe core provides additional benefits that include straightforward magnetic separation of the Fe3O4@SiO2@mSiO2-NH2@Ni catalyst from the electroplating wastewater, a light-activated broadband nano heater to drive the photothermal reverse water-gas shift reaction at higher efficiency, milder conditions, and a lower CO2 footprint than the thermochemical reverse water-gas shift reaction.
Results
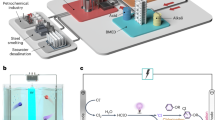
Our strategy to upcycle Ni from real electroplating wastewater can be divided into several steps (Fig. 1). First, a designed adsorbent is suspended in real Ni(II)-containing electroplating wastewater (step 1). After reaching adsorption equilibrium, the adsorbed Ni(II), together with the adsorbent, is collected via a magnet (step 2) and then directly calcined to be transformed into an active catalyst with a nanoreactor-type architecture (step 3). The obtained catalyst is then used in (photothermal) catalytic CO2 hydrogenation reactions (step 4). The architecture of the nanoreactor catalyst was carefully designed to realize the following functionalities: the Fe core of our nanoreactor supplies sustainable photothermal heat, and the Ni within the silicon oxide synthesizes solar fuel.

The Ni resources in the electroplating wastewater were upcycled for photothermal CO2 catalysis applications.
Characterization of the catalysts
The adsorbent used in this work was designed to have a core-mantle-crust structure, with a magnetic core (Fe3O4 colloidal nanocrystal clusters, denoted as CNC) and two layers of silica (Supplementary Fig. 1), employing the first and second most abundant element in the Earth’s crust: O and Si. The strong magnetism of the CNC core ensured easy separation of adsorbents from solution35,36,37. The dense silica in the middle mantle layer prevented the CNC core from being etched in the acidic wastewater. The outmost crust of mesoporous silica served to enlarge the specific surface area (SSA) to benefit adsorption and further catalytic reaction. Besides, the adsorbent particles (denoted as CNC@SiO2@mSiO2) were also grafted with amino groups (denoted as Sad) to facilitate the capture of Ni(II). The Brunauer–Emmett–Teller (BET) surface areas of the amino-grafted adsorbents with and without the mesoporous silica layer were calculated to be 301.7 and 16.3 m2 g−1, respectively, demonstrating the function of mesoporous silica in increasing SSA (Supplementary Fig. 2). A sample prepared by coating CNC with only a mesoporous SiO2 layer (without the middle mantle protecting layer, denoted as CNC@mSiO2) was used as a control sample to verify the acid resistance of the dense mantle SiO2 layer in CNC@SiO2@mSiO2 (Supplementary Fig. 3). The similar particle size distributions of CNC@SiO2@mSiO2 and CNC@mSiO2 suggest that the thickness of SiO2 for these two samples are the same, making the comparison fair. The concentration of Fe in the etching solution for CNC@SiO2@mSiO2 is much lower than that for CNC@mSiO2 under various etching conditions. These results indicate that the dense silica in the middle layer of CNC@SiO2@mSiO2 has strong resistance against acid.
After the treatment of the electroplating wastewater, the recycled Ni2+ together with the adsorbents was directly calcined in air and H2 sequentially (denoted as SFe-Ni), which completed the assembly of the nanoreactor catalyst. Figure 2 shows the morphology and composition of the obtained catalyst. The overall core-shell structure was retained, while the CNC core was cracked (Fig. 2b and Supplementary Fig. 1). Notably, no apparent Ni nanoparticles could be observed, suggesting that Ni should be highly dispersed in SFe-Ni. This was further corroborated by the X-ray diffraction (XRD) results (Supplementary Fig. 4). The patterns for SFe-Ni showed no characteristic peaks of Ni crystals, excluding the generation of large particles and aggregates. Whereas, the characteristic peaks of Fe were clearly observed in the patterns, suggesting that the CNC core had been reduced in the calcination process. The high dispersion of Ni in SFe-Ni was ultimately confirmed by the aberration-corrected high-angle annular darkfield-scanning transmission electron microscopy (HAADF-STEM) images (Fig. 2c), in which no nanoparticles but only bright dots could be observed. Energy Dispersive Spectrometer (EDS) mappings further demonstrated the core-mantle-crust structure (CNC@SiO2@mSiO2) in which Ni was successfully loaded (Fig. 2d–h).

a Schematic of SFe-Ni. The black core, the dark gray middle layer, the light gray porous outer layer and the red spheres represent CNC, the dense silica, the mesoporous silica, and Ni, respectively. b TEM image of SFe-Ni. Scale bar, 50 nm. c HAADF-STEM image of SFe-Ni. Scale bar, 2 nm. d–h EDS mappings of SFe-Ni. Scale bars, 50 nm.
To identify the chemical environment in the sub-nano dimension and the electronic structure of Ni in SFe-Ni, X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectra at the Ni K-edge for SFe-Ni were obtained (Fig. 3 and Supplementary Table 1). In the Ni K-edge XANES spectrum, the near-edge absorption energy of SFe-Ni is located between the Ni foil and NiO, indicating the partially oxidized state of Ni in SFe-Ni. The FT-EXAFS curve for SFe-Ni shows a main peak at ~1.6 Å, which could be attributed to Ni–O scattering38. A very small peak attributed to the Ni–Ni scattering signal (located at ~2.1 Å) for SFe-Ni demonstrates the existence of aggregated Ni is rare38. Fitting the peak of the first shell of Ni–O shows the average coordination number (CN) of oxygen atoms around a Ni atom (CN (Ni–O)) in SFe-Ni is 3.8, indicating that Ni is unconventionally coordinated in comparison to NiO, which has a CN (Ni–O) of 639. The decreased CN (Ni–O) of SFe-Ni should be ascribed to the highly dispersed adsorbed state of Ni on the surface of SiO2 without NiO nanoparticles. The second coordinative shell can be assigned to Ni–Ni coordination, of which the CN (Ni–Ni) is 4.8. The much lower CN (Ni–Ni) of SFe-Ni than that of Ni foil (CN (Ni–Ni) = 12)39, combined with the aberration-corrected HAADF-STEM results, suggests that our catalyst preparation strategy ensured that Ni existed as highly dispersed atoms and ultrasmall clusters, rather than aggregated nanoparticles.

a Ni K-edge XANES spectra of the fresh and used SFe-Ni catalyst, exhibiting the normalized absorption intensity as a function of the X-ray photon energy. The used catalyst is denoted as SFe-Ni (after). Ni foil and NiO were used as references. b Ni K-edge EXAFS fitting results of the fresh and used SFe-Ni catalyst. The black open circles show the original data of the EXAFS spectra, while the lines of other colors are the fitting results of the spectra.
As reported, small-sized Ni catalysts exhibit excellent catalytic performance in CO2 hydrogenation40. Therefore, the unique structure of SFe-Ni should render it a potential candidate for this reaction. Meanwhile, the Fe core, as an active metal and a great photothermal material, could offer additional activity to the system. Therefore, we conducted systematic catalytic tests to evaluate the catalytic performance of this designed catalyst and identify the roles of the key components, under both thermal and photothermal conditions. For comparison, two primary control samples excluding either Fe or Ni were investigated to manifest the rationality of our catalyst design, under the same testing conditions: a control sample prepared by annealing the bare Sad without Ni under the same condition as SFe-Ni (denoted as SFe, Supplementary Fig. 5), and another control sample substituting the CNC core with a SiO2 core (denoted as SNi) by treating the SiO2@mSiO2 nanoparticles with the same amino functionalization, adsorption, and annealing procedure of SFe-Ni (Supplementary Fig. 6). SFe exhibited the same morphology of SFe-Ni, while no Ni could be found from the EDX element maps. Meanwhile, a solid silica core was found for SNi with Ni uniformly dispersed in the outer silica layer, and no Fe was observed in the EDX element maps.
