
Abstract
Li-ion-conducting chloride solid electrolytes receive considerable attention due to their physicochemical characteristics such as high ionic conductivity, deformability and oxidative stability. However, the raw materials are expensive, and large-scale use of this class of inorganic superionic conductors seems unlikely. Here, a cost-effective chloride solid electrolyte, Li2ZrCl6, is reported. Its raw materials are several orders of magnitude cheaper than those for the state-of-the-art chloride solid electrolytes, but high ionic conductivity (0.81 mS cm–1 at room temperature), deformability, and compatibility with 4V-class cathodes are still simultaneously achieved in Li2ZrCl6. Moreover, Li2ZrCl6 demonstrates a humidity tolerance with no sign of moisture uptake or conductivity degradation after exposure to an atmosphere with 5% relative humidity. By combining Li2ZrCl6 with the Li-In anode and the single-crystal LiNi0.8Mn0.1Co0.1O2 cathode, we report a room-temperature all-solid-state cell with a stable specific capacity of about 150 mAh g–1 for 200 cycles at 200 mA g–1.
Similar content being viewed by others

Introduction
Identifying an appropriate solid electrolyte is crucial for enabling the safe, energy-dense all-solid-state Li batteries1,2,3,4,5,6. Recently, chloride superionic conductors were raised as an additional class of promising solid electrolytes7,8. They are ionically conductive and easily deformable like sulfides, but in the meanwhile are not plagued by the poor oxidative stability of sulfides9,10,11,12,13. With Cl being more electronegative than S14,15, the oxidation potential of chlorides is generally much higher (comparable to oxides)16, leading to an excellent compatibility with 4V-class cathodes7,8. The rare combination of these appealing characteristics rapidly attracted intensive research interest8.
Nevertheless, for all the chloride solid electrolytes reported so far, the cost of raw materials is still too high to allow for an efficient industrial application. Unlike sulfide and oxide solid electrolytes, the major materials cost of chloride systems does not lie in the raw materials that contribute Li, but in the non-Li-containing ones. Using a method developed by Hart et al.17, we calculated and compared the bulk prices of Li-containing compounds needed to synthesize sulfide, oxide, and chloride solid electrolytes (Supplementary Table 1); the laboratory-scale prices used to infer these bulk prices are also specified in Supplementary Table 2. According to such cost analysis, Li2S and Li2O are both quite expensive; for certain oxide solid electrolytes this cost can be greatly alleviated by using alternate Li-containing raw materials like Li2CO3, LiNO3, LiOH, or their hydrates18,19,20, which could be as cheap as $10.73/kg, whereas the Li2S with a high price of $654.18/kg seems irreplaceable for nearly all the sulfide solid electrolytes. In contrast, the price of LiCl is only $5.88/kg, much lower than all the Li-containing raw materials mentioned above. From this perspective, chloride solid electrolytes seem quite promising in terms of cost-effectiveness. Unfortunately, this advantage no longer exists when the non-Li-containing raw materials are taken into account. The present chloride solid electrolyte systems can be divided into two types: Li3MCl67,14,21,22,23,24,25,26 and Li2M2/3Cl427 (M is a non-Li element). Elements that had been acting as M in literature are Y7,14, Tb-Lu21, Sc25,27, and In22,23, all of which show very low abundance in Earth’s crust, ranging from 0.25 ppm (In) to 33 ppm (Y)28. Such values are way below those of the non-Li cationic elements in successfully commercialized solid electrolytes like Li7P3S1110,11 (P: 1050 ppm28) and Li1.3Al0.3Ti1.7(PO4)329 (Al: 82,300 ppm, Ti: 5650 ppm28). Consequently, as indicated by the bulk prices in Supplementary Table 3 (inferred from the laboratory-scale prices listed in Supplementary Table 4), the costs for most non-Li-containing chlorides needed to synthesize Li3MCl6 or Li2M2/3Cl4 are way above $1000/kg. Although the corresponding hydrates are often relatively cheap (their bulk prices and the laboratory-scale prices used for estimation are listed in Supplementary Tables 5 and 6, respectively), they cannot be used directly to synthesize any of the reported chloride solid electrolytes other than Li3InCl68, and dehydrating them before synthesis would likely make the total cost no lower than that of anhydrous chlorides. These expensive non-Li-containing raw materials completely offset the cost-effectiveness of LiCl ($5.88/kg). In order for all-solid-state batteries to be competitive against existing technologies, it has been proposed that the cost of solid electrolytes needs to be lower than $10/m2 (note that this includes both raw-material cost and synthesis/processing cost)30. If the solid-electrolyte layer is assumed to exhibit a thickness of 50 μm (which is already quite challenging for inorganic systems31), even Li3YCl6, i.e., the solid electrolyte formed by the least expensive chloride in Supplementary Table 3, exhibits a raw-material cost of $23.05/m2. This is already more than twice the $10/m2 threshold, not to mention the synthesis/processing cost that has not been taken into account here. Therefore, in terms of cost-effectiveness, the present chloride solid electrolytes are far from satisfactory.
This bottleneck arises from the fact that only 3+ cations may act as M in both Li3MCl6 and Li2M2/3Cl4 due to the charge balance requirement7,14,21,22,23,24,25,26. Although cations with other valences may be introduced through aliovalent doping32,33,34, a considerable fraction of M still has to be 3+ ones. Then, after excluding the trivalent cations not sufficiently stable at 3+ and those too large or too small to fit in the crystal structure, the remaining options are almost confined to the aforementioned expensive elements, i.e., Y, Tb-Lu, Sc, and In. This is why the 3+ valence requirement for M makes it very difficult, if not impossible, to lower the cost of Li3MCl6 and Li2M2/3Cl4. In fact, beyond the cost issue, such valence requirement is limiting improvement in many other aspects too. For example, soft acids like Sn4+ and As5+ may result in compounds that are less likely to undergo hydrolysis in humid atmosphere35,36,37 (although this does not necessarily prevent the material from becoming a hydrate with water molecules fitting into the crystal structure). Ge4+ and Sn4+ with higher electronegativity (2.02 and 1.72, respectively, Allred-Rochow scale38) than the aforementioned expensive elements (1.08–1.49) are believed to result in lower electron density on the anions; if other important factors such as the overall crystal structure and anionic species are the same, this effect would supposedly weaken the Coulombic attraction between anions and Li+, enabling more facile ionic transport39,40. Regardless, the 3+ valence requirement allows none of them to dominate the M site. Unless a Li-M-Cl system with M being a non-trivalent cation emerges, exploration in many directions would be quite difficult; lowering the materials cost is a particularly important and formidable one among them.
