
Abstract
Electrochemical reduction of CO2 (CO2R) to formic acid upgrades waste CO2; however, up to now, chemical and structural changes to the electrocatalyst have often led to the deterioration of performance over time. Here, we find that alloying p-block elements with differing electronegativities modulates the redox potential of active sites and stabilizes them throughout extended CO2R operation. Active Sn-Bi/SnO2 surfaces formed in situ on homogeneously alloyed Bi0.1Sn crystals stabilize the CO2R-to-formate pathway over 2400 h (100 days) of continuous operation at a current density of 100 mA cm−2. This performance is accompanied by a Faradaic efficiency of 95% and an overpotential of ~ −0.65 V. Operating experimental studies as well as computational investigations show that the stabilized active sites offer near-optimal binding energy to the key formate intermediate *OCHO. Using a cation-exchange membrane electrode assembly device, we demonstrate the stable production of concentrated HCOO– solution (3.4 molar, 15 wt%) over 100 h.
Similar content being viewed by others

Introduction
Electrochemical CO2 reduction (CO2R) driven by electrical energy converts CO2 into low carbon footprint chemicals and fuels1. Among CO2R products, formic acid (HCOOH) or formate (HCOO–) is used in pharmaceutical, electrolytic metallurgy, leather, and fuel cell applications2,3,4,5,6. HCOO– has a high market value per energy (cents/kWh)7, making it a candidate of particular interest.
Conventionally, HCOO– is produced from the hydrolysis of methyl formate, which uses CH3OH and CO as starting materials. This chemical reforming process requires strict reaction conditions and high energy input8,9,10. In contrast, CO2R requires only CO2, water, and electrical energy7,11,12. Unfortunately, to date, CO2R catalysts and systems favouring HCOO– have not achieved the required combination of high selectivity (Faradaic efficiency (FE))13, high reaction rate (current density)13, high energy efficiency (EE)14, and, in particular, long-term stability.
Among electrocatalytic materials studied, Sn is a promising candidate owing to its low cost and planetary abundance15,16. Sn has strong binding energy for *OCHO, and this favours the first-step CO2 hydrogenation in CO2-to-formate conversion17,18. The second step electron transfer requires high energy to reduce *OCHO to HCOO–, resulting in a large overpotential and consequently a low EE. Sn has medium binding energy to *COOH and *H, which makes it difficult to fully suppress CO and H2 generation; the FE for HCOO– has therefore been limited to 80–85%. Recent studies of Sn-based core-shell structures, including Ag-Sn19, Cu-Sn20, Bi-Sn21, and phase-segregated bimetallic systems22,23, showed improved FE for HCOO–. Further improvement of the HCOO– production rate and cathodic energy efficiency (CEE) relative to current benchmarks (Supplementary Table 4) requires precise control of the elemental distributions in the active sites. In particular, knowledge of the electrochemical stability of Sn and Sn-based materials in aqueous electrolytes at different pH is lacking; indeed, among reported formate catalysts, it has been challenging to combine optimal adsorption energetics for intermediate binding with sites stable against reconstruction13,14,15,16.
Here, we present active Sn-Bi/SnO2 surfaces that are grown conformally on uniformly alloyed Bi0.1Sn crystals. These surfaces support the stable reduction of CO2 to formate over a period in excess of 2400 h (100 days) of continuous operation, with a near-unity formate FE of over 95% and a CEE exceeding 70% at a current density of 100 mA cm−2 in 1 M potassium bicarbonate (KHCO3) and potassium hydroxide (KOH) electrolytes at pH 11. This stability and CEE is significantly improved compared to literature benchmarks13,24,25,26,27. The selectivity and energy efficiency obtained to meet the performance required for a positive net present value in an economic analysis of formate production11. Computational studies reveal that, across a wide range (1.5–12.5%) of Bi incorporation into Sn, the facets of Sn-Bi alloys and Sn-Bi/SnO2 composites provide sites with near-optimal binding energy to *OCHO. This approach lowers the reaction energy in CO2-to-formate conversion. The by-products CO and H2 are suppressed due to their increased reaction energy on Sn-Bi alloys. The redox-modulated Sn-Bi/SnO2 surfaces remain active and protect Bi0.1Sn catalysts against corrosion or reconstruction during extended CO2R. We further demonstrate stable production of 3.4 molar (15 wt%) HCOO– (formate) solution of over 100 h at a constant current density of 60 mA cm−2 with an average full-cell EE of ~27% in a membrane electrode assembly (MEA) system based on a cation-exchange membrane (CEM), highlighting that Bi0.1Sn catalysts are reliable for long-lasting HCOO– (formate) production in different CO2R systems.