Thermocatalytic performance
Before pursuing the sustainable potential in light-driven CO2 reduction, we first evaluated the traditional thermocatalytic performances of the samples to provide fundamental insights into the catalytic sites, product selectivity, and thermal stability of our catalyst. It was found that the CO production rates of SFe-Ni were much higher than those of SFe at all testing temperatures (Supplementary Fig. 7a). Below 400 °C, the control sample SFe hardly even showed observable activity. This indicates that the loading of Ni led to pronounced improvement in catalytic performance for Sad. Notably, the CO rate of SNi was close to that of SFe-Ni. This further demonstrates that the catalytic ability of our designed architecture under pure thermal conditions mainly comes from Ni rather than the Fe core. Only at the relatively high temperatures of 450 and 500 °C, when the diffusion of reactants was greatly enhanced, the Fe component could start to contribute to the yield of CO, indicated by the increasingly observable rate of SFe and the higher rate of SFe-Ni than SNi at these temperatures. Thanks to the sub-nano size of Ni–O species, only a trace amount of CH4 was found in the products (Supplementary Fig. 8), consistent with the literature that ultrasmall Ni if oxidized, would favor the production of CO from hydrogenation of CO238,40,41. Nearly 100% of CO selectivity (99.998%) was thus achieved for SFe-Ni at 500 °C, which is in contrast with traditional supported Ni nanoparticles that often yield a significant proportion of CH4 and would necessitate an energy-intensive separation process for the products. For example, the selectivity of CH4 was found to be 58.6% for commercially available silica-alumina supported Ni nanoparticles (Ni/SiO2·Al2O3) tested in our reactor (Supplementary Table 2).
To examine whether the mesopores contribute to the small size of Ni and, in turn, the great selectivity, another control sample was prepared by using solid SiO2 spheres as the support to graft amino groups, followed by the adsorption and activation of Ni similar to other samples (denoted as SSiO2-Ni). As Supplementary Fig. 9a-b shows, no large Ni particles, and no obvious crystalline Ni signals could be observed from the TEM and XRD results, respectively. Moreover, the thermocatalytic results suggest that CO was the only product for SSiO2-Ni, which is characteristic of small-sized Ni (Supplementary Fig. 9c). The BET surface area of amino-grafted solid SiO2 (SSiO2-NH2) is much lower than that of CNC@SiO2@mSiO2-NH2 (15.9 m2/g vs. 301.7 m2/g, Supplementary Fig. 9d). This leads to the much poorer catalytic activity of SSiO2-Ni, only half of that of SFe-Ni. These results indicate that the mesoporous SiO2 only act as effective support with a high specific surface area.
As Supplementary Table 3 shows, many other ions besides Ni2+ exist in the electroplating wastewater. To verify the influence of these ions on the catalytic performance, a synthetic Ni2+ solution made from reagent Ni(NO3)2·6H2O with only Ni2+ (the Ni2+ concentration is the same as that for the real electroplating wastewater) was used as the precursor to prepare the SFe-Ni sample. Notably, Supplementary Fig. 10 shows that the CO rate for SFe-Ni prepared from real electroplating wastewater is the same as that for SFe-Ni prepared from the synthetic Ni2+ solution at 400 °C, while only a slightly lower CO rate was found for the former than for the latter at 500 °C. Nevertheless, the overall discrepancy between these two SFe-Ni samples is minimal. This should be attributed to the similar loading amount of Ni for these two samples and the trace amount of other ions therein (Supplementary Tables 4 and 5). These results suggest that the existence of the other ions besides Ni2+ in the real electroplating wastewater does not deteriorate the upcycling efficiency of Ni for the CO2 catalysis reported herein. Overall, the above results preliminarily manifest that our designed adsorbent could successfully recycle Ni from the electroplating wastewater into a highly active and selective catalyst for CO2 hydrogenation reactions.
The stability of SFe-Ni was evaluated through testing at the relatively high temperature of 500 °C, shown in Supplementary Fig. 7b. After a slight decrease, the CO rate began to increase slowly and tended to be stabilized above 5 mmol gcat−1 h−1. The mild decrease of yield in the initial stage should arise from the oxidation of Ni, demonstrated by the XAS results (Fig. 3 and Supplementary Table 1). The used catalyst, denoted as SFe-Ni (after), showed a very similar Ni K-edge XANES spectrum with SFe-Ni, but the pattern became slightly closer to that of NiO (Fig. 3a). In the Ni K-edge EXAFS spectrum, a small Ni–O-Ni hump (located at ~2.6 Å) emerged while the small peak for Ni–Ni disappeared41. This indicates that the Ni clusters had been oxidized after the reaction. Meanwhile, the intensity of the Ni–O with low CN was maintained after the stability test, which is in line with the preserved high catalytic activity and selectivity towards CO production. Notably, more Ni atoms and clusters without serious aggregation could be seen in the used catalyst (Supplementary Fig. 11c) than in the fresh samples (Fig. 2c). The high dispersion of Ni in the used samples could also be evidenced by the XRD patterns which show no characteristic signals of crystalline Ni (Supplementary Fig. 12). The migration of Ni from the inner part of the mesopores to the more outer part was beneficial for the mass transfer of the reactants, leading to the gradual increase of production rate seen in Supplementary Fig 7b.
The catalytic pathway was studied through the in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) experiments and kinetic studies (Supplementary Figs. 13–15). The in situ DRIFTS spectra of SFe-Ni (Supplementary Fig. 13a) show weak signals of gaseous CO (2111 and 2174 cm−1)28,42, coinciding well with the products of the reverse water-gas shift reaction. No obvious signals of gaseous CH4 (3016 cm−1) or other intermediate products were observed. The absence of peaks between 2800 and 2900 cm−1 demonstrates the non-existence of formate species28. Millet et al. also reported that only a weak formate signal could be observed for small-sized Ni, which favored the production of CO, whereas, for aggregated Ni nanoparticles that can produce a significant amount of CH4, formate seemed a key intermediate40. Therefore, the absence of formate and selective production of CO corresponds well with our small-sized Ni. The lack of formate species and bridged CO (1800–2000 cm−1) impeded the further hydrogenation into CH428. This again demonstrates the high selectivity of CO for the SFe-Ni catalyst. The reverse water-gas shift (RWGS) reaction commonly follows a direct dissociation route (CO2 → *CO2 → *CO + *O) or a formate route28,43,44,45. Since the signal of *HCOO− is negligible, the RWGS reaction on the SFe-Ni catalyst is more likely to follow a dissociation route. The overall weak signals in the in situ DRIFTS spectra for SFe-Ni are ascribed to its excellent absorption in the infrared region (Supplementary Fig. 13b), but the relatively poor signal-to-noise ratio might cause difficulty for interpretation. Therefore, additionally, we obtained in situ DRIFTS spectra for the SSiO2-Ni sample, which also has the characteristics of small-sized Ni but with more intense reflectance signals in the infrared light region (Supplementary Fig. 13b). It is a perfect substitute to verify the reaction route in this system. The in situ DRIFTS spectra of SSiO2-Ni show distinct signals of gaseous CO and still no other obvious intermediates (Supplementary Fig. 13c). This again suggests that the high possibility of a direct dissociation route for small-sized Ni in our catalysts as depicted above. Moreover, another control sample was prepared by impregnating Ni(NO3)2 • 6H2O on solid SiO2 spheres (Niim/SiO2) to show the difference between the small-sized Ni and Ni nanoparticles. The characteristic signals of crystal Ni in the XRD pattern (Supplementary Fig. 13d) confirm the nanoparticle state of Ni in Niim/SiO2, which showed low selectivity towards CO (only 55.8%, Supplementary Table 2). The in situ DRIFTS spectra of Niim/SiO2 show obviously characteristic peaks of gaseous CH4 and linearly absorbed CO (2030 cm−1)46, which are not seen in the samples with small-sized Ni (Supplementary Fig. 13e). Therefore, the absence of intermediates verified from the in situ DRIFTS spectra of the samples with small-sized Ni can explain their much higher selectivity towards CO production, and accordingly insignificant production of CH4. As to the basic kinetic studies, the results suggest that the CO production rate over the SFe-Ni catalyst exhibits higher dependence on CO2 (0.8) than H2 (0.08), as shown by Supplementary Figs. 14 and 15, respectively. This indicates that the Ni surface is mostly covered by hydrogen instead of carbon species, which is in good agreement with the in situ DRIFTS results. When the pressure of CO2 increases, the dissociation of CO2 becomes easier, leading to promoted CO production.