In this work, a cost-effective Li-M-Cl solid electrolyte is successfully constructed using a 4+ cation as M. Described by the chemical formula Li2ZrCl6, this solid electrolyte is synthesized using ZrCl4, which is orders of magnitude cheaper than the raw materials of all the present chloride solid electrolytes (Fig. 1). The raw-material cost of Li2ZrCl6 at 50 μm thickness is found to be $1.38/m2, which is much lower than that for Li3YCl6 ($23.05/m2), i.e., the cheapest chloride system in literature, and is also far below the $10/m2 threshold for cost-competitive all-solid-state batteries30. With such advantage in cost-effectiveness, Li2ZrCl6 not only preserves the desirable properties of other chloride solid electrolytes, e.g., high ionic conductivity (0.81 mS cm–1), deformability, excellent compatibility with 4V-class cathodes, etc., but also displays a unique stability against moisture. After being exposed to the atmosphere with 5% relative humidity, Li2ZrCl6 undergoes neither moisture uptake nor conductivity degradation, while Li3InCl6, i.e., the chloride system considered most humidity tolerant in literature8,41, partially becomes Li3InCl6·2H2O and shows significantly decreased conductivity under the same condition.

a Estimated unit prices of different chlorides when purchased in the quantity of 1000 kg. These chemicals are the raw materials needed to synthesize different chloride solid electrolytes. The bulk prices plotted here are also listed in Supplementary Table 3, and the laboratory-scale prices used to estimate these bulk prices are provided in Supplementary Table 4. b Abundance of the corresponding non-Li element in Earth’s crust28.
Results and discussion
Li2ZrCl6 (LZC) was mechanochemically synthesized from a stoichiometric mixture of LiCl and ZrCl4. At 25 °C, the as-milled LZC was found to exhibit a drastically different ionic conductivity from that annealed at elevated temperatures. As indicated by the electrochemical impedance spectroscopy (EIS) measurement (Fig. 2a), the as-milled LZC shows a high ionic conductivity of 8.08 × 10–4 S cm–1 at 25 °C. However, annealing at 350 °C for 5 h decreases the ionic conductivity at 25 °C by two orders of magnitude, which reaches 5.81 × 10–6 S cm–1 as indicated in Fig. 2b (the Bode plots corresponding to these Nyquist plots are shown in Supplementary Fig. 1). With the annealing temperature further increased to 450 °C, the material begins to melt. For both the as-milled and 350 °C-annealed LZC, the electronic conductivities determined by the Hebb–Wagner polarization method7,42 (9.22 × 10–8 S cm–1 and 3.59 × 10–8 S cm–1 for the as-milled and 350 °C-annealed LZC, respectively, as shown in Fig. 2c) are much lower than the ionic conductivities mentioned above. In particular, the electronic conductivity of the as-milled LZC is about four orders of magnitude lower than its ionic conductivity, which should allow us to safely consider this material as a Li-ion conductor for Li-based batteries. In addition to the room-temperature ionic conductivities, the activation energies were evaluated through the Arrhenius plot (Fig. 2d). Consistent with the measured ionic conductivities, the activation energy of the as-milled LZC (0.35 eV) is much lower than that of the 350 °C-annealed one (0.50 eV), and is comparable with other high-performance chloride solid electrolytes like Li3YCl6 (0.40 eV7) and Li3InCl6 (0.347 eV22). It should be pointed out that all the conductivity measurements described above were performed directly on cold-pressed pellets, and no semicircle, associated with the presence of grain boundaries in the solid electrolyte, is identified in the Nyquist plot of Fig. 2a. This result seems to suggest that the as-milled LZC might be easily deformable and could favor the formation of an intimate electrode-electrolyte contact in all-solid-state cells. In addition to LZC, preliminary attempts were made to explore two similar halides, Li2ZrF6 and Li2ZrBr6. The phase-pure Li2ZrF6 samples can be successfully synthesized using the similar method as that for LZC (Supplementary Fig. 2). However, as shown in Supplementary Fig. 3, both the as-milled and the annealed Li2ZrF6 exhibit very low ionic conductivities of only 10−9−10−8 S cm−1 and electronic conductivities quite close to these values. Therefore, they do not seem to be ideal solid electrolytes for Li batteries. As for Li2ZrBr6, we calculated its electrochemical stability using the established scheme16,43,44 based on Materials Project45, and found that the oxidation potential is only 3.14 V vs. Li/Li+ (Supplementary Fig. 4). That is, this bromide, unlike the chloride solid electrolytes, seems unstable against the 4 V-class cathodes. Consequently, the investigation below is still focused on LZC.

a, b Nyquist plots of the as-milled (a) and 350 °C-annealed LZC (b) at 25 °C. Note that the data here are not plotted in the unit of resistance, but in the unit of resistivity, i.e., the reciprocal of conductivity, which is calculated using the resistance and sample dimension. The diameters of the pellets used for measurement are 10.8 mm, while the pellet thicknesses are 0.90 and 0.92 mm for the as-milled and 350 °C-annealed LZC, respectively. c The transient current behavior under DC bias for the as-milled and 350 °C-annealed LZC with stainless-steel (SS) electrodes. Note that the data of the as-milled LZC here is vertically offset by 5 µA cm−2. d Arrhenius plots of the as-milled and 350 °C-annealed LZC.
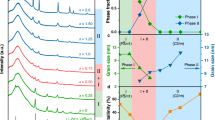
While LZC inherits the excellent deformability and ionic conductivity of the state-of-the-art chloride solid electrolytes, the evolution of its crystal structure with processing conditions is quite different from these materials. The X-ray diffraction (XRD) patterns of the as-milled and 350 °C-annealed LZC are displayed in Fig. 3a. The former shows weak and diffuse reflections, suggesting that the intense planetary mill led to a low crystallinity. The Bragg reflections of the as-milled LZC very much resemble those of Li3YCl6 with the P\(\overline{3}\)m1 symmetry7,14. This phase is referred to as α-LZC below. In contrast, the 350 °C-annealed LZC, besides showing improved crystallinity and sharper reflections, exhibits a completely different crystal structure. The reflections appear to match well with those of Li3InCl6 crystalizing in the C2/m space group22,23. This phase is referred to as β-LZC hereafter. Clearly, unlike other chloride solid electrolytes, LZC undergoes changes not only in crystallinity, but also in crystal structures after being annealed at high temperatures.

a XRD patterns of the as-milled and 350 °C-annealed LZC. The broad hump below 30° comes from the Kapton film that was used to prevent air exposure. The Bragg positions of Li3InCl6 and Li3YCl6 correspond to the structures reported in references 22 and 7, respectively. b Two-dimensional intensity color map of temperature-dependent NPD patterns for the as-milled LZC. The data were obtained in-situ between 27 and 427 °C.
