
Abstract
How natural selection facilitates sexually dimorphic evolution despite a shared genome is unclear. The patrilineal inheritance of Y-chromosomes makes them an appealing solution. However, they have largely been dismissed due to their gene-poor, heterochromatic nature and because the additive genetic variation necessary for adaptive evolution is theoretically difficult to maintain. Further, previous empirical work has revealed mostly Y-linked sign epistatic variance segregating within populations, which can often impede adaptive evolution. To assess the evolutionary impact of Y-linked variation, we established replicate populations in Drosophila simulans containing multiple Y-chromosomes (YN populations) or a single Y-chromosome variant (Y1 populations) drawn from a single population. We estimated male and female heritabilities for several traits known to be influenced by Y-chromosomes, including the number of sternopleural bristles, abdominal bristles, sex comb teeth, and tibia length. A decrease in YN heritabilities compared with Y1 would be consistent with Y-chromosome variation being sign epistatic. A decrease in Y1 heritabilities would be consistent with Y-chromosome variation being additive, though additive-by-additive epistatic variation cannot be entirely dismissed. Female heritability estimates served as controls and were not expected to differ. We found male Y1 populations exhibited lower heritabilities for all traits except tibia length; consistent with Y-linked additivity (on average YN trait heritabilities were 25% greater than Y1). Female estimates showed no difference. These data suggest Y-chromosomes should play an important role in male trait evolution and may even influence sexually dimorphic evolution by shaping traits shared by both sexes.
Similar content being viewed by others
Introduction
Sexual dimorphism is a pervasive feature of animal life. But how natural selection overcomes the constraints of a shared genome to facilitate dimorphic evolution is not fully understood (Dean and Mank 2014; Stewart and Rice 2018). This constraint is manifested by sexually antagonistic alleles (SA) that are beneficial in one sex but detrimental in the other, causing a perpetual tug-of-war over SA allele frequencies. Theoretically, this tension can be minimized if male-benefit alleles reside on the X-chromosome and are recessive to female-benefit alleles (Fry 2010). The extent to which this mechanism facilitates dimorphism, however, remains unresolved (Ruzicka and Connallon 2020). Dimorphism can also be facilitated via sex-specific modifiers that alter an allele’s dominance to benefit the sex in which it resides (Spencer and Priest 2016). Recent evidence suggests this is a viable mechanism (Barson et al. 2015; Grieshop and Arnqvist 2018), although the prevalence of its impact is unclear.
The simplest solution to the shared-genome constraint is for male-benefit alleles to reside on the Y-chromosome, as it is patrilineal in inheritance and free from female counter selection (assuming the alleles do not reduce female or offspring fitness; Lund-Hansen et al. 2021). Traditionally, however, Y-chromosomes have been dismissed as unimportant because they tend to be highly heterochromatic, comprised largely of repetitive sequences, harbor few protein coding genes, decay over evolutionary time, and have been lost entirely in some species (Brown et al. 2020; Burgoyne 1998; Carvalho et al. 2009; Castillo et al. 2010; Cheng et al. 2019). Moreover, in many species that harbor Y-chromosomes, males are perfectly viable without them (Carvalho et al. 2015). Despite these limitations, numerous studies have reported that Y-chromosome variants differentially influence a broad range of behavioral, physiological, and morphological characters (Supplemental Table 1), including human disease (Prokop and Deschepper 2015). This impact is likely due to the ability of some Ys to alter the expression of autosomal and X-linked loci by modulating the genome’s chromatin landscape (Lemos et al. 2008; Brown et al. 2020). Furthermore, Y-chromosomes appear to rapidly diverge between species in part due to positive selection (Chang et al., (2022)). Thus, contrary to its inherent limitations, Y-chromosomes might have the capacity to adaptively shape male characters under autosomal influence including those that are sexually dimorphic.