Results
Characterization
We used thermal evaporation to produce BixSn (x = 0.1, 0.2, 0.3), Bi, and Sn precatalysts on polytetrafluoroethylene (PTFE) gas diffusion substrates (Supplementary Fig. 1). Dense and compact particle-film layers were formed for all BixSn (x = 0.1, 0.2, 0.3), Bi, and Sn catalysts, ensuring electrical conductivity for CO2R (Supplementary Fig. 2). The particle size is 200–300 nm for the Bi and 1–1.5 μm for the Sn catalyst. Bi0.2Sn and Bi0.3Sn possess similar particle sizes of 1–1.5 μm to that of Sn, but they fail to form a uniform alloy; Bi precipitates over a large area (Supplementary Figs. 3, 4). Bi0.1Sn forms uniform crystals with particle sizes of 700–1000 nm, as confirmed by scanning electron microscopy (SEM), transmission electron microscopy (TEM) images, and scanning transmission electron microscopy–energy dispersive spectroscopy (STEM-EDS) elemental mapping in Fig. 1b, c, e. The selected area electron diffraction (SAED) pattern of Bi0.1Sn exhibits distinct single-crystal diffraction spots corresponding to the Sn plane indexes (Fig. 1d), suggesting that Bi is incorporated into the Sn crystals. The facets exposed are primarily (200) and (101), consistent with the observations from high-resolution TEM analysis (Fig. 1c). We carried out STEM-EDS, SEM-EDS, and XPS analyses, and obtained similar Bi concentrations of ~10 at.% for the Bi0.1Sn catalyst (Supplementary Figs. 5–9 and Table 1), suggesting that Bi and Sn are uniformly distributed from the surface to the bulk.

a Schematic of Bi0.1Sn electrocatalyst on a polytetrafluoroethylene gas diffusion layer for electroreduction of CO2. b SEM, c HRTEM, d TEM, SAED, and, e STEM, EDS analyses of Bi0.1Sn electrocatalyst. The high-resolution TEM image in (c) shows the lattice fringes corresponding to the Sn (200) facet. The dashed box in (d) is the location to take the HRTEM.
Density functional theory (DFT) calculations
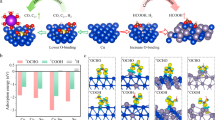
In light of the above characterizations, we built models and performed DFT studies to calculate the reaction energy on Bi, Sn, and BiySn64 (y = 1, 2, 4, 8) surfaces (details in Supplementary Figs. 10–15 and Supplementary Table 1). As shown in Fig. 2a, the production of CO, CH4, and other C2+ hydrocarbons generally proceeds via the *COOH intermediate pathway28,29. *COOH adsorption requires the outer shell electrons of catalysts to have both energy that matches the LUMO energy of CO2, and electronic orbitals that overlap with molecular orbitals of the C–O π bond. Production of HCOO– often occurs via the *OCHO intermediate pathway14,18. This requires catalysts to possess suitable binding energy to O. Bi and Sn are both p-block metals that, as shown in Fig. 2b, d, display significantly uphill reaction energy to *COOH compared to that to *OCHO, indicating that HCOO– is the major product.

a Possible pathways of electrochemical CO2 reduction. The top panel shows the *COOH pathway to produce CO, CH4, or C2+ hydrocarbons. The bottom panel shows the *OCHO pathway to produce HCOO–. Bi, Sn, C, O, and H atoms are represented by green, yellow, grey, red, and white spheres, respectively. b Reaction energy for CO formation on Bi (003), Sn (200), and Bi4Sn64 (200) surfaces without applying any external potential (U = 0 eV). c The volcano plot describing *OCHO adsorption energy versus CO2-to-HCOO– reaction energy. d Reaction energy for HCOO– formation on Bi (003), Sn (200), and BiySn64 (y = 1, 2, 4, 8) surfaces without applying any external potential (U = 0 eV).
Since the intermediate *OCHO is energetically more favourable than *COOH on Bi, Sn, and Bi0.1Sn surfaces, we examined their reaction energy for HCOO– production in detail. Figure 2d reveals that Bi (003) tends to bind *OCHO too weakly, that Sn (002) may bind *OCHO too strongly, and that of the calculated BiSn slabs, Bi4Sn64 and Bi8Sn64 show the optimal binding energy to *OCHO of near to –0.13 eV, indicating low reaction energy for the CO2-to-HCOO– conversion. We plotted the reaction energy of different catalyst archetypes as a function of their adsorption energy to *OCHO at 298 K and 1 atm. (Fig. 2c). We took an average of the binding energy on enumerated possible configurations and present the averaged binding energy in Fig. 2d. BiySn64 (y = 1, 2, 4, 8), with a relatively wide range of Bi ratios in different Sn facets, exhibits an abundance of adsorption sites with improved *OCHO binding energy that enhances CO2 reduction (DFT calculation details are given in Supplementary Figs. 12–15). The volcano relationship predicts that Bi4Sn64 (incorporation of 6.25% Bi into Sn) may be the most active (Supplementary Table 2). We further find that Bi4Sn64 elevates the reaction energy to CO and H2, suppressing the generation of CO, H2, and C2+ hydrocarbons (Fig. 2b and Supplementary Fig. 11). In summary, our computational simulations point to the alloying of Sn with Bi as a strategy to enhance HCOO– production.
CO2R in aqueous flow cells
The CO2R performance of BixSn alloys (x = 0.1, 0.2, 0.3), as well as pure Bi or Sn control samples, was first evaluated in 1 M KOH electrolyte (pH = 14) in a flow-cell electrolyser. Ag/AgCl electrodes were used as the reference electrodes, and commercial Ni foams were used as the anodes for the water oxidation reaction in a three-electrode setup. Linear sweep voltammetry (LSV) curves obtained from 0 V to –2 V vs. reversible hydrogen electrode (VRHE) show that Bi0.1Sn has the most positive onset potential for CO2R (Fig. 3a and Supplementary Fig. 16a). This is consistent with the DFT calculations that incorporating 1.5–12.5% Bi into Sn lowers the reaction energy in the electrochemical CO2-to-formate conversion (Fig. 2). A rapid increase of the cathodic current is observed with Bi0.1Sn at potentials more negative than –0.5 VRHE. To study the electrochemical characteristics of the Bi0.1Sn, Bi, and Sn catalysts, we carried out the electrochemical active surface area (ECSA), and electrochemical impedance spectroscopy (EIS) analyses30. The estimated ECSAs of Bi and Bi0.1Sn are twice as large as that of Sn (Supplementary Fig. 17). Also, Bi0.1Sn has the lowest interface resistance (Supplementary Fig. 18). These results confirm that Bi0.1Sn reduces the potential loss and, consequently, improves the CEE.