The phase evolution of LZC was investigated in further detail by in-situ neutron powder diffraction (NPD). The measurement was performed on the as-milled LZC between 27 and 427 °C in argon atmosphere. As shown in Fig. 3b, at first the reflections corresponding to α-LZC persisted with increasing temperature. Starting from 277 °C, the β-LZC reflections (representative ones include 29.4°, 34.1°, 48.7°, and 58°) emerged and grew stronger in the consumption of the α-LZC reflection intensities. The α- and β-LZC phases coexisted between 277 and 350 °C. At higher temperatures, β-LZC is the only phase identified in the NPD pattern. Considering that the 350 °C-annealed (and then furnace-cooled) material still shows pure β-LZC phase (Fig. 3a) instead of becoming α-LZC again, the latter should likely be a meta-stable phase resulting from the intense planetary mill. This is drastically different from other chloride solid electrolytes, where planetary mill may only change the crystallinity, but not the crystal structure7,15,23.
Since the as-milled and 350 °C-annealed LZC differ in not only the crystallinity but also the crystal structure, the contribution of these two factors to the ionic conductivity needs to be discussed separately. To begin with, the influence of crystal structure was investigated through Rietveld refinement. The structure of the as-milled LZC is determined directly from its NPD data. The analysis of the 350 °C-annealed LZC is based on the XRD pattern, but the initial structure used for this refinement is obtained from the NPD pattern of the as-milled sample at 427 °C (Supplementary Fig. 5), which shows the β-LZC structure just like the 350 °C-annealed LZC at room temperature. In this way, only the minor structural variations caused by the different temperatures and processing conditions need to be adjusted when refining the XRD pattern. The initial models used for refining the α- and β-LZC phases are the crystal structures of Li3YCl6 (space group P\(\overline{3}\)m1) and that of Li3InCl6 (space group C2/m), respectively, with necessary adjustment of Li contents and the non-Li cation. During the refinement, compositional constraints were applied to ensure that the overall Li:Zr:Cl ratio remains as 2:1:6, while the occupancy of each atomic site was still allowed to vary freely. Such refinement shows excellent agreement between the experimental and calculated data (Supplementary Figs. 5–7), suggesting that α- and β-LZC are indeed structurally similar with Li3YCl6 and Li3InCl6, respectively. Using the theoretical density calculated from the refined structure in Supplementary Table 7 (2.561 g cm–3) and the prices listed in Supplementary Tables 1 and 3, the raw-material cost of LZC can be estimated as $1.38/m2 at 50 μm thickness. This is much more affordable even than the chloride system with the lowest raw-material cost in literature, Li3YCl6 ($23.05/m2), and far below the $10/m2 threshold for ensuring the competitiveness of all-solid-state batteries30. Beyond such cost-effectiveness, the difference between LZC and the state-of-the-art isostructural chloride systems mainly lies in two aspects. First of all, the lattice parameters of LZC (Supplementary Tables 7 and 8) are generally smaller. This is consistent with the fact that Zr4+ is smaller than both Y3+ and In3+ at the same coordination number46. Secondly, the site occupancies are very different too. For example, the 2a and 4g sites (the Zr ones) in the 350 °C-annealed LZC are much less occupied than those in Li3InCl622. In addition, while the two Li sites (6g and 6h) in Li3YCl6 are both well occupied (occupancy > 0.50)7, the Li ions in the as-milled LZC seem to preferentially reside in the 6h site. In order to probe the influence of these structural characteristics on ionic transport, we calculated the migration pathways and the associated energy barriers using a bond valence site energy (BVSE) method developed by Adams et al.47,48. For the α-LZC structure (schematically shown in Fig. 4a), the [Li1–Li2–Li1] chain along the [001] direction was found to be the most favorable 1D migration pathway (marked in red in Fig. 4b–d). In the meanwhile, Li ions may also migrate along [Li1–i1–Li1-i2-Li1] and [Li2–i3–Li2] (marked in blue and green, respectively, in Fig. 4b–d), which interconnect the [Li1–Li2–Li1] chains to form a 3D percolating network with an effective migration barrier of 0.803 eV (Fig. 4d). For the β-LZC structure (schematically shown in Fig. 4e), the pathways within the a–b plane (marked in red in Fig. 4f–h) are relatively favorable for Li-ion transport, and exhibit an effective barrier of 0.809 eV (Fig. 4h). In comparison, the barrier for migration between two neighboring a–b planes (marked in blue in Fig. 4g, h) are much higher (2.987 eV). It should be emphasized that such results do not necessarily entail a 2D ionic transport. As a matter of fact, the Zr sites hindering the Li-ion migration between two a–b planes are not fully occupied (Supplementary Table 8). In BVSE analysis, their contribution is overly simplified as an average potential that hinders Li-ion migration at each Zr site, no matter whether the particular Zr site is occupied or not. However, in reality the unoccupied ones among these Zr sites could still allow for the local occurrence of Li-ion migration between the neighboring a-b planes. If such migration is too sluggish or too infrequent to make meaningful contribution to the overall ionic transport, the highest barrier of 0.809 eV within the a–b plane should be regarded as the effective migration barrier of the structure48. If not, β-LZC would be a 3D ionic conductor, and there might exist more facile percolation pathways than those within the a–b plane, making the effective migration barrier of the entire structure lower than 0.809 eV. As such, the 0.809 eV barrier resulting from BVSE analysis should not be directly considered as the effective migration barrier for β-LZC, but only its highest possible value.

a, e The crystal structures of α-LZC (a) and β-LZC (e) superimposed with the Li-ion potential map. b, c, f, g Li-ion migration pathways of α-LZC (b,c) and β-LZC (f, g). d, h Energy profiles of the migration pathways in α-LZC (d) and β-LZC (h). Each pathway in b–c and f–g corresponds to the energy profile of the same color in d and h, respectively. The crystal structures and potential isosurfaces are visualized using VESTA56.