For natural selection to adaptively shape Y-linked variation, and hence the quantitative traits they affect, additive variation must exist among Y-chromosomes of a local gene pool (Fig. 1A, B). However, theoretical work predicts that additive genetic variation on Y-chromosomes is difficult to maintain (Clark 1987, 1990) and most empirical studies assessing Y-linked effects have been unable to assess the potential for additive variation. That is because these studies generally sample Ys from geographically distinct populations (Supplemental Table 1) and placed them into isogenic and/or non-coevolved genetic backgrounds. This methodology creates several limitations when attempting to assess the evolutionary impact of Y-linked effects. First, Y-chromosomes sourced from distant populations provide little information regarding the variation segregating within local populations where selection operates. Second, non-coevolved backgrounds can induce artificial epistatic interactions. Epistasis occurs when an allele’s contribution to the phenotype is contingent upon alleles at other loci (Wolf et al. 2000). In other words, non-coevolved backgrounds can create Y-linked effects that do not occur in natural populations (for example, Fig. 1B if genotype A1A1 represented a coevolved background and A2A2 a novel background; see also Stoltenberg and Hirsch 1997). Third, when Ys are assessed in an isogenic background, Y-chromosome by genetic background epistatic variation is eliminated. Removal of this variation can deceptively generate Y-linked additive effects in populations where none functionally exist (for example, Fig. 1C if A1A1 was the only genetic background examined).

A The background alleles and Y-chromosomes exhibit pure additivity, creating a phenotype heritability equal to 1. B Significant Y-chromosome by background epistasis exists, reducing phenotype heritability compared with (A). C The background alleles and Y-chromosomes exhibit pure sign epistasis, reducing phenotype heritability to zero. In all three hypothetical examples, dominance and environmental deviations are ignored and allele frequencies are assumed to be equal.
Only a handful of animal studies have assessed the adaptive potential of Y-chromosome variants sampled from a single population and placed within coevolved, genetically variable backgrounds. Chippindale and Rice (2001) found Y-linked additive variation for male fitness in D. melanogaster when Ys were placed in unique backgrounds (i.e., backgrounds with minimal variation). However, this variation disappeared once multiple Ys and backgrounds were considered. Similarly, Kutch and Fedorka (2017) found significant Y-chromosome effects for D. melanogaster immune function within coevolved isogenic backgrounds that vanished when multiple backgrounds were incorporated into the analysis. The elimination of Y-linked additive variation once multiple genetic backgrounds were examined (Chippindale and Rice 2001; Kutch and Fedorka 2017) is indicative of sign epistasis, which occurs when alleles produce opposite phenotypic effects in different genetic backgrounds (Fig. 1C). Therefore, the Y-linked effects reported for numerous traits over the years (Supplemental Table 1) may represent sign epistatic variation circulating in natural populations and might not actively contribute to the adaptive evolution of the traits they affect.
To determine if Y-linked variation contributes to adaptive evolution, Kutch and Fedorka (2018) selected for improved male geotaxis in D. melanogaster populations possessing multiple Y-chromosomes (i.e., possessing Y-linked variation) or a single Y-chromosome variant (i.e., possessing no Y-linked variation). Geotaxis was chosen because it is a quantitative trait expressed by both sexes and previously shown to be affected by Y-chromosome variants (Stoltenberg and Hirsch 1997). They found that after 20 generations, single-Y populations responded to selection while multi-Y populations did not. This pattern implies that Y-chromosome by background epistasis constrained the rate of adaptive evolution, most likely by reducing male trait heritability.
It should be noted that epistasis can either increase or decrease the heritability of quantitative traits (Cheverud and Routman 1995). Studies to date, however, suggest Y-chromosome by genetic background interactions tends to minimize heritable variation and obscure Y-chromosome variants from selection’s view (Chippindale and Rice 2001; Kutch and Fedorka 2017). Thus, contemporary Y-chromosome variants within a population may generally act as evolutionary hinderances for male quantitative traits instead of as fuel for adaptive evolution. If a trait is expressed by both sexes (as with geotaxis), then a Y-linked reduction in male heritability could also slow the female selection response if they share a similar phenotypic optimum. This could have significant implications for how a population responds to a novel selective pressure like climate change or an invading pathogen / species. To better understand the evolutionary impact of Y-chromosome variation, more studies that assess Y-linked effects in their appropriate and variable genetic backgrounds are needed; especially considering that most studies reporting Y-linked effects also noted significant Y-by-background effects (Supplemental Table 1).
Here we address the potential for Y-chromosome variation in D. simulans to influence male trait heritability. Using similar methodology to Kutch and Fedorka (2018), we created replicate populations containing multiple Y-chromosomes and populations with a single Y-chromosome variant, with other genetic elements being homogenized within replicates. We then assessed the male and female heritabilities for sternopleural bristles, abdominal bristles, tibia length, and sex comb morphology; the former three traits being expressed by both sexes and the latter trait being male-only. If Y-chromosomes positively contribute to the additive genetic variation of a shared trait and hence its heritability, then Y-linked effects would be an effective way to shape sexual dimorphism. In contrast, if Y-chromosomes induce significant sign epistasis, then male trait heritability could be reduced, which would constrain adaptive trait evolution.