Photothermal catalytic performance
It was found that SFe-Ni and SFe appeared to be black while the color of SNi is pale (Supplementary Fig. 16). The diffuse reflectance spectra of these samples show that SFe-Ni and SFe have similar reflectance, which is less than 15%, spanning the ultraviolet, visible-light, and part of the near-infrared region (Fig. 4a). Even in the further end with the longer wavelength, the reflectance of these two samples is not beyond 25%. In contrast, SNi exhibits much higher reflectance throughout the full solar spectrum from 200 to 2500 nm. These results manifest that the Fe core plays an important role in improving the light-harvesting ability of SFe-Ni. The strong light absorption of SFe-Ni should render it an excellent photothermal material, which allows the CO2 hydrogenation reaction to be more sustainable with the aid of light28,47,48,49,50,51,52,53,54,55.

a Diffuse reflectance spectra of different catalysts. b CO production rates of different samples illuminated with a Xe arc lamp supplemented with a light concentrator in the flow reactor. The power of the lamp was set to be 190, 200, and 210 W, corresponding to the light intensities of 28.3, 30.7, and 33.3 suns, respectively. Three data points were collected to obtain the average CO rate after 10 min illumination for each sample. c Surface temperature profiles of different samples illuminated with concentrated light from a Xe arc lamp. The power of the lamp was set to be 210 W. The equilibrium temperatures under illumination for SFe-Ni, SFe, SNi, and without catalyst were 401.8, 389.6, 295.3, and 180.8 °C, respectively. d CO production rates of different samples in the batch reactor illuminated with a Xe arc lamp supplemented with a light concentrator. The light intensity was 28 suns. The CO rates with the unit of ‘mmol·gNi−1·h−1’ for SFe-Ni in b, d were calculated based on the mass of Ni, and the activity of the isolated Fe component was deducted by subtracting the CO rate of SFe under the same testing condition. The red data points in b, d correspond to the selectivity towards production of CO.
The photothermal catalytic performances of the samples were first determined under illumination with a Xe arc lamp and without any heat supplied by the heating unit of the bulk reactor. Surprisingly, differently in the thermal test results, the yield of CO was only observed for SFe-Ni while the other two samples had no catalytic activity (Supplementary Fig. 17). With the further assistance of a light concentrator, the production of CO could finally be detected for SFe and SNi (Fig. 4b). However, their CO yields were still very low, in contrast to the pronounced rates for SFe-Ni under the same testing conditions. Typically, CO rates of 9.73, 3.59, and 1.35 mmol·gcat−1·h−1 were obtained for SFe-Ni, SFe, and SNi, respectively, under concentrated 210-W illumination. Meanwhile, the production of CH4 was still negligible in photothermal catalytic reactions as well (Supplementary Fig. 18), ensuring ultrahigh selectivity towards CO (>99.99%). These results indicate that both the Fe core and Ni sites are indispensable components in the nanoreactor for achieving considerable CO rates in photothermal catalytic CO2 hydrogenation. Compared with SNi that is devoid of Fe, the CO production rate of SFe-Ni was significantly boosted, around 7 times, which can be ascribed to the excellent light-harvesting ability and thereby high photo-to-thermal conversion efficiency of the Fe core. These are manifested by the fact that the equilibrium temperature of SFe-Ni could be achieved at the high value of 402 °C under the illumination of concentrated light (Fig. 4c). The surface temperature of SFe is close to that of SFe-Ni, which is far beyond 295 °C, the equilibrium temperature of SNi. This trend was also found under other illumination conditions (Supplementary Fig. 19). Therefore, the observed high rate is largely attributed to the heat provided by the Fe core under such light-only conditions. In comparison, SFe, which is with the Fe core but without Ni sites, could reach a similar high temperature under light but did not show a high reaction rate. This echoes with the thermocatalytic result that Ni was the major active site for the hydrogenation of CO2. Notably, SFe-Ni could achieve similar CO rates of ~5 mmol·g−1·h−1 in either the thermocatalytic process (500 °C) or the photothermal catalytic (190 W illumination assisted with a concentrator) process. This indicates the real local temperature of SFe-Ni under this illumination was much higher than the apparent surface temperature of 361 °C measured by the thermocouple, further demonstrating the excellent photothermal conversion property of SFe-Ni. The increased local temperature and its catalytic boosting effect induced by SiO2 encapsulation have also recently been discovered and discussed in detail by Cai et al.56,57. The heat insulation and infrared shielding effects of the SiO2 sheath confine the photothermal energy of the Fe core within the catalyst, enabling a supra-photothermal effect. The thermocouple could only reach the surface of the outer-layer mesoporous silica, the temperature of which was lower than that inside the catalyst. The fact that SNi only exhibited nearly 1/7 of the CO production rate of SFe-Ni under the same light illumination condition (33.3 suns) indicates that photochemical contribution if there was any, was trivial. The significant CO rate of SFe-Ni under light was still driven by the thermochemical reaction pathway, supported by the same product selectivity and similar activation energy as shown in thermal catalytic test results, shown in Supplementary Table 6 and Supplementary Fig. 20. Besides, the exponential dependence of the reaction rate on illumination intensity in Fig. 4b rather than a linear dependence, is a characteristic feature of thermally driven transformation58. The excellent photothermal catalytic performance of SFe-Ni was further demonstrated by using commercial Cu-ZnO-Al2O3 as a control sample which shows a lower CO rate than SFe-Ni under similar testing conditions (Supplementary Fig. 21). Another control experiment was also conducted by using only CO2 and N2 as the reactants, under which the production of CO is negligible in comparison to the H2-existing atmosphere (Supplementary Fig. 22). This demonstrates that the production of CO originates from the reverse water-gas shift reaction rather than the Boudouard reaction (C + CO2 → 2CO).