a Linear sweep voltammetry curves of Bi0.1Sn, Bi, and Sn in 1 M KOH electrolyte. b The corresponding formate FEs under different current densities. c Linear sweep voltammetry curves. d The corresponding formate FEs in 1 M KHCO3 and KOH electrolytes at different pH (pH = 8, 10, 11, 12, 13, and 14) at a current density of 100 mA cm−2. e The CO2-reduction chronopotentiometry curve (blue line), CO2-to-formate FE (blue dots), and CO2-to-formate half-cell energy conversion efficiency (purple dots) in 1 M KHCO3 and KOH electrolyte at pH = 11 at an applied current density of 100 mA cm−2. f Comparison of our work with previously published data. Error bars correspond to the standard deviation of five independent measurements.
To quantify the FEs of CO2R products, BixSn (x = 0.1, 0.2, 0.3), Bi, and Sn catalysts were evaluated in a chronopotentiometry mode by applying current densities of 100, 200, and 300 mA cm−2. Gaseous and liquid products were quantified by gas chromatography (GC) and nuclear magnetic resonance (NMR), respectively. As shown in Fig. 3b and Supplementary Fig. 16b, the Bi0.1Sn catalyst shows over 95% FE for HCOO– in a wide range of current densities. Only a small amount of H2 and CO was detected, and no other liquid products were formed (Supplementary Figs. 19, 20). A CO2-to-formate CEE of ~75% was achieved at pH 14 at 100 mA cm−2. Under similar conditions, Bi0.2Sn, Bi0.3Sn, and Bi catalysts produce 95% FE for HCOO–, but they require a more negative potential for the same current density (Supplementary Fig. 16b, 21 and Supplementary Table 3). The Sn catalyst produces an FE of ~85% for HCOO–, along with a constant total FE of ~15% for H2 and CO. It also requires a more negative potential to reach 100 mA cm−2, and thus the CEE for HCOO– is limited to 50%.
To assess the stability of the Bi0.1Sn catalyst during CO2R, carbon nanoparticles (NPs) and graphite were coated on the Bi0.1Sn/PTFE electrode to form a hybrid structure of graphite/carbon NPs/Bi0.1Sn/PTFE. This structure enables uniform distribution of the electrolyte and electrical current over the Bi0.1Sn surfaces31,32. Note that the coated carbon nanoparticles and graphite are not CO2R electrocatalysts and they don’t produce any formate under our CO2R operating conditions. Importantly, we coated carbon nanoparticles and graphite for all the Bi, Sn, and Bi0.1Sn catalysts, comparing their CO2R activity under the same working conditions. We are therefore certain that the performance obtained solely represents the properties of each catalyst. We also kept the electrolyte pH unchanged during the long-term CO2R stability test so that the CO2R results are all compared under the same pH conditions. In our studies, Bi0.1Sn catalyst produces HCOO– with an FE exceeding 95% for over 170 h at 100 mA cm−2 and an overpotential of ~–0.5 VRHE in 1 M KOH electrolyte (Supplementary Fig. 22). Control experiments for Bi and Sn samples using similar carbon/graphite coatings led to both lower CEEs and worse stability within 50–70 h (see Supplementary Methods and Supplementary Figs. 23–25).
To evaluate the effect of pH on the activity and stability of CO2R, we studied the performance of Bi0.1Sn catalysts in KHCO3 and KOH electrolytes at different pH levels (see the Supplementary Methods for details). As shown in Supplementary Fig. 26, the formate FEs are above 90% at all pH (8–14) at current densities from 25 to 300 mA cm−2. This indicates that the formate FE is directly related to the properties of catalysts because the CO2R kinetics are more favourable than those of the competing hydrogen evolution reaction under these working conditions. The use of alkaline electrolytes also shifts the working potential positively with the increase of electrolyte pH. As shown in Figs. 3c, 3d and Supplementary Fig. 27, the CEEs for HCOO– are above 70% at 100 mA cm−2 at pH greater than 11. We performed 100-hour stability tests at varying pH (11–14) at a current density of 100 mA cm−2 and analysed their surface morphology, composition, and other material properties after the reaction by SEM, XRD, XPS, and EDX (Supplementary Figs. 28–31). At pH 11, the morphology change is minor compared to those at higher pH levels after 100 h of operation. This is in line with the Pourbaix diagram (Supplementary Fig. 32), in which both metallic Bi and Sn are cathodically protected at potentials more negative than –0.35 VRHE at pH 11. The self-corrosion potentials of Bi0.1Sn catalysts at pH 11–14 in Supplementary Fig. 34 indicate that, thermodynamically, the tendency of corrosion increases with the increase of pH.