The effective migration barriers obtained this way provide an important clue about the origin for the high ionic conductivity in the as-milled LZC; admittedly, BVSE analysis cannot be expected to precisely repeat the experimentally measured activation energy, so the discussion here focuses only on the calculated migration barriers of different structures. To begin with, it should first be noted that BVSE analysis only describes ionic transport within crystals, but cannot account for contribution from factors beyond the ideal, periodic atomic arrangements, such as defects, surface structure, particle size, strain, amorphous phase, etc. (they are referred to as nonperiodic features below). The 350 °C-annealed LZC is dominated by the highly crystalline phase, so its ionic transport can be better described by BVSE. In contrast, the as-milled LZC contains not only the crystalline phase, but also significant amount of nonperiodic features due to the intense ball milling. According to BVSE analysis, its crystalline part merely allows for a Li-ion migration comparable to or even slower than that in the 350 °C-annealed LZC (the effective migration barrier of the former, 0.803 eV, is close to the highest possible effective migration barrier of the latter, 0.809 eV), and obviously cannot account for the orders of magnitude higher conductivity in the as-milled LZC. As such, the observed fast ionic transport may only arise from the nonperiodic features created by planetary mill. In order to verify this scenario, we annealed the as-milled LZC at 215 °C for 5 h. Such treatment leaves the crystal structure unchanged, but suppresses the metastable nonperiodic features to a great extent, as reflected by the sharper, stronger reflections (Supplementary Fig. 8). With the amount of nonperiodic features reduced this way, the ionic conductivity decreases from 8.08 × 10–4 to 3.22 × 10–5 S cm–1, and the activation energy increases from 0.35 to 0.43 eV (Supplementary Figs. 9 and 10). Therefore, the high ionic conductivity of the as-milled LZC should primarily originate from the noperiodic features induced by the intense ball milling; in fact, similar phenomena had also been observed in other solid electrolytes7,8,24,49. It should be pointed out that the results here cannot identify which specific nonperiodic feature is mainly responsible for the high ionic conductivity, because it is very difficult, if not impossible, to separate the influence of each nonperiodic feature on the diffraction patterns. The achievement of this goal demands systematic investigation using advanced characterization methods with ultrahigh spatial resolution, such as aberration-corrected transmission electron microscopy. If the microscopic origin of the facile ionic transport can be precisely identified, an ionic conductivity even higher than that reported here (0.81 mS cm–1) might also be realized through a rational optimization.
With the as-milled LZC confirmed to be highly conductive and deformable, its electrochemical stability was evaluated both theoretically and experimentally. Using the established scheme16,43,44 based on Materials Project45, the electrochemical stability window (ESW) of LZC was calculated to be 1.75–4.25 V vs. Li/Li+ (Supplementary Fig. 11); the reductions originate from Zr4+ becoming Zr metal in multiple steps, while the oxidation happens through Cl– becoming Cl2. The ESW predicted above is verified by the cyclic voltammetry (CV) measurement, which was performed using a Li | Li7P3S11-LZC | LZC + C cell; the LZC + C part in such a cell contains 70 wt% of as-milled LZC and 30 wt% of carbon to make the redox peaks more easily detectable, and the purpose of incorporating a Li7P3S11 layer is to prevent the reaction between LZC and Li. Consistent with the calculated ESW, the CV measurement (Supplementary Fig. 12) reveals multiple reduction peaks below 2 V, along with one oxidation peak above 4 V. These corroborating results convey important information about the electrodes that can be contacted directly with LZC in all-solid-state cells. First of all, the non-zero reduction potential of LZC indicates that this material is unstable against Li reduction. Besides, considering that the reduction products are suggested to contain both the electronic conductor (Zr) and ionic conductor (LiCl), the reaction between LZC and Li metal may not be self-limited, but would likely proceed continuously, similar to the situation for Li3YCl6 and Li3InCl650,51. This scenario is supported by the electrochemical measurements carried out in the Li | LZC | Li symmetric cell, whose Li stripping/plating voltages increase constantly without any sign of stabilizing and reach the instrument limit of 5 V in only 88 h (Supplementary Fig. 13a). Besides, LZC would significantly discolor after being contacted directly with Li (Supplementary Fig. 13b−c), further confirming the severe reaction between these two materials. Consequently, like other chloride solid electrolytes50,51, LZC should not be in direct contact with Li in all-solid-state cells. Nevertheless, this material does exhibit a rather high oxidation potential beyond 4 V, which could entail a good compatibility with the 4 V-class cathodes. This promising characteristic warrants more detailed investigation of LZC in all-solid-state cells.
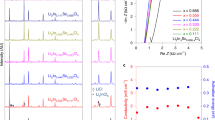
To this end, the as-milled LZC was integrated as the solid electrolyte into all-solid-state cells with LiCoO2 (LCO) or single-crystal LiNi0.8Mn0.1Co0.1O2 particles (scNMC811) powder as the cathode and Li-In alloy as the anode; to prevent the reaction between the anode and LZC, they are separated by a thin layer of Li6PS5Cl (abbreviated as LPSCl below), which was applied to the surface of the LZC layer before attaching the anode (further details in Methods). Such a cell was fabricated simply by cold pressing, and bare cathode particles were in direct contact with the as-milled LZC, without any extra coating that is typically needed for sulfide solid electrolytes. When cycled at 0.1 C (1 C = 140 mA g–1) between 1.9 and 3.6 V, the cell with LCO cathode (referred to as the LCO/LZC cell below) showed an initial Coulombic efficiency of 97.9% and a discharge capacity of 137 mAh g–1 (Fig. 5a), comparable to those delivered by similar cells constructed using Li3InCl623 (92% and 127 mAh g–1) and Li3YCl67 (94.8% and 118 mAh g–1). The rate capability of the LCO/LZC cell is shown in Fig. 5b and c. When the rate increased step-by-step from 0.2 C to 0.5 C, the capacity only dropped slightly; the average capacities at 0.2 C, 0.33 C, and 0.5 C are 136, 130, and 124 mAh g–1, respectively. Even at 2 C, a capacity of 68 mAh g–1 on average was still retained. Beyond the rate capability, the cycling stability is also examined. The LCO/LZC cell maintained a Coulombic efficiency of 99.9% and a discharge capacity of 114 mAh g–1 after 100 cycles at 0.5 C. The ex-situ SEM observation suggests that long-term cycling barely compromised the contact between LCO and LZC (Supplementary Fig. 14), while the energy dispersive spectroscopy (EDS) mapping (Supplementary Fig. 14) and XRD (Supplementary Fig. 15) conducted post-mortem to the composite cathodes after 100 cycles do not indicate any reaction or inter-diffusion happening between the two aforementioned materials either. When LCO was replaced by scNMC811 in the all-solid-state cell (this cell is referred to as the scNMC811/LZC cell below), comparable performances were still observed (Fig. 6). Being cycled at 0.1 C (1 C = 200 mA g–1) between 2.2 and 3.8 V, the cell delivered an initial Coulombic efficiency of 90.3% and a discharge capacity of 181 mAh g–1 (Fig. 6a). The rate capability of the scNMC811/LZC cell is shown in Fig. 6b and c; the average discharge capacities at 0.2 and 2 C are 176 and 96 mAh g–1, respectively. The long-term cycling data is displayed in Fig. 6d, which demonstrates a 99.9% Coulombic efficiency and a 149 mAh g–1 discharge capacity after 200 cycles at 1 C. According to the ex-situ SEM (Supplementary Fig. 16) and XRD (Supplementary Fig. 17), the positive electrode and solid electrolyte were still intimately contacted with each other without noticeable reaction or inter-diffusion after prolonged cycling.