Methods
Experimental design overview
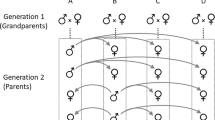
To examine how the Y-chromosome influences trait heritability, two types of populations were created. YN populations were assumed to contain numerous Y-chromosome variants and Y1 populations contained a single Y-chromosome variant. Two replicate pair of YN and Y1 populations were created and population size, allele frequencies, and cytoplasmic elements were equalized within each pair (Fig. 2). Heritabilities were calculated for left and right sternopleural bristle number, abdominal bristle number on sternites 4 and 5, left and right tibia length, and number of teeth on left and right sex comb. Trait heritabilities were calculated separately for each population type, replicate, and sex. If trait heritability is lower in the Y1 population, it would suggest Y-chromosomes contribute additive genetic variation. However, if YN population exhibits lower heritability, it would suggest Y-chromosomes reduce heritable variation through epistasis, which would constrain the rate of trait evolution. For each trait, female heritabilities served as controls. This is because YN and Y1 female heritabilities are not expected to differ, as their allele frequencies were equalized, and they do not contain Y-chromosomes. If female differences in YN and Y1 heritabilities were found for a given trait, differences in male YN and Y1 trait heritabilities would be difficult to interpret.

P1 and F1 represent the parental and first filial generation, respectively. Female offspring in Y1 crosses were discarded in generations G1–G4 and replaced with YN females. In G6, all female offspring were swapped between YN and Y1 to homogenize alleles. In G7-11, half of the female offspring were swapped. See methods for details.
YN and Y1 population creation
All flies were maintained on a cornmeal medium at 25 °C, under a 12 h:12 h light: dark photoperiod in Percival incubators. During winter 2017, 351 female D. simulans were collected within 6 miles of the University of Central Florida and isofemale lines were established for species identification. After species identification was confirmed via male offspring, the isofemale offspring were homogenized and two independent YN replicate populations were established by randomly assigning half of the homogenate to a YN-α and YN-β population. This approach likely created YN replicates with slightly different allele frequencies and/or private alleles (including Ys) due to random sampling bias. From each YN population, a paired Y1 population was established such that YN-α created Y1-α and YN-β created Y1-β. This was accomplished by isolating a single YN male and mating him with three YN virgin females (generation 0; Fig. 2). The newly established Y1 populations were then expanded by mating Y1 sons with YN virgin females until their population size reached a stable 1000 individuals (generations 3–5). All populations were maintained at 500 males and 500 females per generation in 12in3 population cages (except in the first two Y1 generations, which had smaller population sizes).
Considering that Y1 populations were completely derived from the YN populations, their genetic similarity to YN was likely well above 99% by generation 5. Still, a small number of private alleles and/or allele frequency imbalances could persist between the populations due to founder effects. To minimize this, all newly eclosed virgin females from YN populations were swapped with the paired Y1 females at generation 6 (i.e., YN-α females were swapped with Y1-α females and YN-β with Y1-β). If both males and females of a population simultaneously possessed a particular private allele, that allele would now be represented in both populations. Any minor allele frequencies differences between Y1 and YN would also be further homogenized with this swap. However, if unique cytoplasmic elements existed in Y1, they would now exist only in YN (and vice versa), as only females transmit these elements. To minimize this, half of the females from each YN and Y1 paired populations were swapped in generation 7 while half remained in their original population. To further minimize sampling error and homogenize allele frequencies, half of newly eclosed virgin females were swapped among paired YN and Y1 populations an additional 4 times (Generations 8–11). Female swapping between populations represents a powerful migratory force that homogenizes allele frequencies and minimizes divergence through founder effects or drift while keeping Y-chromosome gene pools separate in each population.
In generation 12, virgin flies were collected and mating pairs established in vials for 24 h. After 24 h the flies were removed from the vials. The next generation was collected as 3-day-old flies, with all flies within a vial being full-siblings. For each vial, males and females were placed in separate, labeled microcentrifuge tubes which were placed in a −80 °C freezer for subsequent trait measurement (Generation 13). The flies were frozen dry.
While the diversity of Y-chromosome variants in YN was not confirmed in this study, previous work using D. melanogaster drawn from the same geographic location (Orlando, FL) showed Y-chromosome diversity existed in a laboratory population established from just 40 gravid females (Kutch and Fedorka 2018). D. simulans tend to exhibit similar, if not higher, amounts of genetic diversity than D. melanogaster (Langley et al. 2012). Given that our D. simulans laboratory population was established from an order of magnitude greater number of gravid females (n = 351), we feel the assumption of multiple Ys within the YN populations is reasonable.