Notably, the photothermal catalytic CO rate for SFe-Ni could be further improved to be 44.1 mmol·g−1·h−1 by using a batch reactor, which is about 9 times the rate in the flow reactor under the same light intensity (Fig. 4b, d). At the same time, the selectivity was maintained to be nearly 100%. This improvement can be attributed to the sealed environment and the catalyst bed geometry that can reduce loss of heat (reduced thermoconvection compared with the flow reactor) and enhance light absorption (larger absorption area due to the better dispersion of catalyst on the glass fiber filter). The photothermal catalytic activity of SFe-Ni was far beyond that of SFe (5.9 mmol·g−1·h−1) in the batch reactor, again demonstrating that Ni is the major active component. This allows the CO production rate per weight of Ni to be calculated, which turned out to be as high as 1.9 mol·gNi−1·h−1. Meanwhile, the activity of the control sample SNi in the batch reactor was also improved compared to the flow reactor result, but only around half of the activity of SFe-Ni, due to the lack of the central Fe heating core. The photothermal activity of our catalyst was also compared with those of various reported photothermal catalysts (Supplementary Table 7). The SFe-Ni samples have higher or at least the same level of production rate compared with the listed catalysts. Overall, the intentionally designed architecture of our nanoreactor catalyst and control tests have pinpointed the functions of the Fe core and the well-distributed Ni sites, and exhibited extraordinarily high catalytic selectivity at a high production rate.
Cycle experiment
The concentration of Ni in the electroplating wastewater was 6576 mg L−1 according to the inductively coupled plasma mass spectrometry (ICP-MS) results. Obviously, the Ni resources in the wastewater were affluent. In this work, the Ni wastewater could be utilized as an abundant source for preparing large amounts and multiple batches of SFe-Ni catalysts. To confirm this concept, a batch of wastewater was treated by Sad repeatedly to determine how many times it could serve for the preparation of SFe-Ni. For each cycle of treatment, the fresh adsorbent was used, and the catalyst thus prepared was named as SFe-Nin, in which n represents the cycle number. Supplementary Fig. 23 shows the concentration of the remaining Ni in the wastewater after numerous typical cycles (abbreviated as CNi-n; n represents the cycle number). After 15 cycles, CNi−15 was detected to be 224 mg L−1, about 3% of the original value (6676 mg L−1). Namely, 97% of the Ni resources in the wastewater were upcycled to prepare 15 batches of the active catalyst SFe-Ni. The loading amounts of Ni onto SFe-Ni after these cycles were determined by ICP-MS (Supplementary Fig. 24). In the initial few cycles, the loading ratios of Ni for different SFe-Ni samples were similar: ~2%. After the 9th cycle, the Ni loading began to decrease, which is ascribed to the lower maximum adsorption capacity under equilibrium when CNi-n drops. The loading ratio of Ni was 1% for SFe-Ni15, which is half of that for SFe-Ni1 (Supplementary Fig. 24 and Supplementary Table 4). The thermocatalytic property for three typical SFe-Nin (n = 3, 9, and 15) samples at 500 °C was evaluated (Supplementary Fig. 25). The CO production rates of SFe-Ni3 and SFe-Ni9 approached that of SFe-Ni, which should be attributed to their similar loading amount of Ni and catalyst structure. In contrast, SFe-Ni15 showed a much lower CO production rate (3.12 mmol gcat−1 h−1), which was expected since its Ni loading was only half that of SFe-Ni. Nevertheless, this rate is still higher than that of SFe, again demonstrating the function of adsorbed Ni as the active sites. Besides, only a trace amount of CH4 could be detected for all these samples, marking good repeatability in achieving high product selectivity via this cycling approach. Overall, 97% of Ni in the electroplating wastewater could be recycled for the preparation of SFe-Ni catalysts through cycling adsorption and calcination, realizing the sustainable upcycling of waste Ni.
Despite the overall high upcycling efficiency through cycle adsorption, the drop of activity for the samples in the later cycles due to the lower loading of Ni might be a problem. We have clarified this in the revised manuscript, as the reviewer suggested. Moreover, we further propose a possible remedial strategy regarding this problem: performing adsorption in fresh wastewater again using the sample in the later cycles to achieve improved Ni loading. As a simple demonstration, we prepared a SFe-Ni sample using diluted Ni wastewater (1 mL of wastewater diluted by 29 mL of milli-Q water) to simulate SFe-Ni in the later cycles. Next, a second adsorption process in the fresh Ni wastewater was operated on this sample. The CO rate of the treated sample achieved was 5.3 mmol gcat−1 h−1 which is similar to that of SFe-Ni prepared using fresh Ni wastewater under the same testing condition, demonstrating the feasibility of the remedial strategy. Nevertheless, the second adsorption also made the upcycling procedure more time-consuming. Simpler and more cost-effective upcycling strategies should be developed in future works.
CO2 footprint and cost evaluation
Carbon neutrality is an urgent goal in the current age59,60,61,62,63,64,65. Many technologies for producing value-added products claim using CO2 as the feedstock can reduce CO2 emission, but this may not be necessarily true, since the power consumed to run the facility, to produce H2, and to drive the chemical reaction may generate an even larger CO2 footprint than the one the technology aims to reduce66. Chen’s group has recently demonstrated that using CO2 to synthesize methanol either via thermocatalysis or electrocatalysis is still generating several mol of new CO2 when one mol of CO2 is consumed under the current US energy structure, far from reaching carbon neutrality67. Inspired by his quantitative study, we recently developed a universal evaluation approach to compare the net CO2 emission rates between thermal and photothermal CO2 hydrogenation using typical small-scale fix-bed reactors68. As shown in Fig. 5, net CO2 emission rates (denoted as dM/dt) for SFe-Ni under the thermocatalytic and photothermal conditions were calculated based on the catalytic results from this work and the criterions built in our previous work. To be specific, the value of M equals the amount of emitted CO2 after subtracting the consumed CO2 in a catalytic process. The derivatives of M in terms of t (dM/dt, representing net CO2 emission rate) for flow reactors are shown as follows:
in which, a, c, m, n, x1, x2, and k2T+b2 represent the power consumption of the lamp (kW), the conversion rate of CO2 (mol·g−1·h−1), the mass of the catalyst (g), the original feed amount of H2 in a cycle (mol), CO2 emission per kWh of electricity according to the US 2014 energy structure (mol), CO2 produced per mol of H2 (mol), and the electricity consumed to maintain the reactor at T per hour (kWh), respectively. The subscript ‘kp’ denotes that the H2 feed is calculated based on the kinetic parameters. Detailed calculation and value assignment are listed in the supplementary notes.

The dependence of net CO2 reduction rate on CO2 emission per kWh of electricity (x1) for flow reactors based on the assumption that H2 is CO2-cost-free. The values of a, c, m, and k2T+b2 were set to be 0.19, 0.005, 0.03, and 0.224, respectively, according to the catalytic results and reactors used in this work. In the inset, the vertical dashed line shows the minimum x1 which can achieve net CO2 reduction while the horizontal dashed line represents the net CO2 emission rates when the production of electricity is CO2-free.
Owing to the high photothermal activity of SFe-Ni, the net CO2 emission rates for the photothermal catalytic process (denoted as dM1kp/dt) were obviously lower than that for the thermocatalytic process (denoted as dM2kp/dt) when electricity was used to power the facilities (either the Xe arc lamp or the heating system), as shown in Fig. 5. For example, when considering the CO2 emission per kWh of electricity according to the reported US 2014 energy structure, the net CO2 emission rates of using 30 mg of the SFe-Ni catalyst was significantly reduced by 15% from 2.473 mol h−1 (500 °C) to 2.097 mol h−1 (190 W illumination supplemented with a light concentrator), while maintaining the same production rate of CO. Despite that these electricity-powered catalytic processes are still generating CO2 rather than reducing the amount of CO2, the values of dM/dt for the photothermal catalytic process could be greatly reduced by increasing the usage of sustainable energy (sunlight, tide, wind, geothermal energy, etc.) to reduce x1 (Fig. 5). When x1 is 0.00079 mol CO2 kWh−1, the photothermal route can veritably achieve carbon neutrality, and at the same time produce fuel.