a The initial charge/discharge curves at 0.1 C, with the Coulombic efficiency ηCoulomb denoted. b,c Rate capability at 0.2, 0.33, 0.5, 1 and 2 C. d Long-term cycling performance at 0.5 C.

a The initial charge/discharge curves at 0.1 C, with the Coulombic efficiency ηCoulomb denoted. b,c Rate capability at 0.2, 0.33, 0.5, 1 and 2 C. d Long-term cycling performance at 1 C.
In addition to these appealing characteristics, another interesting phenomenon about LZC is its variation with stoichiometry. To study this effect, two off-stoichiometric materials, LiZrCl5 and Li3ZrCl7, were synthesized using the same conditions as the as-milled Li2ZrCl6. In spite of such large composition variation, these two off-stoichiometric materials remained phase-pure with the α-LZC structure (Supplementary Fig. 18). Besides, their microstructures (Supplementary Figs. 19–21) and calculated ESWs (Supplementary Fig. 22) are also nearly identical with those for the stoichiometric Li2ZrCl6. However, LiZrCl5 and Li3ZrCl7 exhibit inferior electrochemical properties compared to Li2ZrCl6. First of all, they are less stable in contact with Li metal electrode; as shown in Supplementary Fig. 23, the Li | LiZrCl5 | Li and Li | Li3ZrCl7 | Li cells reached 5 V Li stripping/plating overpotential in a much shorter time (14.7 h and 48.4 h, respectively) than the Li | Li2ZrCl6 | Li symmetric cell (88.0 h). Secondly, according to the EIS measurement (Supplementary Fig. 24), the off-stoichiometric materials possess lower ionic conductivity (0.15 and 0.27 mS cm−1 for LiZrCl5 and Li3ZrCl7, respectively) than the as-milled Li2ZrCl6 (0.81 mS cm−1). Last but not least, the all-solid-state cell formed by Li2ZrCl6 (cell configuration Li-In | LPSCl-Li2ZrCl6 | LCO) deliver higher capacity than those formed by LiZrCl5 and Li3ZrCl7, although all three of them exhibit satisfactory cycling stability (Supplementary Fig. 25). Generally speaking, despite the fact that the material can remain phase-pure in a relatively large stoichiometry range, Li2ZrCl6 is still the one with the optimal electrochemical performances.
Beyond the desirable characteristics shared by most chloride solid electrolytes, LZC also shows a unique advantage in humidity tolerance. The chloride systems reported so far are generally sensitive to moisture. The most humidity tolerant one among them, i.e., Li3InCl6, will still degenerate even in atmosphere with a very low relative humidity of 1%; its humidity tolerance is in fact a good recoverability after absorbing moisture23. In contrast, LZC is truly moisture resistant at relative humidity even above 1%. To demonstrate this point, the as-milled LZC powder was exposed to nitrogen with 5% relative humidity for 24 h. For comparison, Li3InCl6 powder of the same mass was placed alongside LZC within the same container for the same period of time. According to the XRD patterns (Fig. 7a), the as-milled LZC remained unchanged after such treatment, but Li3InCl6 partially became Li3InCl6·2H2O. This result is also corroborated by the X-ray photoelectron spectroscopy (XPS) data, where neither the Zr-3d nor the Cl-2p spectra of the as-milled LZC exhibits any noticeable variation after humidity exposure (Fig. 7b and c). Therefore, the change caused by the moisture, if any, is too trivial to be detected by either XRD or XPS. Consistent with these observations, the as-milled LZC very well preserved its high conductivity after the humidity treatment described above (Fig. 7d), while Li3InCl6 underwent a conductivity degradation by nearly an order of magnitude (Fig. 7e). The slight difference between the Nyquist plots of LZC before and after its humidity exposure should result from the measurement error of pellet dimensions; with the moisture sensitivity tests performed to loose powders, the EIS measurement before and after humidity exposure cannot be conducted using the same pellet, whereas the conductivities determined using different pellets would inevitably vary slightly due to the measurement error of thicknesses and diameters. Therefore, the conductivity of the as-milled LZC should be considered largely unchanged after humidity exposure. Clearly, in the atmosphere with 5% relative humidity, LZC shows better stability than Li3InCl6, even though the latter is considered the most humidity tolerant chloride system in literature8,41. Since LZC is truly moisture resistant and thus does not need to be recovered after humidity exposure, the environment for its manufacturing and storage does not necessarily possess the low dew point that is typically required by sulfide solid electrolytes. Considering that this material is also quite cost-competitive (Fig. 1), it seems very suitable for large-scale industrial production. The advantages of LZC over state-of-the-art chlorides essentially arise from the fact that a tetravalent cation Zr4+, instead of the trivalent ones forming Li3MCl6 or Li2M2/3Cl4, is acting as the non-Li cation. If 4+ cations other than Zr4+ are acting as M in Li2MCl6, chloride systems showing other interesting properties may very likely arise too.

a XRD patterns of the as-milled LZC and Li3InCl6 before and after being exposed to the atmosphere with 5% relative humidity. b, c Zr-3d (b) and Cl-2p (c) X-ray photoelectron spectra of the as-milled LZC before and after being exposed to the atmosphere with 5% relative humidity. d, e Nyquist plots of the as-milled LZC (d) and Li3InCl6 (e) before and after being exposed to the atmosphere with 5% relative humidity. Note that the data here are not plotted in the unit of resistance, but in the unit of resistivity, i.e., the reciprocal of conductivity, which is calculated using the resistance and sample dimension. The diameters of the pellets used for measurement are all 10.8 mm. The pellet thicknesses are 1.12, 1.10, 1.50, and 1.00 mm for the non-exposed as-milled LZC, humidity-exposed as-milled LZC, non-exposed Li3InCl6, and humidity-exposed Li3InCl6, respectively.