Trait measurement
Morphological traits were measured rather than life-history, behavioral, or physiological traits because of their generally higher heritabilities, smaller associated standard errors (Roff and Mousseau 1987), and ease of measurement at large sample sizes. The morphological traits chosen were assumed to be polygenic, which would increase the probability that the Y-chromosome affected their expression through autosomal chromatin modification. To this end, we measured left and right sternopleural bristles, sternite 4 and 5 abdominal bristles, left and right front leg tibia length, and left and right sex comb number (males only). Abdominal and sternopleural bristle numbers were counted under a dissecting microscope. Front legs were mounted on slides, from which tibia length and sex comb number were assessed using a compound microscope equipped with an ocular camera (Dino-Lite Dino-Eye model AM-423X). Bristle and sex comb tooth number were count measurements that have very high repeatability. Tibia length repeatability was 0.92, calculated via the intraclass correlation (Zar 1984) using 240 tibias measured twice.
Heritability analysis
A full-sibling design was used to calculate heritabilities (h2) and associated standard errors, which were derived from one-way ANOVA model parameters (Roff 1997, equations 2.27 and 2.28). Unlike half-sibling estimates, full-sibling estimates carry ¼ of the dominance variance. Note that Y-chromosomes are haploid and do not contribute dominance variance per se. The comparison of YN and Y1 heritability differences should be minimally affected by the added ¼ dominance variance as this addition would affect YN and Y1 equally, considering their allele frequencies were homogenized prior to assessment and they only differed in the abundance of Y-chromosomes. Thus, any difference in heritability due to the removal of Y-chromosomes in Y1 is likely due to a change in (i) Y-additive variation, (ii) Y-additive by genome-additive epistatic variation, or (iii) Y-additive by genome-dominance epistatic variation.
To statistically test for differences between YN and Y1 heritability estimates, we employed 95% confidence intervals. Specifically, we calculated the heritability and standard error (SE) for each YN and Y1 pair. We then determined if the 95% confidence interval associated with the YN – Y1 difference included zero using the following test: |YN – Y1 | - 1.96 * \(\sqrt {\left( {{{{\mathrm{YN}}}}SE^2 + {{{\mathrm{Y}}}}1SE^2} \right)}\). If the YN-Y1 difference minus the 95% confidence interval was negative (i.e., included zero), the null hypothesis (i.e., no difference) was accepted. If the difference was positive (i.e., did not include zero), the null hypothesis was rejected (Schenker and Gentleman 2001).
Heritabilities were calculated for each of the 8 groups (male/female, YN/Y1, replicate 1/2). Full-sib family size ranged between 8 and 10 individuals per sex. For abdominal bristle number, tibia length, and sex comb number, if the area associated with the trait appeared damaged, then the measurement was excluded from analysis. For sternopleural bristle number, measurements were only included in the analysis if all three macro bristles were present or if two macro bristles were present and the other bristles appeared undisturbed; in which case one bristle was added to the count to account for the missing macro bristle.
Genetic correlations were estimated among all traits using Pearson product moment correlations among family means (Via 1984). The same statistical test employed above was used to determine if two genetic correlations were different. All analyses were done using R version 4.0.0 (R Core Team 2016).
Results
In total, 3348 flies were analyzed: 1688 male and 1660 female. Summary statistics are provided for each of the 56 trait heritabilities that were calculated (Supplemental Table 2). The average number of families (±SE) per heritability estimate was 32.7 ± 0.5, with an average family size of 9.15 ± 0.07 flies. Average, maximum, and minimum morphological trait values resemble values seen in previous studies (Capy et al. 1993; Macdonald and Goldstein 1999). Accordingly, females were shown to have a higher number of sternopleural bristles (♂average = 9.68 ± 0.03; ♀average = 10.51 ± 0.03) and abdominal bristles (♂average = 15.55 ± 0.04; ♀average = 19.75 ± 0.05; calculated as average of 4th and 5th sternite), as well as longer tibias (♂average = 0.453 ± 0.001 mm; ♀average = 0.464 ± 0.001 mm).
To assess the influence of Y-chromosome variation on male trait heritability, we first determined if YN and Y1 paired populations differed for female trait heritabilities. If differences existed, it would suggest that the YN and Y1 populations differed in their allele frequencies, which could cause divergent heritabilities not due to the Y-chromosome and weaken our assessment of male differences. However, no significant differences were detected for any female trait between YN and Y1 paired populations (Fig. 3; Supplemental Table 3).