Remarkably, we achieved efficient CO2R over 2400 h (100 days) at pH 11 with stable 70% CEE (Fig. 3e). A full-cell EE of 35% (without IR correction) was obtained using unmodified, commercial Ni foams as anodes for the water oxidation reaction in a flow-cell system (Supplementary Fig. 35). This is significant in developing CO2R electrolysers that meet the long operational stability requirements for commercialization12 of at least 1000 h with current densities exceeding 100 mA cm−2. Ours is the first demonstration of CO2R stability achieving this goal. To put our results in a broader context, we plotted our data with reported values13,14,15,19,21,22,23,25,26,27,33,34,35,36,37 in Fig. 3f. The stability and CEE at current densities ≥ 100 mA cm−2 we achieved outperform literature benchmarks by two orders of magnitude, indicating a critical milestone in the field of CO2R field.
We compared the CO2R performance of Bi and Sn at 100 mA cm−2 and in 1 M KHCO3 and KOH electrolytes at pH 11. Sn produces ~80% FE for HCOO– with a constant total FE of ~20% for H2 and CO. The hydrogen evolution reaction increases and HCOO– decreases throughout electrolysis (Supplementary Fig. 37). Bi produces 95% FE for HCOO–, but the CEE is much lower (Supplementary Figs. 36, 37). We found that the stability of Bi was poor: We performed 10 independent tests for Bi samples in an aqueous system with a continuous operation of 100 h. We observed performance decay and peeling off of Bi in all tests (Supplementary Fig. 38).
To diagnose the origins of catalyst instability during CO2R, we compared Bi and Bi0.1Sn catalysts before and after the reactions. After the 70-h test, the original 200–500 nm Bi crystals were completely reconstructed to 20–50 nm nanoparticles with a large amount of O on the surfaces (Fig. 4a–c). SEM-EDX and HRTEM analyses reveal that polycrystalline Bi2O3 was formed on Bi, which is in line with the XRD results for Bi after the reaction; the yellow line in the HRTEM image indicates the boundary between the Bi bulk and the Bi2O3 surface (Fig. 4b, c and Supplementary Figs. 39–40). This chemical and structural transformation is associated with effects involving (i) the non-uniform distribution of electrical potential on the surfaces during the reaction38,39 and (ii) the existence of OH– near Bi at pH 11 to make redox reactions between Bi and Bi2O3 continuously occur. Metastable Bi failed to form equilibrated phases on the surfaces and, thus, Bi was continuously reconstructed during the reaction. The reconstructed Bi2O3/Bi nanoparticles show increased electrical resistance to decrease the overall performance. As shown in the XPS depth profile studies, Bi3+ was detected on the 50h-reaction Bi throughout three rounds of the 3–5 nm soft Ar etching (Fig. 4h). This gives evidence that Bi3+ is presented in the bulk of the 50h-reaction Bi sample. In contrast, when we studied the evaporated Bi catalyst before reaction, Bi3+ disappeared after one round of ~3–5 nm soft Ar etching, indicating that the top surface Bi was oxidized in air and no bulk oxidation was observed (Supplementary Fig. 41). DFT studies in Supplementary Fig. 42 reveal that the Bi2O3 (002) and (210) facets largely shift the *OCHO reaction energy to ~–0.83 and ~0.30 eV, which is either too strong or too weak for the reaction as predicted by the volcano relationship in Fig. 2c. Taking these together with SEM, TEM, and electrochemical analyses for the Bi control samples before and after the reaction in Fig. 4b, c and Supplementary Figs. 38, 39, we conclude that Bi reconstructed to Bi2O3/Bi during the reduction, which leads to performance degradation and eventually catalyst peeling off.

a Schematic of Bi reconstruction during the electrochemical reaction. b SEM results of the as-prepared Bi nanoplates, and the reconstructed Bi/Bi2O3 core/shell nanoparticles after 70 h of reaction. c SEM-EDX elemental mapping results of the Bi/Bi2O3 catalysts after 70 h of reaction. d Schematic of a redox-modulated Bi0.1Sn/SnO2 surface on Bi0.1Sn during the electrochemical reaction. e SEM results of the as-prepared Bi0.1Sn crystals, and the (Bi0.1Sn/SnO2)/Bi0.1Sn particles over 100 h of reaction. f SEM-EDX elemental mapping results of the (Sn-Bi/SnO2)/Bi0.1Sn catalyst over 100 h of reaction. g The SAED pattern, bright-field and dark-field TEM images of the Sn-Bi/SnO2 catalysts over 100 h of reaction. h XPS depth profiles of the post-reaction Bi after 50-h CO2 reduction at 100 mA cm−2 in 1 M KHCO3 and KOH electrolytes at pH = 11. i XPS depth profile of the post-reaction Bi0.1Sn after 100-h CO2 reduction at 100 mA cm−2 in 1 M KHCO3 and KOH electrolytes at pH = 11. The black, red, green and blue lines represent the XPS data before etching (black) and after different time courses of soft Ar etching (red, green, blue), respectively. The etched depth in each soft Ar etching is ~3–5 nm.