When it comes to practical utilization, the cost of Ni during catalyst preparation is also very important in addition to the CO2 footprint. The common Ni resources used in the preparation of Ni catalysts include Ni(NO3)2·6H2O, NiCl2·6H2O, and NiSO4·6H2O. The prices of these Ni resources range from 1.7 to 9.84 USD g−1 (Supplementary Table 8). In contrast, the electroplating industries need to pay for the treatment of the Ni wastewater, contributing to a negative cost of Ni (−$0.0019 g−1 to −$0.0028 g−1) from it. This makes the wastewater from electroplating an ideal precursor for the preparation of the SFe-Ni catalysts.
Discussion
The essence of the work presented herein is inspired by the prescience of Buckminster-Fuller, who in a 1971 Life Magazine interview said, “There is no energy crisis, just a crisis of ignorance.” He imagined pollution as a feedstock to be harvested for its value rather than treated as the waste of a consumer society. This is the underpinning of our carefully designed adsorbent for the Ni tailings in electroplating wastewater. They can be given new life in a recycle-upcycle process that takes the Ni from a toxic grave to a sustainable cradle, a kind of inverse life cycle paradigm to create a cost-effective eco-friendly photothermal CO2 catalysis process. We envision many ‘pollution solutions’ that could benefit from the recycle-upcycle concepts and principles of the genre delineated herein, turning plastic, paper, mining, food, and electronic waste into sustainable consumer products.
Methods
Materials and chemicals
All the chemicals were used as received without further purification. Commercial Cu-ZnO-Al2O3 was purchased from Sichuan Shutai Chemical Technology Co., Ltd. Tetraethyl orthosilicate (TEOS, > 96%), N,N-diisopropylethylamine (>99%), and hexadecyl trimethyl ammonium bromide (CTAB, >98%) were purchased from TCI. Ferric chloride hexahydrate (FeCl3•6H2O, reagent grade), nickel nitrate hexahydrate (Ni(NO3)2•6H2O, ≥97%), N1-(3-trimethoxysilylpropyl) diethylenetriamine (≥98%), and PSSMA [poly(4-styrenesulfonic acid-co-maleic acid) sodium salt] were purchased from Sigma–Aldrich. Sodium acetate anhydrous (99%), hydrochloric acid (36%–38%, analytical reagent), ethylene glycol (99.5%), ethanol (GR, ≥99.8%), and ammonium hydroxide solution (NH3•H2O, 28 wt %) were purchased from Energy Chemical, Enox, J&K scientific, Sinopharm Chemical Reagent Co., Ltd, and Macklin, respectively. Milli-Q water (Millipore, 18.2 MΩ cm at 25 °C) was used in all experiments.
Synthesis of Fe3O4 colloidal nanocrystal clusters (CNC)
Fe3O4 CNCs were synthesized according to a reported recipe69,70. For a typical batch, 7.5 g of PSSMA was dissolved in 300 mL of ethylene glycol with magnetic stirring. 8.1 g of FeCl3•6H2O and 22.5 g of sodium acetate were then added to the mixture under continuous stirring until the solution turned homogeneously red-brown. The mixture was then sealed in a Teflon-lined stainless-steel autoclave and heated at 200 °C for 10 h. When cooled to room temperature, the dark precipitates were isolated from the solution by a magnet and washed with Milli-Q water and ethanol alternately six times, and finally dispersed in ethanol to form a suspension (concentration: 10 mg mL−1).
Synthesis of CNC@SiO2@mSiO2-NH2
The CNC nanoparticles were coated with a thin layer of dense silica via a modified Stöber method71,72. Briefly, 20 mL of the CNC suspension was diluted by ethanol (60 mL) and Milli-Q water (12 mL). The mixture was then sonicated for 30 min, followed by the addition of ammonium hydroxide solution (10 mL), and TEOS (500 μL) sequentially. The reaction vessel was then stirred in a shaking bed (400 rpm, 30 °C) for 1 h. The obtained nanoparticles (denoted as CNC@SiO2) were washed twice with ethanol under centrifugation and redispersed in Milli-Q water to form a suspension (concentration: 40 mg mL−1). 2.5 mL of the CNC@SiO2 suspension was added to 20 ml of the prepared CTAB solution (0.9 g of CTAB dissolved in a mixture of 100 mL of ethanol and 300 mL of Milli-Q water). The suspension was sonicated and stirred, each for 20 min, followed by the sequential addition of ammonium hydroxide solution (125 μL), and TEOS (175 μL). The mixture was stirred for another 4 h in a shaking bed (400 rpm, 30 °C). The obtained nanoparticles were then separated and redispersed in acetone and refluxed at 80 °C for 48 h. The refluxing procedure was repeated 3 times. The obtained products (denoted as CNC@SiO2@mSiO2) were then washed with ethanol several times and dried in a vacuum oven. One hundred milligrams of the CNC@SiO2@mSiO2 powder was dispersed in ethanol (40 mL). 0.5 ml of N1-(3-trimethoxysilylpropyl) diethylenetriamine and 0.1 ml of N,N-diisopropylethylamine were then added to the suspension. The mixture was stirred in a shaking bed (400 rpm, 30 °C) for 12 h and then washed with Milli-Q water and ethanol six times alternately. The obtained nanoparticles (denoted as CNC@SiO2@mSiO2-NH2, abbreviated as Sad) were dried under vacuum.
Synthesis of SiO2@mSiO2-NH2
Silica nanospheres with a similar size of CNC@SiO2 were prepared via a modified Stöber method71,72. 3.6 mL of Milli-Q water, 24 mL of ethanol, and 800 μL of ammonium hydroxide solution were mixed, followed by the addition of 265 μL of TEOS. The mixture was stirred in a shaking bed (400 rpm, 30 °C) for 1 h. The resulting nanoparticles were washed twice with ethanol and were finally dispersed in Milli-Q water to form a suspension (concentration: 26 mg mL−1). 2.5 mL of the SiO2 suspension was added to 20 ml of the prepared CTAB solution (0.9 g of CTAB dissolved in a mixture of 100 mL of ethanol and 300 mL of Milli-Q water). The suspension was sonicated and stirred, each for 20 min, followed by the sequential addition of ammonium hydroxide solution (125 μL), and TEOS (175 μL). The mixture was stirred for another 4 h in a shaking bed (400 rpm, 30 °C). The obtained nanoparticles were then separated and redispersed in acetone and refluxed at 80 °C for 48 h. The refluxing procedure was repeated three times. The obtained products (denoted as SiO2@mSiO2) were then washed with ethanol several times and dried in a vacuum oven. 100 mg of the SiO2@mSiO2 powder was dispersed in ethanol (40 mL), followed by the addition of N1-(3-trimethoxysilylpropyl) diethylenetriamine (0.5 ml) and N,N-diisopropylethylamine (0.1 ml). The mixture was stirred in a shaking bed (400 rpm, 30 °C) for 12 h and then washed with Milli-Q water and ethanol 6 times alternately. The obtained nanoparticles (denoted as SiO2@mSiO2-NH2) were dried under vacuum.