Male YN populations exhibited a significantly greater heritability for AB4, AB5, TLR, and SCL. Females showed no heritability differences between YN and Y1 for any trait. Black bar represents mean YN-Y1 difference among all traits. Error bars are 95% CIs. ‘*’ represents statistical significance at α = 0.05. SP = sternopleural bristle, AB = abdominal bristle, TL = tibia length, SC = sex comb, L = left, R = right, 4 = 4th sternite, 5 = 5th sternite. All heritabilities and standard errors appear in the supplemental material.
Regarding males, significant differences were found for 4th and 5th sternite abdominal bristle number, right tibia length and left sex comb (Fig. 3; Supplemental Table 3). All significant differences indicate that Y1 populations had lower heritabilities than YN populations. When the heritability differences between YN and Y1 were averaged across all traits, we found that males from the YN populations exhibited on average a 0.16 ± 0.06 (mean ± SE) greater heritability than the Y1 populations. In contrast, females exhibited an average heritability difference of −0.01 ± 0.04 (mean ± SE). This approach provides a conservative method for assessing an overall difference between the YN and Y1 heritabilities that included all traits.
Although significant YN Y1 differences were detected, these differences were not perfectly consistent among replicates α and β. For instance, the 5th sternite abdominal bristle exhibited a significant YN-Y1 difference for α but not β. Further, α and β tibia length heritabilities were statistically different from each other for both left and right tibia (Supplemental Table 3). Such inconsistency likely stems from the replicate populations containing different allele frequencies and private alleles established at the founding of YN-α and YN-β. However, it may also be due to the limited number of families used to estimate heritabilities. To alleviate this concern, we combined α and β replicate populations and recalculated our estimates, with replicate included as a covariate in our models. This recalculation increased the average number of families examined for each trait from 32.7 ± 0.5 to 65.6 ± 1.4. Again, we found no significant difference between YN and Y1 female estimates (Fig. 4, Supplemental Table 4). Male YN heritability estimates were greater than Y1 estimates for 4th sternite abdominal bristle number, 5th sternite abdominal bristle number, right sternopleural bristle number, and left sex comb tooth number. Left sternopleural number was significant at the α = 0.1 level. Moreover, the YN populations exhibited on average a 0.15 ± 0.04 (mean ± SE) greater heritability than the Y1 populations for males, but only a 0.01 ± 0.03 greater heritability for females. In no instance were heritability estimates significantly different between the left and right side of a trait when measured within the same replicate population (Fig. 3; Supplemental Table 3) or when replicates were combined (Fig. 4; Supplemental Table 4).

Male YN populations exhibited a significantly greater heritability for SPL, SPR, AB4, AB5, and SCL, as well as for the average of all traits (\(\bar X\)). Females showed no heritability differences between YN and Y1 for any trait. Error bars are 95% CIs. ‘*’ and ‘**’ represent statistical significance at the α = 0.1 and α = 0.05, respectively. SP = sternopleural bristle, AB = abdominal bristle, TL = tibia length, SC = sex comb, L = left, R = right, 4 = 4th sternite, 5 = 5th sternite. All heritabilities and standard errors appear in the supplemental material.
Genetic correlations between left and right sides of the same trait were high for all populations and sexes (all r > 0.74; Supplemental Table 5) and male and female correlations appeared similar. No statistical difference between YN and Y1 correlations was detected for females (Fig. 5). In males, YN populations exhibited higher correlations between the 4th and 5th sternite abdominal bristles, as well as between the 4th sternite abdominal bristle and right sternopleural bristle. Overall, the average genetic correlation in the YN population significantly increased compared to the Y1 population for males but not for females (Fig. 5; mean increase ± SE: 0.08 ± 0.02 vs −0.02 ± 0.04, respectively).

The difference in genetic correlations between YN and Y1 populations appears above the diagonal. The statistical test appears below the diagonal. If |YNcorr – Y1corr | - 1.96 * \(\sqrt {\left( {{{{\mathrm{YN}}}}SE^2 + {{{\mathrm{Y}}}}1SE^2} \right)}\) > 0, then the correlations are statistically different at α = 0.05. A YN males exhibited a greater genetic correlation between AB4 and AB5, as well as between AB4 and SPR, than did Y1 males. B No difference in YN and Y1 was detected among female genetic correlations. All correlations and standard errors appear in the supplemental material.