In contrast, the Bi0.1Sn catalyst remains stable during the long CO2R operation under the same working conditions. After our tests, Bi0.1Sn remains intact on PTFE with morphology preserved (Fig. 4d–f and Supplementary Fig. 43). Dark-field and bright-field TEM images with SAED patterns and HRTEM visualized conformal ~20–50 nm SnO2/Sn-Bi mixture layers on Bi0.1Sn surfaces (Fig. 4g and Supplementary Figs. 44–46). The SAED patterns show a polycrystalline nature of surface SnO2, in line with the XRD results (Supplementary Figs. 47, 44c). We carried out calculations of different initial configurations with O involved in the Sn-Bi system on the surface (Supplementary Fig. 48). The results reveal that the homogeneously alloyed Bi0.1Sn crystal with few O atoms involved on the surfaces shows similar catalytic performance to that of the metallic Bi0.1Sn crystal. DFT studies also reveal that the SnO2 (110) facet remains active for HCOO– production with the optimal binding energy of ~–0.15 eV for *OCHO (Supplementary Fig. 42).
As shown in the XPS depth profiles (Fig. 4i and Supplementary Fig. 49), after a round of ~ 3–5 nm soft Ar etching, Bi0, Sn0 and Sn4+ were observed in the consequent three rounds of the 3–5 nm soft Ar etching. The electrochemical redox potentials of Bi/Bi3+ and Sn/Sn4+ are
This suggests that Sn should be oxidized before Bi. A redox-modulated balance between Sn and SnO2 was formed on the bimetallic active sites on the surface during CO2R. The redox modulation between Sn/Sn4+ protects the active BiSn:SnO2 against corrosion. We, therefore, suggest that we in situ formed immobilized, conformal and active BiSn:SnO2 surfaces during CO2 reduction, being stable against chemical and structural change throughout extended CO2R reaction over 2400 h of continuous operation.
After 2400 h, we observed densely packed Bi0.1Sn particles over the electrode with clear surface Bi and Sn signals identified by XPS (Supplementary Fig. 50). No significant leaching of Bi and Sn into solution was detected via inductively coupled plasma atomic emission spectroscopy (ICP-AES) analysis (Supplementary Fig. 51). We also provide SEM images and performance curves of Bi0.1Sn and Bi after different times taken from the stability tests as shown in Supplementary Fig. 52. We witness no peeling of the catalyst during the 2400 h test.
Note that a stable and efficient CO2R device requires stable and efficient reactions of CO2R and water oxidation. On the cathode side, Bi0.1Sn catalysts deliver excellent CO2R stability and activity via redox-modulated stabilization of active sites. On the anode side, it is vital to improving the stability and efficiency of the catalysts.
CO2R in MEA systems
To produce concentrated HCOO– solutions, we assessed the CO2R performance of Bi, Sn, and Bi0.1Sn catalysts in an MEA system (Fig. 5a, b). We first used an anion-exchange membrane (AEM) to test the MEA performance. As shown in Fig. 5c, d, and Supplementary Table 5, Bi0.1Sn produced over 90% FE for HCOO– in a wide range of current densities from 30 to 180 mA cm−2. A peak FE for HCOO– of 97.8% was achieved at a current density of 120 mA cm−2 with a full-cell potential (without IR correction) of –3.6 V and a full-cell EE of 36%. As control experiments, pristine Bi and Sn delivered peak HCOO– FEs of 95.8% and 93.5% at the same current density of 120 mA cm−2, but they required more negative potentials of –4.1 V and –4.3 V. As a result, the full-cell EEs for the MEAs using Bi and Sn catalysts were both lower, 31% and 29%, respectively. LSV curves confirmed the CO2R performance of Bi0.1Sn surpassed those of Bi and Sn in AEM-based MEA (Supplementary Fig. 53).

a Schematic of the MEA configuration. b The optical image of an MEA device. c Current densities vs. applied potentials of Bi, Sn, and Bi0.1Sn in AEM-based MEA systems. d HCOO– FEs and EEs of Bi, Sn, and Bi0.1Sn at different current densities in AEM-based MEA systems. e Comparison of our data with literature benchmarks (We plot the concentration values obtained in the longest duration tests in each report).
We then sought to translate the champion performance of Bi0.1Sn to a CEM-based MEA electrolyser as a strategy to minimize the crossover of HCOO– (formate) to the anode side to producing a concentrated HCOO– solution at the cathodic stream (HCOO–: 3.4 molar, 15 wt%). Replacing AEM with CEM likely changed the local pH near the catalyst surfaces during the reaction, leading to a shift of peak HCOO– FE to relatively lower current densities7,40. By using a high anolyte flow rate of 15 mL/min., we obtained a peak HCOO– FE of ~90% at 60 mA cm−2 with Bi0.1Sn (Supplementary Fig. 54 and Supplementary Table 5). We then optimized the single-pass flow rate of anolyte (0.1 M KHCO3) to 1.5 mL/min. to promote the HCOO– concentration at the cathodic liquid stream. This optimization established a balance between membrane hydration and K+ availability near the catalyst surface, enabling the collection of concentrated HCOO– directly from the cathodic stream. We obtained an average HCOO– FE of 82% (or 4.49 mmol/h) at 60 mA cm−2 during 100 h of CO2R with an average HCOO– concentration of 3.4 molar (15 wt%) (Supplementary Fig. 55). This represents a 30-fold improvement in HCOO– concentration with 100-hour stability along with a 2-fold increase in current density compared to the literature benchmarks reported in duration tests6,27,41,42 (Fig. 5e).