Synthesis of amino-grafted solid SiO2 (SSiO2-NH2)
Solid silica nanospheres (SSiO2) were prepared via a modified Stöber method71,72. Milli-Q water (3.6 mL), ethanol (24 mL), and ammonium hydroxide solution (3 mL) were mixed, followed by the addition of 150 μL of TEOS. The mixture was stirred in a shaking bed (400 rpm, 30 °C) for 1 h. The resulting nanoparticles were washed twice with ethanol and were finally dried under vacuum. 100 mg of the SSiO2 powder was dispersed in ethanol (40 mL), followed by addition of N1-(3-trimethoxysilylpropyl) diethylenetriamine (0.5 ml) and N,N-diisopropylethylamine (0.1 ml). The mixture was stirred in a shaking bed (400 rpm, 30 °C) for 12 h and then washed with Milli-Q water and ethanol six times alternately. The obtained nanoparticles (SSiO2-NH2) were dried under vacuum.
Preparation of the catalysts
Two hundred milligrams of the dried Sad or SiO2@mSiO2-NH2 or SSiO2-NH2 powder was mixed with 10 mL of the electroplating wastewater, the compositions of which are listed in Supplementary Table 3. The suspension was sonicated and then transferred to a shaking bed (400 rpm, 30 °C), subjected to stirring overnight. The adsorbents together with the adsorbed metal ions were collected via magnetic separation and then dried in a vacuum oven. The dried samples were calcined at 500 °C for 1 h in a muffle furnace to remove the organic pollutants, followed by reduction at 600 °C for 2 h in H2 atmosphere. The reduction temperature, under which the metal precursors could be fully reduced, was determined by the TPR results (Supplementary Fig. 26). The final products were denoted as SFe-Ni, SNi, and SSiO2-Ni, corresponding to the Sad, SiO2@mSiO2-NH2 and the SSiO2-NH2 support, respectively. The loading amounts of Ni and Fe for different samples are listed in Supplementary Table 4. A control sample was prepared by replacing the electroplating wastewater with manmade Ni2+ solution from reagent Ni(NO3)2·6H2O. The compositions of the prepared catalysts are listed in Supplementary Tables 4 and 5.
In a cycle adsorption process, 10 mL of wastewater was mixed with Sad. After adsorption equilibrium, the spent Sad was separated from the solution for the following calcination process, and the same amounts of new Sad were put into the treated wastewater. The procedure was repeated multiple times.
Thermocatalytic tests
Thermocatalytic CO2 hydrogenation under atmospheric pressure was performed in a quartz tube flow reactor (CEL-GPPCM, BEIJING CHINA EDUCATION AU-LIGHT CO., LTD., Supplementary Fig. 27) at a certain temperature (300, 350, 400, 450, or 500 °C). Prior to the reaction, the powder catalysts (<80 mesh) were reduced under H2 (20 mL min−1) at 600 °C for 2 h. The flow rates of CO2, H2, and N2 were set at ~2.5, 2.5, and 5 mL min−1, respectively. The products at the reactor outlet were detected with an online gas chromatographer (Agilent 8860) equipped with a thermal conductivity detector (TCD) and a flame ionization detector (FID).
In-situ DRIFTS experiments were performed on a Bruker VERTEX 70 v Spectrometer with a mercury cadmium telluride detector cooled with liquid nitrogen. Approximately 20 mg of as-reduced SFe-Ni, SSiO2-Ni, or Niim/SiO2 catalyst (<80 mesh) was packed into an in situ cell (BEIJING OPERANDO TECHNOLOGY CO., LTD.). Prior to the reaction, the catalyst was reduced in a H2/N2 mixture gas (2.5/17.5 mL/min) at 500 °C for 30 min. The inlet flow in the cell was then switched to N2 (17.5 mL/min) for 30 min, and the temperature was kept at 500 °C. After obtaining the background spectra, the inlet flow was switched to a CO2/H2 mixture gas (2.5/2.5 mL/min). The adsorption species on the surface of catalysts were detected online.
Rate-order experiments were carried out with 30 mg of the SFe-Ni catalyst (<80 mesh) at 500 °C under a constant flow rate (40 mL/min). The internal and external diffusions were eliminated based on a reported recipe (see the details in the supplementary notes, Supplementary Figs. 28 and 29). To study the rate order for H2, the concentration of feed CO2 was kept at 12.5%, while the concentration of feed H2 gradually decreased from 67.5% to 42.5% and then gradually increased back to 67.5%, and N2 was used as the balance component. The CO rates in the downslope and uphill stages under the same conditions were averaged for the calculation of rate order for H2. Similarly, to study the rate order for CO2, the concentration of feed H2 was kept at 75%, while the concentration of feed CO2 was gradually increased from 7.5 to 11.25% and then gradually decreased to 7.5%, and N2 was used as the balance component. The CO rates in the upslope and downslope stages under the same conditions were averaged for the calculation of the rate order for CO2. In all rate-order experiments, the conversions for H2 and CO2 were always kept below 5%.
Photothermal catalytic tests in the flow reactor
The photothermal tests were conducted similarly to the thermocatalytic tests, except that a 300-W Xe arc lamp (PF300-T8E, BEIJING CHINA EDUCATION AU-LIGHT CO., LTD.) rather than heating modulate was used as the energy source. The light intensity was measured using an optical power meter (PL-MW2000, Beijing Perfectlight Technology Co., Ltd.). The relationship between the power of the lamp and light intensity is listed in Supplementary Fig. 30
Photothermal catalytic tests in the batch reactor
The gas-phase photothermal catalytic experiments were conducted in a batch reactor (CEL-HPR100T+) with an inner volume of 100 mL. A group of glass slides was put inside the reactor to support the catalyst. The final effective volume of the reactor was 57.77 mL. A 300 W Xe arc lamp was used to illuminate the catalysts with the assistance of a concentrator. For all the catalysts, Samples (9 mg) were dispersed in ethanol and then transferred into a glass fiber filter through drop-casting. The catalyst film was then dried under a vacuum. After the loading of the catalyst film, the reactor was degassed first. A mixture of CO2 and H2 (1:1) was used to purge the reactor three times. The reactor was sealed when the pressure reached 1 bar. The lamp was then turned on to initiate the photothermal catalytic reaction. After 10 min illumination, product gases were analyzed with a gas chromatographer (Agilent 8860) equipped with a thermal conductivity detector (TCD) and a flame ionization detector (FID).
Characterization
STEM images and EDX mappings were obtained in a double aberration-corrected transmission electron microscope (FEI Titan Themis G2) operated at 300 kV with a HAADF detector (collection angle range of 48–200 mrad) and Super-X EDX detector. Transmission electron microscopy (TEM) images were obtained with an FEI-Tecnai F20 (200 kV) transmission electron microscope. The XAS experiments at the Ni K-edge were performed at the Shanghai Synchrotron Radiation Facility (SSRF,11B), and the intensities of the two SFe-Ni samples were multiplied by five for the EXAFS spectra. The metal content of different samples was measured by an Inductively coupled plasma optical emission spectrometer (ICP-OES) (i CAP Pro X, Thermofisher) or an Inductively coupled plasma source mass spectrometer (ICP-MS) (Aurora M90, Jenoptik). The temperature-programmed reduction (TPR) results were recorded with an infrared spectrometer (ThermoFisher Nicolet iS 50) equipped with a Deuterated Triglycine Sulfate (DTGS) detector.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request. Source data are provided with this paper.
References
Lu, H., Wang, Y. & Wang, J. Recovery of Ni2+ and pure water from electroplating rinse wastewater by an integrated two-stage electrodeionization process. J. Clean. Prod. 92, 257–266 (2015).
Qasem, N. A. A., Mohammed, R. H. & Lawal, D. U. Removal of heavy metal ions from wastewater: a comprehensive and critical review. npj Clean. Water 4, 36 (2021).
Nriagu, J. O. & Pacyna, J. M. Quantitative assessment of worldwide contamination of air, water and soils by trace metals. Nature 333, 134–139 (1988).