Discussion
Understanding how sexual dimorphism evolves has been of great interest to evolutionary biologists (Dean and Mank 2014). Although Y-chromosomes are gene-poor and highly heterochromatic by nature, they have been shown to influence the expression of hundreds of autosomal and X-linked genes (Lemos et al. 2008) and offer a simple mechanism by which dimorphism can evolve. Nevertheless, the potential for Y-chromosomes to contribute to sexually dimorphic evolution via Y-linked additive variation has gone largely unstudied. If Y-chromosomes contribute to a trait’s additive variation, they can help shape dimorphism, permitting the sexes to reach separate phenotypic optima. Here we show that populations with a single Y-chromosome variant (Y1) exhibited lower male heritabilities compared to populations with multiple Y-chromosomes (YN). Further, female heritabilities did not significantly differ between these populations (females served as controls since they do not possess Y-chromosomes). In short, our data are consistent with Y-chromosomes contributing additive variation to three of the four traits examined.
The overall observation that gene-poor, highly heterochromatic Y-chromosomes contribute substantial genetic variation to male trait heritabilities is profound. However, it is important to note that our experimental design cannot determine if the decrease in Y1 heritabilities was due to a loss from Y-linked additive variation (VA) or additive-by-additive epistatic variation (VAA). Regardless, both types of variation should have important implications to male trait evolution. For instance, if the Y’s contribution is mostly additive, then short-term responses to selection and sexual dimorphic evolution is easily facilitated. If the variation is mostly VAA, contributions to medium and long-term selection can be facilitated via VAA conversion into VA over time (Hill 2016; Barton 2017). This conversion would be substantial given the size of the Y-linked contribution found here, which on average exceeds 25% of YN trait heritabilities. In other words, VAA would serve as a deep well from which new VA is continually drawn at rates that far exceed contributions from novel mutation (Hill 2016). Future work should elucidate the type of variation contributed by Y-chromosomes; if it is mostly additive, mostly additive-by-additive epistatic, or a mix thereof.
The contribution of Y-linked additive variation to complex quantitative traits is unexpected, as theoretical models predict such variation is hard to maintain (Clark 1987) and the only known function of the 16 known protein coding genes are associated with male fertility (Hafezi et al. 2020). Furthermore, previous empirical work suggests that Y-chromosome variation within D. melanogaster populations may be largely sign epistatic (Chippindale and Rice 2001; Kutch and Fedorka 2017), which likely decreases trait heritability and can impair populations from quickly responding to selective pressures (Kutch and Fedorka 2018). However, several recent studies support the maintenance of VA on Y-chromosomes that can impact sexual dimorphism. Wittenburg et al., (2010) found that Y-chromosomes contributed to sexual dimorphism in the birth weights of piglets (Sus domesticus). Milsted et al., (2010) found that variable Y-linked SRY alleles in rats (Rattus norvegicus) contributed to sexual dimorphism in hypertension. Last, Kaufmann et al., (2021) showed that Y-linked variation in seed beetles (Callosobruchus maculatus) significantly contributed to the evolution of sexually dimorphic body size under artificial selection. Thus, our findings, coupled with these latter studies, suggest that Ys may play a significant role in the evolution of sexual dimorphism for a variety of traits.
Although we show evidence of Y-linked variation for complex traits, several minor caveats exist. Central to our design was the creation of populations with multiple Y-chromosomes (YN). Although we did not directly identify the existence of multiple Ys (e.g., via sequencing), our experiment was designed to detect the effects of variable Y’s should they exist. Our results indicate they do; especially given that both independent replicates exhibited Y1 and YN differences (suggesting random artifacts are not a concern). Indirect evidence stemming from previous work also suggests the existence of within population Y-chromosome variation. First, a multitude of variable Y’s persist across Drosophila populations that exhibit minimal genetic structure (see Supplemental Table 1). Second, variable Ys were shown to exist within a laboratory population of D. melanogaster established form 40 gravid females from Orlando, FL (Kutch and Fedorka 2015; Kutch et al. 2017). D. simulans tend to exhibit greater genetic diversity than D. melanogaster (Langley et al. 2012) and the current design established the D. simulans laboratory population from 351 gravid female from the same location, Orlando. Therefore, it is reasonable to assume numerous Y-chromosomes are segregating in the YN populations (all else being equal). In addition, the tests for additive variation for each morphological character is not entirely independent, as non-zero genetic correlations exist between these traits. For instance, tibia length showed moderate genetic correlations with abdominal bristles and sex comb in the male YN population. However, correlations between sex comb and both bristle types were minor, suggesting they were relatively independent characters.