Discussion
We present redox-modulated, stable, and active Sn-Bi/SnO2 surfaces on uniformly alloyed Bi0.1Sn crystals, and these catalysts show a combination of high activity and performance stability in the CO2-to-HCOO– reduction exceeding 2400 h (100 days) of continuous operation. We use DFT calculations to explain that the stabilized active sites improve *OCHO binding energy and fine-tune *COOH and *H binding energy for selective HCOO– production. We show stable production of concentrated HCOO– (formate) of 3.4 molar with Bi0.1Sn catalysts in a solid-state CEM-based MEA system over 100 h. Our demonstration of a stable catalyst and system is a crucial step to deliver reliable and long-lasting CO2R technology. Further efforts will be necessary to increase CO2 single-pass yield and to the broader applicability of this system in the production of other C2 + liquid fuels and beyond.
Methods
Synthesis
BixSn (x = 0.1, 0.2, 0.3), Bi, and Sn electrocatalysts were synthesized using thermal evaporation (SKY-RH400). In brief, to fabricate the BixSn (x = 0.1, 0.2, 0.3) catalysts, different amounts of Sn and Bi were co-evaporated onto the PTFE substrates under the pressure of 10−5 Torr. The Bi evaporation rate was set to 0.1 nm s−1, and the Sn evaporation rate was set to 1 nm s−1, 0.5 nm s−1 and 0.3 nm s−1 to make the BixSn (x = 0.1, 0.2, and 0.3) samples. The thickness of the deposited BixSn layers was ~700 nm. The pure Bi and Sn films with the same film thicknesses were prepared at an evaporation rate of ~0.3 nm s−1 under the pressure of 10−5 Torr.
Characterization
SEM images were taken using a Gemini500 SEM at an accelerating voltage of 2 kV. High-resolution transmission electron microscopy (HRTEM) and transmission electron microscopy-energy dispersive X-ray spectroscopy (TEM-EDX), SAED, and bright-field and dark-field TEM analyses were performed in a TEM (Tecn F20) with an accelerating voltage of 200 kV. X-ray photoelectron spectroscopy (XPS) studies were performed using PHI5000 VersaProbe. The binding energy data were calibrated relative to the C 1s signal at 284.6 eV. In the XPS depth profile studies, the etched depth is ~3–5 nm in each round of the soft Ar etching. X-ray powder diffraction (XRD) was carried out with a Bruker D8 Advance X-ray diffractometer using Cu Kα radiation at a scanning rate of 9°/min in the 2θ range from 20° to 80°.
Electrochemical experiments
The CO2R experiments were performed in the KHCO3 and KOH electrolytes in a flow-cell device with a three-electrode setup. Different volumes of 10 M KOH were added to 1 M KHCO3 solution to adjust the pH to 11, 12, 13, and 14, respectively, confirming with a pH meter. An Ag/AgCl (in saturated KCl) electrode was used as a reference electrode; a nickel foam was used as a counter-electrode; Sn, Bi, and BixSn electrodes were used as working electrodes. The three electrodes were connected to an electrochemical workstation (Metrohm Autolab). The flow rate of CO2 was set to 20–50 mL min−1. under standard conditions at the outlet end of the flow cell for all the experiments. The gaseous products were quantified using gas chromatography (GC, PerkinElmer) with a thermal conductivity detector (TCD) and a flame ionization detector (FID). The liquid products were detected using nuclear magnetic resonance (NMR, Bruker 400 M) with water peak suppression. To determine the Faraday efficiency of the liquid products, we quantified the liquid products in both analytes and catholyte by NMR. The electrolytes (on both sides) are also replaced at regular time intervals before the NMR tests. The CO2R performance of Bi, Sn, and Bi0.1Sn catalysts was also evaluated in MEA electrolysers. A commercial CO2R MEA electrolyser (Dioxide Materials) was used to accommodate the electrochemical reactions. The MEA electrolyser was composed of anode and cathode flow field plates with a serpentine-configuration flow field of 5 cm2 for the continuous supply of anolyte (0.1 M KHCO3) and humidified CO2 to each respective electrode. We used Bi/PTFE, Sn/PTFE, and Bi0.1Sn/PTFE as the cathode, an iridium oxide deposited titanium foam as the anode, and a solid-state cation-exchange membrane (CEM) for HCOO– placed in between the cathode and anode. Before the electrochemical testing, the cathode electrodes (Bi, Sn, or Bi0.1Sn on PTFE) were taped to the stainless-steel flow field plate by using a copper frame for homogeneously distributing the electrical current. The anode (IrOx on Ti foam) and cathode were physically separated by CEM (NafionTM 117, Fuel Cell Store) in the solid-state CEM-based MEA experiments and by AEM (Sustainion X37-50 membrane) in the AEM-based MEA experiments. Electrolyser bolts were tightened by applying an equal compression torque. Before experiments, the AEM was activated in 1 M KOH for more than 24 h. Upon completion of the electrolyser assembly, the anolyte (0.1 M KHCO3) flowed through the anode with a constant flow rate of 15 mL min−1, while the humidified CO2 was supplied from the gas diffusion electrode (GDL) back with a constant flow rate of 60 standard cubic centimetres per minute (sccm). The full-cell potentials are presented without IR correction. The gas samples were examined using GC (PerkinElmer Clarus 680) to calculate the FE of gas products. The liquid product, formate (when collected from the anodic stream from the AEM-based MEA) or HCOO– (when collected from the cathodic stream from the solid-state CEM-based MEA), was collected from the anodic and cathodic streams simultaneously and analysed by an NMR spectroscopy (Agilent DD2 600 MHz) by using dimethylsulfoxide (DMSO) as the internal standard. The FE towards formate or formic acid at each current density was calculated by adding up both anodic and cathodic FEs. More details are discussed in the supplementary information.