Raabe, D., Tasan, C. C. & Olivetti, E. A. Strategies for improving the sustainability of structural metals. Nature 575, 64–74 (2019).
Wang, H., Wang, H., Zhao, H. & Yan, Q. Adsorption and Fenton-like removal of chelated nickel from Zn-Ni alloy electroplating wastewater using activated biochar composite derived from Taihu blue algae. Chem. Eng. J. 379, 122372 (2020).
Bolisetty, S., Peydayesh, M. & Mezzenga, R. Sustainable technologies for water purification from heavy metals: review and analysis. Chem. Soc. Rev. 48, 463–487 (2019).
Coman, V., Robotin, B. & Ilea, P. Nickel recovery/removal from industrial wastes: a review. Resour. Conserv. Recycl. 73, 229–238 (2013).
Peydayesh, M. & Mezzenga, R. Protein nanofibrils for next generation sustainable water purification. Nat. Commun. 12, 3248 (2021).
Lu, L. et al. Wastewater treatment for carbon capture and utilization. Nat. Sustain. 1, 750–758 (2018).
Mallapaty, S. How China could be carbon neutral by mid-century. Nature 586, 482–483 (2020).
Global thirst for electricity is fuelling the rise of a potent greenhouse gas. Nature 583, 171–171 (2020).
Yan, Z., Hitt, J. L., Turner, J. A. & Mallouk, T. E. Renewable electricity storage using electrolysis. Proc. Natl Acad. Sci. USA 117, 12558 (2020).
Styring, P., Dowson, G. R. M. & Tozer, I. O. Synthetic fuels based on dimethyl ether as a future non-fossil fuel for road transport from sustainable feedstocks. Front. Energy Res. 9, 240 (2021).
Jiang, Z. et al. Building a bridge from papermaking to solar fuels. Angew. Chem. Int. Ed. 58, 14850–14854 (2019).
Sovacool, B. K. & Griffiths, S. Culture and low-carbon energy transitions. Nat. Sustain. 3, 685–693 (2020).
Qian, H. et al. Air pollution reduction and climate co-benefits in China’s industries. Nat. Sustain. 4, 417–425 (2021).
Peng, Y. et al. A versatile MOF-based trap for heavy metal ion capture and dispersion. Nat. Commun. 9, 187 (2018).
Ali, I. & Gupta, V. K. Advances in water treatment by adsorption technology. Nat. Protoc. 1, 2661–2667 (2006).
Hamza, M. F., Wei, Y., Mira, H. I., Abdel-Rahman, A. A. H. & Guibal, E. Synthesis and adsorption characteristics of grafted hydrazinyl amine magnetite-chitosan for Ni(II) and Pb(II) recovery. Chem. Eng. J. 362, 310–324 (2019).
Juang, R.-S. & Wang, Y.-C. Use of complexing agents for effective ion-exchange separation of Co(II)/Ni(II) from aqueous solutions. Water Res. 37, 845–852 (2003).
Li, X. et al. Porous Fe2O3 microcubes derived from metal organic frameworks for efficient elimination of organic pollutants and heavy metal ions. Chem. Eng. J. 336, 241–252 (2018).
Wang, L. et al. Anionic polypeptide poly(γ-glutamic acid)-functionalized magnetic Fe3O4-GO-(o-MWCNTs) hybrid nanocomposite for high-efficiency removal of Cd(II), Cu(II) and Ni(II) heavy metal ions. Chem. Eng. J. 346, 38–49 (2018).
Rott, E. et al. Removal of phosphonates from synthetic and industrial wastewater with reusable magnetic adsorbent particles. Water Res. 145, 608–617 (2018).
Drenkova-Tuhtan, A. et al. Pilot-scale removal and recovery of dissolved phosphate from secondary wastewater effluents with reusable ZnFeZr adsorbent @ Fe3O4/SiO2 particles with magnetic harvesting. Water Res. 109, 77–87 (2017).
Tennakoon, A. et al. Catalytic upcycling of high-density polyethylene via a processive mechanism. Nat. Catal. 3, 893–901 (2020).
Korley, L. T. J., Epps, T. H. II, Helms, B. A. & Ryan, A. J. Toward polymer upcycling-adding value and tackling circularity. Science 373, 66–69 (2021).
Li, Y. et al. Selective light absorber-assisted single nickel atom catalysts for ambient sunlight-driven CO2 methanation. Nat. Commun. 10, 2359 (2019).
Yan, X. et al. Nickel@siloxene catalytic nanosheets for high-performance CO2 methanation. Nat. Commun. 10, 2608 (2019).
Wang, L., Liu, H., Liu, Y., Chen, Y. & Yang, S. Influence of preparation method on performance of Ni-CeO2 catalysts for reverse water-gas shift reaction. J. Rare Earths 31, 559–564 (2013).
Chen, P. et al. Nano-intermetallic InNi3C0.5 compound discovered as a superior catalyst for CO2 reutilization. iScience 17, 315–324 (2019).
Mikhail, M. et al. Effect of Na and K impurities on the performance of Ni/CeZrOx catalysts in DBD plasma-catalytic CO2 methanation. Fuel 306, 121639 (2021).
Panagiotopoulou, P. Methanation of CO2 over alkali-promoted Ru/TiO2 catalysts: II. Effect of alkali additives on the reaction pathway. Appl. Catal. B Environ. 236, 162–170 (2018).
Ahmad, W., Al-Matar, A., Shawabkeh, R. & Rana, A. An experimental and thermodynamic study for conversion of CO2 to CO and methane over Cu-K/Al2O3. J. Environ. Chem. Eng. 4, 2725–2735 (2016).
Chai, G.-Y. & Falconer, J. L. Alkali promoters on supported nickel: effect of support, preparation, and alkali concentration. J. Catal. 93, 152–160 (1985).
Hu, Y., He, L. & Yin, Y. Magnetically responsive photonic nanochains. Angew. Chem. Int. Ed. 50, 3747–3750 (2011).
He, L. et al. Magnetic assembly of nonmagnetic particles into photonic crystal structures. Nano Lett. 10, 4708–4714 (2010).
He, L., Wang, M., Zhang, Q., Lu, Y. & Yin, Y. Magnetic assembly and patterning of general nanoscale materials through nonmagnetic templates. Nano Lett. 13, 264–271 (2013).
Xiong, X. et al. Photocatalytic CO2 reduction to CO over Ni single atoms supported on defect-rich zirconia. Adv. Energy Mater. 10, 2002928 (2020).
Akri, M. et al. Atomically dispersed nickel as coke-resistant active sites for methane dry reforming. Nat. Commun. 10, 5181 (2019).
Millet, M.-M. et al. Ni single atom catalysts for CO2 activation. J. Am. Chem. Soc. 141, 2451–2461 (2019).
Feng, K. et al. Experimentally unveiling the origin of tunable selectivity for CO2 hydrogenation over Ni-based catalysts. Appl. Catal. B Environ. 292, 120191 (2021).
Chen, Y. et al. Cooperative catalysis coupling photo-/photothermal effect to drive Sabatier reaction with unprecedented conversion and selectivity. Joule 5, 3235–3251 (2021).
Ziemba, M., Weyel, J. & Hess, C. Elucidating the mechanism of the reverse water–gas shift reaction over Au/CeO2 catalysts using operando and transient spectroscopies. Appl. Catal. B Environ. 301, 120825 (2022).
Kattel, S., Liu, P. & Chen, J. G. Tuning selectivity of CO2 hydrogenation reactions at the metal/oxide interface. J. Am. Chem. Soc. 139, 9739–9754 (2017).