In summary, the removal of Y-chromosomes from Y1 populations and subsequent decline in heritabilities is consistent with Ys contributing additive genetic variation for some traits. Considering both sexes express sternopleural and abdominal bristles further suggests that Ys could facilitate sexually dimorphic evolution. This result is both consistent (Kaufmann et al., (2021)) and inconsistent (Chippindale and Rice 2001; Kutch and Fedorka 2017, 2018) with previous works. The lack of consensus likely stems from the traits examined. If the chromatin modification model underlying the Y-chromosome regulatory effect is accurate (Brown et al. 2020), then only some genes will be susceptible to Y-linked variance, depending on where they reside in the genome (e.g., if they exist at euchromatin/herterochromatin boundaries). Furthermore, traits closely associated with fitness or sexual antagonism may limit the Y’s ability to harbor additive variance (Clark 1987). Future research should examine the type of variation (additive or epistatic) induced by the Y-chromosome across these variables. This would make clearer the evolutionary consequences of the Y-chromosome’s influence on hundreds of autosomal genes.
Data availability
Data are available at Dryad: https://doi.org/10.5061/dryad.x69p8cznk.
References
Barson NJ, Aykanat T, Hindar K, Baranski M, Bolstad GH, Fiske P et al. (2015) Sex-dependent dominance at a single locus maintains variation in age at maturity in salmon. Nature 528(7582):405
Barton NH (2017) How does epistasis influence the response to selection? Heredity 188:96–109
Brown EJ, Nguyen AH, Bachtrog D (2020) The Drosophila Y chromosome affects heterochromatin integrity genome-wide. Mol Biol Evol 37(10):2808–2824
Burgoyne PS (1998) The mammalian Y chromosome: a new perspective. Bioessays 20(5):363–366
Capy P, Pla E, David JR (1993) Phenotypic and genetic variability of morphometrical traits in natural populations of Drosophila melanogaster and D simulans. I. Geographic variations. Genet Selection Evol 25(6):517–536
Carvalho AB, Vicoso B, Russo CA, Swenor B, Clark AG (2015) Birth of a new gene on the Y chromosome of Drosophila melanogaster. Proc Natl Acad Sci 112(40):12450–12455
Carvalho AB, Koerich LB, Clark AG (2009) Origin and evolution of Y chromosomes: Drosophila tales. Trends Genet 25(6):270–277
Castillo ER, Marti DA, Bidau CJ (2010) Sex and neo-sex chromosomes in Orthoptera: a review. J Orthoptera Res 19(2):213–231
Chang C, Gregory LE, Gordon KE, Meiklejohn CD, Larracuente AM (2022) Unique structure and positive selection promote the rapid divergence of Drosophila Y chromosomes. eLife 11. https://doi.org/10.7554/eLife.75795
Chang C-H, Larracuente AM (2019) Heterochromatin-Enriched Assemblies Reveal the Sequence and Organization of the Drosophila melanogaster Y Chromosome. Genetics 211(1):333–348
Cheverud JM, Routman EJ (1995) Epistasis and its contribution to genetic variance components. Genetics 139(3):1455–1461
Chippindale AK, Rice WR (2001) Y chromosome polymorphism is a strong determinant of male fitness in Drosophila melanogaster. Proc Natl Acad Sci 98(10):5677–5682
Clark AG (1987) Natural selection and Y-linked polymorphism. Genetics 115(3):569–577
Clark AG (1990) Two tests of Y chromosomal variation in male fertility of Drosophila melanogaster. Genetics 125(3):527–534
Dean R, Mank JE (2014) The role of sex chromosomes in sexual dimorphism: discordance between molecular and phenotypic data. J Evolut Biol 27(7):1443–1453
Fry JD (2010) The genomic location of sexually antagonistic variation: some cautionary comments. Evolution 64:1510–1516
Grieshop K, Arnqvist G (2018) Sex-specific dominance reversal of genetic variation for fitness. PLoS Biol 16:e2006810
Hafezi Y, Sruba SR, Tarrash SR, Wolfner MF, Clark AG (2020) Dissecting Fertility Functions of Drosophila Y Chromosome Genes with CRISPR. Genetics 214:977–990
Hill WG (2016) “Conversion” of epistatic into additive genetic variance in finite populations and possible impact on long-term selection response. J Anim Breed Genet 134:196–201