Density functional theory (DFT) calculation
The facets Bi (003), Sn (200), and Sn (101) are primarily exposed in the Bi, Sn, and Bi0.1Sn catalysts (Fig. 1); these facets were thus used to build the DFT models (Supplementary Fig. 10, Supplementary Table 1). We incorporated 1–8 Bi into 64 Sn crystals, as more than 8 Bi would de-stabilize the Sn crystal structure (Supplementary Figs. 12–15). All DFT calculations were performed with the Vienna ab initio simulation package (VASP)43. The DFT calculation details are included in the Supplementary Information.
Data availability
Source data to generate figures and tables are available from the corresponding authors.
References
Bushuyev, O. S. et al. What should we make with CO2 and how can we make it? Joule 2, 825–832 (2018).
De Luna, P. et al. What would it take for renewably powered electrosynthesis to displace petrochemical processes? Science 364, 350 (2019).
Grasemann, M. & Laurenczy, G. Formic acid as a hydrogen source—recent developments and future trends. Energy Environ. Sci. 5, 8171–8181 (2012).
Mellmann, D., Sponholz, P., Junge, H. & Beller, M. Formic acid as a hydrogen storage material - development of homogeneous catalysts for selective hydrogen release. Chem. Soc. Rev. 45, 3954–3988 (2016).
Ross, M. B. et al. Designing materials for electrochemical carbon dioxide recycling. Nat. Catal. 2, 648–658 (2019).
Fan, L. et al. Electrochemical CO2 reduction to high-concentration pure formic acid solutions in an all-solid-state reactor. Nat. Commun. 11, 3633 (2020).
Kibria, M. G. et al. Electrochemical CO2 reduction into chemical feedstocks: from mechanistic electrocatalysis models to system design. Adv. Mater. 31, 1807166 (2019).
Liu, X.-Q., Wu, Y.-T., Chen, W.-K. & Yu, Z.-L. Concurrent synthesis of methanol and methyl formate catalyzed by copper-based catalysts in a slurry phase. In Natural Gas Conversion V, Proc. 5th International Natural Gas Conversion Symposium 119, 557–560 (1998).
Rong, L., Xu, Z., Sun, J. & Guo, G. New methyl formate synthesis method: coal to methyl formate. J. Energy Chem. 27, 238–242 (2018).
Boddien, A. et al. Kohlenstoffdioxid-neutrale Wasserstoffspeicherung basierend auf Bicarbonaten und Formiaten. Angew. Chem. 123, 6535–6538 (2011).
Jouny, M., Luc, W. & Jiao, F. General techno-economic analysis of CO2 electrolysis systems. Ind. Eng. Chem. Res. 57, 2165–2177 (2018).
Verma, S. et al. A gross-margin model for defining technoeconomic benchmarks in the electroreduction of CO2. ChemSusChem 9, 1972–1979 (2016).
Gong, Q. et al. Structural defects on converted bismuth oxide nanotubes enable highly active electrocatalysis of carbon dioxide reduction. Nat. Commun. 10, 2807 (2019).
He, S. et al. The p-orbital delocalization of main-group metals to boost CO2 Electroreduction. Angew. Chem. Int. Ed. Engl. 57, 16114–16119 (2018).
Zhang, S., Kang, P. & Meyer, T. J. Nanostructured tin catalysts for selective electrochemical reduction of carbon dioxide to formate. J. Am. Chem. Soc. 136, 1734–1737 (2014).
Wen, G. et al. Ternary Sn-Ti-O electrocatalyst boosts the stability and energy efficiency of CO2 reduction. Angew. Chem. Int. Ed. Engl. 59, 12860–12867 (2020).
Yoo, J. S. et al. Theoretical insight into the trends that guide the electrochemical reduction of carbon dioxide to formic acid. ChemSusChem 9, 358–363 (2016).
Feaster, J. T. et al. Understanding selectivity for the electrochemical reduction of carbon dioxide to formic acid and carbon monoxide on metal electrodes. ACS Catal. 7, 4822–4827 (2017).
Luc, W. et al. Ag-Sn bimetallic catalyst with a core-shell structure for CO2 reduction. J. Am. Chem. Soc. 139, 1885–1893 (2017).
Ye, K. et al. In situ reconstruction of a hierarchical Sn-Cu/SnOx core/shell catalyst for high-performance CO2 electroreduction. Angew. Chem. Int. Ed. Engl. 59, 4814–4821 (2020).
Xing, Y. et al. Bi@Sn core-shell structure with compressive strain boosts the electroreduction of CO2 into formic acid. Adv. Sci. 7, 1902989 (2020).
Wen, G. et al. Orbital interactions in Bi-Sn bimetallic electrocatalysts for highly selective electrochemical CO2 reduction toward formate production. Adv. Energy Mater. 8, 1802427 (2018).
Tang, J. et al. Advantages of eutectic alloys for creating catalysts in the realm of nanotechnology-enabled metallurgy. Nat. Commun. 10, 4645 (2019).
Yang, H., Kaczur, J. J., Sajjad, S. D. & Masel, R. I. Electrochemical conversion of CO2 to formic acid utilizing Sustainion™ membranes. J. CO2 Util. 20, 208–217 (2017).
Lei, F. et al. Metallic tin quantum sheets confined in graphene toward high-efficiency carbon dioxide electroreduction. Nat. Commun. 7, 12697 (2016).