In summary, Li2ZrCl6, a cost-effective but high-performance chloride solid electrolyte, is reported. Its raw materials are orders of magnitude cheaper than those for other chloride solid electrolytes, which makes Li2ZrCl6 presently the only chloride solid electrolyte with raw-material cost ($1.38/m2) below $10/m2 (the threshold that ensures the competitiveness of all-solid-state batteries30). In the meanwhile, the electrochemical performances of the state-of-the-art chloride solid electrolytes are still well preserved. The Li2ZrCl6 material directly produced by planetary mill shows the P\(\overline{3}\)m1 symmetry, and it undergoes a phase transition into the C2/m structure between 277 and 350 °C. Although neither crystal structure seems to favour Li-ion transport according to the BVSE analysis, the as-milled Li2ZrCl6 was found to possess a desirable room-temperature ionic conductivity of 0.81 mS cm−1 due to the nonperiodic features introduced by the intense milling. Besides, this material is easily deformable and compatible with the 4V-class cathode materials. Consequently, without any extra cathode coating that are typically needed for sulfide solid electrolytes, all-solid-state cells with a direct contact between Li2ZrCl6 and the 4V-class cathode particles can deliver initial Coulombic efficiencies of 97.9% and 90.3% for LiCoO2 and scNMC811, respectively. Beyond these common advantages of chloride solid electrolytes, Li2ZrCl6 also displays humidity tolerance. Instead of being well recoverable after humidity exposure but still moisture sensitive (like Li3InCl6), Li2ZrCl6 is truly moisture resistant at 5% relative humidity. Such humidity tolerance, along with the cost-effectiveness, removes two major obstacles to the industrial application of chloride solid electrolytes.
Methods
Cost analysis
The prices of the chemicals in bulk purchase were estimated from the laboratory-scale prices using the method proposed by Hart et al.17. This method relies on the fact that the unit price, P, and the purchase quantity, Q, of the chemicals satisfy this general relationship:
where a and b are constants for a given chemical. If this equation is written in the logarithmic form, it becomes
Therefore, based on a series of unit prices P and quantities Q for the laboratory-scale purchase, the unit price for the purchase in bulk quantity (the amount of 1000 kg was used for the calculations here) can be estimated using Eq. (2) through linear extrapolation, and the reliability of this estimation is reflected by the absolute value of the linear correlation coefficient, |r | , between log10P and log10Q (|r | closer to unity means higher reliability). The laboratory-scale prices used for such estimation are mostly taken from the official website of Alfa Aesar; the only exception is TmCl3, whose price is taken from Fisher Scientific. Following the advice by Hart et al.17, the laboratory-scale chemicals with the lowest purity were selected for the price estimation. More details about these chemicals, such as the stock number, purchase quantity, and purity, are provided in Supplementary Tables 2, 4 and 6.
Materials synthesis
Li2ZrCl6 were synthesized from LiCl (Alfa Aesar, 99.9%) and ZrCl4 (Acros Organics BVBA, 98%). The stoichiometric amount of the starting materials were mechanochemically milled in the WC pot using WC balls (5 mm diameter) with a ball-to-powder mass ratio of 10:1. The milling was performed in a planetary mill (FRITSCH, Pulverisette 7 premium line) at 500 rpm for 45 h. Such milling directly yields the as-milled LZC in the main text, while annealing this powder at different temperatures for 5 h leads to other LZC materials discussed in the present study. In order to prevent air exposure, all the annealing experiments were performed with the sample sealed in a vacuum quartz tube. The Li3InCl6 material used for humidity tolerance test was synthesized by the same method with the annealing temperature of 350 °C; the starting materials were LiCl (Alfa Aesar, 99.9%) and InCl3 (Alfa Aesar, 99.99%). The Li2ZrF6 materials were synthesized by the same method too with the annealing temperature of 500 °C; LiF (Alfa Aesar, 99.98%) and ZrF4 (Sigma-Aldrich, 99.9%) were used as the starting materials.
Structural characterization
The XRD was performed using a Rigaku Ultima IV diffractometer with Cu Kα1 radiation; the powder was sealed in Kapton film to avoid air exposure. The neutron diffraction study was performed on high intensity neutron diffractometer, WOMBAT, at the OPAL Reactor (Lucas Heights, Australia)52 and the General Purpose Powder Diffractometer, GPPD, at the China Spallation Neutron Source. The in-situ NPD experiments were performed at WOMBAT, where around 5 g of the as-milled LZC were loaded in a vanadium can and sealed under an argon atmosphere during measurement. The data were collected at temperatures from 27 to 427 °C with the wavelength of 1.5451 Å. Rietveld refinement was performed using GSAS II53,54.
Conductivity measurements
Prior to the EIS measurement, the powders were cold pressed into pellets at 380 MPa without any heat treatment, and then Au electrodes were sputtered on the pellet surfaces; the sputter coater was placed in the glove box, so that air exposure can be strictly prevented in the entire procedure. The EIS measurement was performed on the cold-pressed pellets acquired above without any external pressure using a MTZ-35 impedance analyzer (Bio-Logic) in the frequency range between 1 Hz and 35 MHz with 10 mV driving potential amplitude. The electronic conductivity was determined by the direct current (DC) polarization measurement on cold-pressed pellets with the applied voltage of 1 V.
Humidity tolerance test
The humidity tolerance test was conducted by placing 0.5 g as-milled LZC and 0.5 g Li3InCl6 powders within the same vacuum desiccator filled with N2 of 5% relative humidity at 25 °C for 24 h. In order to create this humid environment, we first let dry N2 flow through ultrapure water to make it moist. Then, this moisture-containing N2 was mixed with dry N2 in a certain ratio to reach the desired humidity, which was ensured by a humidity sensor (Shenzhen Everbest Machinery Industry, DT-83, accuracy ±3% relative humidity) placed in the desiccator mentioned above.
Bond valence site energy (BVSE) calculations
BVSE calculations were performed with the softBV program47,48 using structural models obtained from the Rietveld refinement. The energies of different Li sites in the crystal structure were calculated against a 3D grid of points with 0.1 Å resolution using the transferable Morse-type softBV force field. Li-ion migration pathways were identified with the regions of low BVSE.
Electrochemical characterizations
The ESW of LZC was evaluated by the CV measurement on a Li | Li7P3S11-LZC | LZC+C (weight ratio: LZC/C = 70/30) cell between −0.25 and 5 V at 0.1 mV s–1. The cathode composite in the all-solid-state cell for charge/discharge test was prepared by mixing the commercial LiCoO2 (Alfa Aesar, 99.5%) or single-cystal LiNi0.8Mn0.1Co0.1O2 (Hunan Shanshan Energy Technology, 99.9%) powders with the as-milled LZC (sieved beforehand through 10 μm, 5 μm, and 3 μm meshes) using a vortex mixer (Haimen Kylin-Bell Lab Instruments, QL-866) at 1500 rpm for 10 min in a weight ratio of 75:25. In order to assemble the all-solid-state cell, 60 mg of the as-milled LZC powder was first placed into a polyetheretherketone (PEEK) mold with 10 mm diameter and pressed at 1 ton to form the solid electrolyte layer. Then, 7 mg of the cathode composite powder was dispersed evenly on one side of this solid electrolyte layer and pressed again at 1.2 tons. To avoid reaction between LZC and the anode, a thin layer of Li6PS5Cl was applied at the other side of the LZC layer in a similar manner: 35 mg of Li6PS5Cl powder (Shenzhen Kejing Star Technology, 99%) was dispersed evenly on the solid electrolyte layer, and then pressed at 1.2 tons. Afterwards, a piece of indium foil (0.1 mm thick, 10 mm diameter) was placed at the surface of the Li6PS5Cl layer, and a piece of lithium foil (0.03 mm thick, 10 mm diameter) was subsequently attached to this indium foil. They were then pressed at 1.5 tons to form the Li-In alloy anode7,55. The cells were cycled under an external pressure of ~1.5 tons at 25 °C using a LAND CT2001A battery testing system within the voltage ranges of 1.9–3.6 V and 2.2–3.8 V for LiCoO2 and scNMC811, respectively; the temperature of 25 °C was ensured by placing the cells in an incubator (Tianjin Hongnuo Instrument, SPX-250B, temperature accuracy ±1 °C) during cycling.