Kaufmann P, Wolak ME, Husby A, Immonen E (2021) Rapid evolution of sexual size dimorphism facilitated by Y-linked genetic variance. Nat Ecol Evol 9:1–9
Kutch IC, Fedorka KM (2015) Y-linked variation for autosomal immune gene regulation has the potential to shape sexually dimorphic immunity. Proc R Soc B: Biol Sci 282(1820):20151301
Kutch IC, Fedorka KM (2017) A test for Y‐linked additive and epistatic effects on surviving bacterial infections in Drosophila melanogaster. J Evolut Biol 30(7):1400–1408
Kutch IC, Fedorka KM (2018) Y-chromosomes can constrain adaptive evolution via epistatic interactions with other chromosomes. BMC Evolut Biol 18(1):1–8
Langley CH, Stevens K, Cardeno C, Lee YCG, Shrider DR, Pool JE et al. (2012) Genomic variation in natural populations of Drosophila melanogaster. Genetics 192:533–598
Lemos B, Araripe LO, Hartl DL (2008) Polymorphic Y chromosomes harbor cryptic variation with manifold functional consequences. Science 319(5859):91–93
Lund-Hansen KK, Olito C, Morrow EH, Abbott JK (2021) Sexually antagonistic coevolution between the sex chromosomes of Drosophila melanogaster. Proc Natl Acad Sci USA 118:e2003359
Macdonald SJ, Goldstein DB (1999) A quantitative genetic analysis of male sexual traits distinguishing the sibling species Drosophila simulans and D. sechellia. Genetics 153(4):1683–1699
Milsted A, Underwood AC, Dunmire J, DelPuerto HL, Martins AS, Ely DL, Turner ME (2010) Regulation of multiple renin–angiotensin system genes by Sry. J Hypertens 28:59–64
Prokop JW, Deschepper CF (2015) Chromosome Y genetic variants: impact in animal models and on human disease. Physiol Genom 47:525–537
R Core Team (2016). R: A language and environment for statistical computing. Vienna, Austria. https://www.R-project.org/
Roff DA (1997). Evolutionary Quantitative Genetics. Chapman and Hall. https://doi.org/10.1007/978-1-4615-4080-9
Roff DA, Mousseau TA (1987) Quantitative genetics and fitness - lessons from Drosophila. Heredity 58:103–118
Ruzicka F, Connallon T (2020) Is the X chromosome a hot spot for sexually antagonistic polymorphisms? Biases Curr Empir Tests Classical Theory Proc R Soc, B 287:20201869
Schenker N, Gentleman JF (2001) On judging the significance of differences by examining the overlap between confidence intervals. Am Statistician 55(3):182–186
Spencer HG, Priest NK (2016) The evolution of sex-specific dominance in response to sexually antagonistic selection. Am Naturalist 187:658–666
Stewart AD, Rice WR (2018) Arrest of sex-specific adaptation during the evolution of sexual dimorphism in Drosophila. Nat Ecol Evol 2:1507–1513
Stoltenberg SF, Hirsch J (1997) Y-chromosome effects on Drosophila geotaxis interact with genetic or cytoplasmic background. Anim Behav 53(4):853–864
Via S (1984). The quantitative genetics of polyphagy in an insect herbivore. II. Genetic correlations in larval performance within and among host plants. Evolution 38(4):896–905
Wittenburg D, Teuscher F, Reinsch N (2010) Statistical tools to detect genetic variation for a sex dimorphism in piglet birth weight. Anim Sci 89:622–629
Wolf JB, Brodie ED, Wade MJ (eds) (2000). Epistasis and the evolutionary process. Oxford University Press, New York
Zar JH (1984) Biostatistical Analysis 2nd edn. 324. Prentice Hall, Englewood Cliffs, New Jersey
Acknowledgements
We thank Megan Danis, Kevin Cedeno, Saara Rasool, Mariam Sleem, Karishma Santadasani, Connor McDonnald, and Naiomy Gonzalez Carrero for assistance with sample processing.
Author information
Authors and Affiliations
Contributions
The study was conceived and designed by TMN and KMF, data collection was overseen by TMN and JB, data analysis was conducted by TMN and KMF, and the paper was authored by TMN and KMF.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Associate editor: Aurora Ruiz-Herrera.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Nielsen, T.M., Baldwin, J. & Fedorka, K.M. Gene-poor Y-chromosomes substantially impact male trait heritabilities and may help shape sexually dimorphic evolution. Heredity 130, 236–241 (2023). https://doi.org/10.1038/s41437-023-00596-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-023-00596-8