Li, F. et al. Hierarchical mesoporous SnO2 nanosheets on carbon cloth: a robust and flexible electrocatalyst for CO2 reduction with high efficiency and selectivity. Angew. Chem. Int. Ed. Engl. 56, 505–509 (2017).
Xia, C. et al. Continuous production of pure liquid fuel solutions via electrocatalytic CO2 reduction using solid-electrolyte devices. Nat. Energy 4, 776–785 (2019).
Vasileff, A. et al. Surface and interface engineering in copper-based bimetallic materials for selective CO2 electroreduction. Chem 4, 1809–1831 (2018).
Wang, Y. et al. Defect and interface engineering for aqueous electrocatalytic CO2 reduction. Joule 2, 2551–2582 (2018).
Zhong, M. et al. Surface modification of CoOx loaded BiVO4 photoanodes with ultrathin p-type NiO layers for improved solar water oxidation. J. Am. Chem. Soc. 137, 5053–5060 (2015).
Zhong, M. et al. Accelerated discovery of CO2 electrocatalysts using active machine learning. Nature 581, 178–183 (2020).
Dinh, C. T. et al. CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface. Science 360, 783–787 (2018).
Koh, J. H. et al. Facile CO2 electro-reduction to formate via oxygen bidentate intermediate stabilized by high-index planes of Bi dendrite catalyst. ACS Catal. 7, 5071–5077 (2017).
Li, F. et al. Towards a better Sn: efficient electrocatalytic reduction of CO2 to formate by Sn/SnS2 derived from SnS2 nanosheets. Nano Energy 31, 270–277 (2017).
Yang, H. et al. Selective CO2 reduction on 2D mesoporous Bi nanosheets. Adv. Energy Mater. 8, 1801536 (2018).
Han, N. et al. Ultrathin bismuth nanosheets from in situ topotactic transformation for selective electrocatalytic CO2 reduction to formate. Nat. Commun. 9, 1320 (2018).
Yang, Q. et al. Novel Bi-doped amorphous SnOx nanoshells for efficient electrochemical CO2 reduction into formate at low overpotentials. Adv. Mater. 32, 2002822 (2020).
García de Arquer, F. P. et al. CO2 electrolysis to multicarbon products at activities greater than 1 A cm−2. Science 367, 661–666 (2020).
Yang, K., Kas, R. & Smith, W. A. In situ infrared spectroscopy reveals persistent alkalinity near electrode surfaces during CO2 electroreduction. J. Am. Chem. Soc. 141, 15891–15900 (2019).
Wang, G., Pan, J., Jiang, S. P. & Yang, H. Gas phase electrochemical conversion of humidified CO2 to CO and H2 on proton-exchange and alkaline anion-exchange membrane fuel cell reactors. J. CO2 Util. 23, 152–158 (2018).
Zheng, X. et al. Sulfur-modulated tin sites enable highly selective electrochemical reduction of CO2 to formate. Joule 1, 794–805 (2017).
Garcia de Arquer, F. P. et al. 2D metal oxyhalide-derived catalysts for efficient CO2 electroreduction. Adv. Mater. 30, e1802858 (2018).
Kresse, G. & Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B 49, 14251–14269 (1994).
Acknowledgements
This work was supported financially by the National Key R&D Program of China (No. 2020YFA0406102), the National Natural Science Foundation of China (grant number 91963121), the Natural Science Foundation of Jiangsu Province (Grant No. BK20190056), and the Frontiers Science Center for Critical Earth Material Cycling of Nanjing University. Parts of the calculations were performed using computational resources on an IBM Blade cluster system from the High-Performance Computing Center (HPCC) of Nanjing University. We thank Yimeng Min, Christine Gabardo, and Ziyun Wang for discussions during the study.
Author information
Authors and Affiliations
Contributions
M.Z. and E.H.S. supervised the project. M.Z. conceived the idea and designed the experiments. L.L. conducted the synthesis, characterizations, and flow-cell tests. L.L., A.O., F.P.G.A., Ming.Z., J.Z., H.J., C.W. and M.Z. discussed the experiment results. A.O., F.P.G.A. and D. S. designed and carried out the MEA tests. S.G., W.W., and H.D. carried out the DFT calculations. S.G., L.L., M.Z., W. W. and H.D. discussed the DFT results. M.Z., A.O., F.P.G.A, L.L. and E.H.S. wrote the manuscript. All authors discussed the results and assisted during manuscript preparation.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks the other anonymous reviewers for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, L., Ozden, A., Guo, S. et al. Stable, active CO2 reduction to formate via redox-modulated stabilization of active sites. Nat Commun 12, 5223 (2021). https://doi.org/10.1038/s41467-021-25573-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-021-25573-9
This article is cited by
-
Spatial decoupling of bromide-mediated process boosts propylene oxide electrosynthesis
Nature Communications (2024)
-
A critical review of operating stability issues in electrochemical CO2 reduction
Science China Materials (2024)
-
Electronic structural engineering of bimetallic Bi-Cu alloying nanosheet for highly-efficient CO2 electroreduction and Zn-CO2 batteries
Nano Research (2024)
-
Surface iodine and pyrenyl-graphdiyne co-modified Bi catalysts for highly efficient CO2 electroreduction in acidic electrolyte
Nano Research (2024)
-
Progress of CO2 Electrochemical Methanation Using a Membrane Electrode Assembly
Electrocatalysis (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