Data availability
The data that support the findings of this study are available within the article (and its Supplementary Information files) and from the corresponding author upon reasonable request.
References
Janek, J. & Zeier, W. G. A solid future for battery development. Nat. Energy 1, 16141 (2016).
Manthiram, A., Yu, X. & Wang, S. Lithium battery chemistries enabled by solid-state electrolytes. Nat. Rev. Mater. 2, 16103 (2017).
Tan, D. H. S., Banerjee, A., Chen, Z. & Meng, Y. S. From nanoscale interface characterization to sustainable energy storage using all-solid-state batteries. Nat. Nanotechnol. 15, 170–180 (2020).
Zhao, Q., Stalin, S., Zhao, C. Z. & Archer, L. A. Designing solid-state electrolytes for safe, energy-dense batteries. Nat. Rev. Mater. 5, 229–252 (2020).
Famprikis, T., Canepa, P., Dawson, J. A., Islam, M. S. & Masquelier, C. Fundamentals of inorganic solid-state electrolytes for batteries. Nat. Mater. 18, 1278–1291 (2019).
Chen, J., Wu, J., Wang, X., Zhou, A. & Yang, Z. Research progress and application prospect of solid-state electrolytes in commercial lithium-ion power batteries. Energy Storage Mater. 35, 70–87 (2021).
Asano, T. et al. Solid halide electrolytes with high lithium-ion conductivity for application in 4 V class bulk-type all-solid-state batteries. Adv. Mater. 30, 1803075 (2018).
Li, X. et al. Progress and perspectives on halide lithium conductors for all-solid-state lithium batteries. Energy Environ. Sci. 13, 1429–1461 (2020).
Wang, Y. et al. Design principles for solid-state lithium superionic conductors. Nat. Mater. 14, 1026–1031 (2015).
Yamane, H. et al. Crystal structure of a superionic conductor, Li7P3S11. Solid State Ion. 178, 1163–1167 (2007).
Kudu, Ö. U. et al. A review of structural properties and synthesis methods of solid electrolyte materials in the Li2S−P2S5 binary system. J. Power Sources 407, 31–43 (2018).
Dawson, J. A. et al. Toward understanding the different influences of grain boundaries on ion transport in sulfide and oxide solid electrolytes. Chem. Mater. 31, 5296–5304 (2019).
Campanella, D., Belanger, D. & Paolella, A. Beyond garnets, phosphates and phosphosulfides solid electrolytes: new ceramic perspectives for all solid lithium metal batteries. J. Power Sources 482, 228949 (2021).
Wang, S. et al. Lithium chlorides and bromides as promising solid-state chemistries for fast ion conductors with good electrochemical stability. Angew. Chem. Int. Ed. 58, 8039–8043 (2019).
Li, X. et al. Origin of superionic Li3Y1-xInxCl6 halide solid electrolytes with high humidity tolerance. Nano Lett. 20, 4384–4392 (2020).
Zhu, Y., He, X. & Mo, Y. First principles study on electrochemical and chemical stability of solid electrolyte–electrode interfaces in all-solid-state Li-ion batteries. J. Mater. Chem. A 4, 3253–3266 (2016).
Hart, P. W. & Sommerfeld, J. T. Cost estimation of specialty chemicals from laboratory-scale prices. Cost Eng. 39, 31–35 (1997).
Xu, B. et al. Influence of sintering additives on Li+ conductivity and electrochemical property of perovskite-type Li3/8Sr7/16Hf1/4Ta3/4O3. Electrochim. Acta 234, 1–6 (2017).
Gao, J. et al. Rational design of mixed electronic-ionic conducting Ti-doping Li7La3Zr2O12 for lithium dendrites suppression. Adv. Funct. Mater. 31, 2001918 (2021).
Ren, Y., Shen, Y., Lin, Y. & Nan, C. W. Direct observation of lithium dendrites inside garnet-type lithium-ion solid electrolyte. Electrochem. Commun. 57, 27–30 (2015).
Bohnsack, A. et al. Ternäre halogenide vom typ A3MX6. VI [1]. ternäre chloride der selten-erd-elemente mit lithium, Li3MCl6 (M = Tb-Lu, Y, Sc): synthese, kristallstrukturen und ionenbewegung. Z. anorg. allg. Chem. 623, 1067–1073 (1997).
Li, X. et al. Water-mediated synthesis of a superionic halide solid electrolyte. Angew. Chem. Int. Ed. 58, 16427–16432 (2019).
Li, X. et al. Air-stable Li3InCl6 electrolyte with high voltage compatibility for all-solid-state batteries. Energy Environ. Sci. 12, 2665–2671 (2019).
Schlem, R. et al. Mechanochemical synthesis: a tool to tune cation site disorder and ionic transport properties of Li3MCl6 (M = Y, Er) superionic conductors. Adv. Energy Mater. 10, 1903719 (2019).
Liang, J. et al. Site-occupation-tuned superionic LixScCl3+x halide solid electrolytes for all-solid-state batteries. J. Am. Chem. Soc. 142, 7012–7022 (2020).
Park, D. et al. Theoretical design of lithium chloride superionic conductors for all-solid-state high-voltage lithium-ion batteries. ACS Appl. Mater. Interfac. 12, 34806–34814 (2020).
Zhou, L. et al. A new halospinel superionic conductor for high-voltage all solid state lithium batteries. Energy Environ. Sci. 13, 2056–2063 (2020).
Haynes, W. M., Lide, D. R. & Bruno, T. J. Abundance of elements in the earth’s crust and in the sea. CRC Handbook of Chemistry and Physics, 97th edn, CRC Press, 14–17 (2016–2017).
Duluard, S. et al. Lithium conducting solid electrolyte Li1.3Al0.3Ti1.7(PO4)3 obtained via solution chemistry. J. Eur. Ceram. Soc. 33, 1145–1153 (2013).