Vogt, C. et al. Unravelling structure sensitivity in CO2 hydrogenation over nickel. Nat. Catal. 1, 127–134 (2018).
Galhardo, T. S. et al. Optimizing active sites for high CO selectivity during CO2 hydrogenation over supported nickel catalysts. J. Am. Chem. Soc. 143, 4268–4280 (2021).
Feng, K. et al. Cobalt plasmonic superstructures enable almost 100% broadband photon efficient CO2 photocatalysis. Adv. Mater. 32, 2000014 (2020).
Sun, W. et al. Heterogeneous reduction of carbon dioxide by hydride-terminated silicon nanocrystals. Nat. Commun. 7, 12553 (2016).
Wu, Z. et al. Niobium and titanium carbides (MXenes) as superior photothermal supports for CO2 photocatalysis. ACS Nano 15, 5696–5705 (2021).
Li, Z. et al. Fe-based catalysts for the direct photohydrogenation of CO2 to value-added hydrocarbons. Adv. Energy Mater. 11, 2002783 (2021).
Zhang, D. et al. All-earth-abundant photothermal silicon platform for CO2 catalysis with nearly 100% sunlight harvesting ability. Sol. RRL 5, 2000387 (2021).
Liu, L. et al. Sunlight-assisted hydrogenation of CO2 into ethanol and C2+ hydrocarbons by sodium-promoted Co@C nanocomposites. Appl. Catal. B Environ. 235, 186–196 (2018).
Chen, G. et al. Alumina-supported CoFe alloy catalysts derived from layered-double-hydroxide nanosheets for efficient photothermal CO2 hydrogenation to hydrocarbons. Adv. Mater. 30, 1704663 (2018).
Qian, C. et al. Catalytic CO2 reduction by palladium-decorated silicon–hydride nanosheets. Nat. Catal. 2, 46–54 (2019).
Wan, L. et al. Cu2O nanocubes with mixed oxidation-state facets for (photo)catalytic hydrogenation of carbon dioxide. Nat. Catal. 2, 889–898 (2019).
Cai, M. et al. Greenhouse-inspired supra-photothermal CO2 catalysis. Nat. Energy 6, 807–814 (2021).
Cai, M., Li, C. & He, L. Enhancing photothermal CO2 catalysis by thermal insulating substrates. Rare Met. 39, 881–886 (2020).
Mateo, D., Cerrillo, J. L., Durini, S. & Gascon, J. Fundamentals and applications of photo-thermal catalysis. Chem. Soc. Rev. 50, 2173–2210 (2021).
Styring, P. & Dowson, G. R. M. Oxygenated transport fuels from carbon dioxide driving towards net zero. Johns. Matthey Technol. Rev. 65, 170–179 (2021).
Peng, L. et al. Co–Fe clusters supported on N-doped graphitic carbon as highly selective catalysts for reverse water gas shift reaction. ACS Sustain. Chem. Eng. 9, 9264–9272 (2021).
Albero, J., Peng, Y. & García, H. Photocatalytic CO2 reduction to C2+ products. ACS Catal. 10, 5734–5749 (2020).
Kan, M. et al. 2-aminobenzenethiol-functionalized silver-decorated nanoporous silicon photoelectrodes for selective CO2 reduction. Angew. Chem. Int. Ed. 59, 11462–11469 (2020).
Yan, Z., Hitt, J. L., Zeng, Z., Hickner, M. A. & Mallouk, T. E. Improving the efficiency of CO2 electrolysis by using a bipolar membrane with a weak-acid cation exchange layer. Nat. Chem. 13, 33–40 (2021).
Loh, J. Y. Y., Kherani, N. P. & Ozin, G. A. Persistent CO2 photocatalysis for solar fuels in the dark. Nat. Sustain. 4, 466–473 (2021).
Wang, T. et al. Health co-benefits of achieving sustainable net-zero greenhouse gas emissions in California. Nat. Sustain. 3, 597–605 (2020).
McCord, S., Armstrong, K. & Styring, P. Developing a triple helix approach for CO2 utilisation assessment. Faraday Discuss. 230, 247–270 (2021).
Tackett, B. M., Gomez, E. & Chen, J. G. Net reduction of CO2 via its thermocatalytic and electrocatalytic transformation reactions in standard and hybrid processes. Nat. Catal. 2, 381–386 (2019).
Wang, S. et al. CO2 footprint of thermal versus photothermal CO2 catalysis. Small 17, 2007025 (2021).
Gao, J. et al. One-step solvothermal synthesis of highly water-soluble, negatively charged superparamagnetic Fe3O4 colloidal nanocrystal clusters. Nanoscale 5, 7026–7033 (2013).
Wang, S. et al. Anomalous effect of the aging degree on the ionic permeability of silica shells. RSC Adv. 8, 38499–38505 (2018).
Zhu, Z. et al. A mechanistic study of silica-etching by hot water. Phys. Chem. Chem. Phys. 20, 1440–1446 (2018).
Wang, S., Yu, Y., He, L., Zhang, D. & Ye, M. Design of magnetic nanoparticles with high magnetic separation efficiencies and durability for Cu2+ adsorption. Nanotechnology 31, 085710 (2019).
Acknowledgements
W.S. thanks the support from the National Key R&D Program of China (2021YFF0502000), National Natural Science Foundation of China (51902287), the U of T-ZJU Joint Seed Fund, the Fundamental Research Funds for the Central Universities (226-2022-00159, 226-2022-00200), Jiangsu Key Laboratory for Carbon Based Functional Materials & Devices, Soochow University, and State Key Laboratory of New Textile Materials and Advanced Processing Technologies (FZ2020020). D.Y. acknowledges the financial support from National Natural Science Foundation of China (61721005). L.H. thanks the support by National Natural Science Foundation of China (52172221 and 51920105005), the Natural Science Foundation of Jiangsu Province (BK20200101), 111 Project, and the Collaborative Innovation Centre of Suzhou Nano Science & Technology. G.A.O. is the Government of Canada Tier 1 Research Chair in Materials Chemistry and Nanochemistry, and he acknowledges the financial support provided by the Natural Sciences and Engineering Research Council of Canada (NSERC). The authors thank the support from SSRF (11B) for the XAS experiments.
Author information
Authors and Affiliations
Contributions
W.S., S.W., D.Z., L.H., D.Y., and G.A.O. conceived and designed the experiments. S.W. and D.Z. carried out the synthesis and catalysis experiments. W.W. and J.H. carried out the HAADF-STEM experiments. J.Z. carried out and analyzed the XAS experiments. K.F. analyzed the XAS results. Z.W. carried out the TEM characterization. B.D. carried out the ICP experiments. Z.L. carried out part of the thermocatalytic experiments. W.S., G.A.O., and S.W. wrote the paper. All authors commented on the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Hermenegildo Garcia and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, S., Zhang, D., Wang, W. et al. Grave-to-cradle upcycling of Ni from electroplating wastewater to photothermal CO2 catalysis. Nat Commun 13, 5305 (2022). https://doi.org/10.1038/s41467-022-33029-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-33029-x
This article is cited by
-
A nonmetallic plasmonic catalyst for photothermal CO2 flow conversion with high activity, selectivity and durability
Nature Communications (2024)
-
Photocatalytic CO2 reduction
Nature Reviews Methods Primers (2023)
-
Effect of cyclic voltammetry on the deposition of Ni cocatalyst on CuInS2 photoelectrode for water splitting under solar light irradiation
Research on Chemical Intermediates (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