McCloskey, B. D. Attainable gravimetric and volumetric energy density of Li–S and Li ion battery cells with solid separator-protected Li metal anodes. J. Phys. Chem. Lett. 6, 4581–4588 (2015).
Yang, X., Adair, K. R., Gao, X. & Sun, X. Recent advances and perspectives on thin electrolytes for high-energy-density solid-state lithium batteries. Energy Environ. Sci. 14, 643–671 (2021).
Park, K. H. et al. High-voltage superionic halide solid electrolytes for all-solid-state Li-ion batteries. ACS Energy Lett. 5, 533–539 (2020).
Wu, E. A. et al. A stable cathode-solid electrolyte composite for high-voltage, long-cycle-life solid-state sodium-ion batteries. Nat. Commun. 12, 1256 (2021).
Schlem, R., Banik, A., Eckardt, M., Zobel, M. & Zeier, W. G. Na3–xEr1–xZrxCl6—a halide-based fast sodium-ion conductor with vacancy-driven ionic transport. ACS Appl. Energy Mater. 3, 10164–10173 (2020).
Pearson, R. G. Hard and soft acids and bases, HSAB, part I: fundamental principles. J. Chem. Educ. 45, 581–587 (1968).
Pearson, R. G. Hard and soft acids and bases, HSAB, part II: underlying theories. J. Chem. Educ. 45, 643–648 (1968).
Sahu, G. et al. Air-stable, high-conduction solid electrolytes of arsenic-substituted Li4SnS4. Energy Environ. Sci. 7, 1053–1058 (2014).
Allred, A. L. & Rochow, E. G. A scale of electronegativity based on electrostatic force. J. Inorg. Nucl. Chem. 5, 264–268 (1958).
Strangmüller, S. et al. Modifying the properties of fast lithium ion conductors—the lithium phosphidotetrelates Li14SiP6, Li14GeP6 and Li14SnP6. Chem. Mater. 32, 6925–6934 (2020).
Krauskopf, T., Culver, S. P. & Zeier, W. G. Bottleneck of diffusion and inductive effects in Li10Ge1–xSnxP2S12. Chem. Mater. 30, 1791–1798 (2018).
Zhu, Y. & Mo, Y. Materials design principles for air-stable lithium/sodium solid electrolytes. Angew. Chem. Int. Ed. 132, 17625–17629 (2020).
Kamaya, N. et al. A Lithium superionic conductor. Nat. Mater. 10, 682–686 (2011).
Zhu, Y., He, X. & Mo, Y. Origin of outstanding stability in the lithium solid electrolyte materials: insights from thermodynamic analyses based on first-principles calculations. ACS Appl. Mater. Interfac. 7, 23685–23693 (2015).
Zhu, Y., He, X. & Mo, Y. Strategies based on nitride materials chemistry to stabilize Li metal anode. Adv. Sci. 4, 1600517 (2017).
Jain, A. et al. Commentary: The Materials Project: A materials genome approach to accelerating materials innovation. APL Mater. 1, 011002 (2013).
Shannon, R. D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. A 32, 751–767 (1976).
Chen, H. & Adams, S. Bond softness sensitive bond-valence parameters for crystal structure plausibility tests. IUCrJ 4, 614–625 (2017).
Chen, H., Wong, L. L. & Adams, S. SoftBV—a software tool for screening the materials genome of inorganic fast ion conductors. Acta Crystallogr., Sect. B: Struct. Sci. 75, 18–33 (2019).
Famprikis, T. et al. Under pressure: mechanochemical effects on structure and ion conduction in the sodium-ion solid electrolyte Na3PS4. J. Am. Chem. Soc. 142, 18422–18436 (2020).
Fu, Y. & Ma, C. Interplay between Li3YX6 (X = Cl or Br) solid electrolytes and the Li metal anode. Sci. China Mater. 64, 1378–1385 (2021).
Riegger, L. M., Schlem, R., Sann, J., Zeier, W. G. & Janek, J. Lithium-metal anode instability of the superionic halide solid electrolytes and the implications for solid-state batteries. Angew. Chem. Int. Ed. 60, 6718–6723 (2021).
Studer, A. J., Hagen, M. E. & Noakes, T. J. Wombat: the high-intensity powder diffractometer at the OPAL reactor. Phys. B 385–386, 1013–1015 (2006).
Toby, B. H. & Von Dreele, R. B. GSAS-II: the genesis of a modern open-source all purpose crystallography software package. J. Appl. Crystallogr. 46, 544–549 (2013).
Von Dreele, R. B. Small-angle scattering data analysis in GSAS-II. J. Appl. Crystallogr. 47, 1784–1789 (2014).
Busche, M. R. et al. In situ monitoring of fast Li-ion conductor Li7P3S11 crystallization inside a hot-press setup. Chem. Mater. 28, 6152–6165 (2016).
Momma, K. & Izumi, F. VESTA3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 44, 1272–1276 (2011).
Acknowledgements
C.M. acknowledges the financial support from the National Key R&D Program of China (2018YFA0209600, 2017YFA0208300), the National Natural Science Foundation of China (51802302), and the Fundamental Research Funds for the Central Universities (WK3430000006, WK2060190085). J.M. thanks the financial support from the National Natural Science Foundation of China (11774223, U1732154) and a Shanghai talent program. Y.L. acknowledges the financial support from the National Key R&D Program of China (2018YFA0209600) and the National Natural Science Foundation of China (22022813, 21878268).
Author information
Authors and Affiliations
Contributions
K.W. and C.M. conceived the research. K.W. synthesized the materials, analyzed the crystal structures, conducted the electrochemical measurements, and performed the humidity stability test. Z.G., F.Z., Y.F., and J.H. conducted the X-ray diffraction experiments and conductivity measurements. C.D., J.W., and Y.L. assisted in electrochemical tests. Q.R., J.Z, L.H., C.-W.W., and J.M. conducted the neutron diffraction study. C.M. directed the entire study.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Andrea Paolella and the other anonymous reviewers for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, K., Ren, Q., Gu, Z. et al. A cost-effective and humidity-tolerant chloride solid electrolyte for lithium batteries. Nat Commun 12, 4410 (2021). https://doi.org/10.1038/s41467-021-24697-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-021-24697-2
This article is cited by
-
Promoting high-voltage stability through local lattice distortion of halide solid electrolytes
Nature Communications (2024)
-
Designing lithium halide solid electrolytes
Nature Communications (2024)
-
From Liquid to Solid-State Lithium Metal Batteries: Fundamental Issues and Recent Developments
Nano-Micro Letters (2024)
-
Enhancement of –OH content on mechanical properties of antiperovskite solid electrolytes
Nano Research (2024)
-
From atomistic modeling to materials design: computation-driven material development in lithium-ion batteries
Science China Chemistry (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
