
Abstract
Drought and salt stress are important factors that affect plant growth and development and cause crop yield reductions worldwide. Phospholipase C is a class of enzymes that can hydrolyze phospholipids, and it has been shown to play an important role in plant growth regulation and stress response. We used rice as a model to investigate the function of the wheat TaPI-PLC1-2B gene in salt and drought tolerance. For this purpose, we heterologously expressed the TaPI-PLC1-2B gene in rice and studied the transcriptional differences in transgenic and wide-type rice plants in the presence and absence of drought and salt stress. Our results showed that 2130 and 1759 genes expressed differentially in the TaPI-PLC1-2B overexpression rice line under salt and drought stress, respectively. Gene ontology enrichment results showed that differentially expressed genes (DEGs) were significantly enriched in cellular process, metabolic process, stimulus-response, cell, organelle, catalytic activity, and other functional processes under salt and drought stress. In addition, the Kyoto Encyclopedia of Genes and Genomes pathway analysis showed DEG enrichment in plant-pathogen interaction, phosphoinositol, plant hormones, and other signaling pathways under the two stress treatments. Furthermore, the chromosomal localization of salt and drought stress-responsive DEGs showed a clear distribution pattern on specific rice chromosomes. For instance, the greatest number of drought stress-responsive genes mapped to rice chromosomes 1 and 6. The current analysis has built the basis for future explorations to decipher the TaPI-PLC1-2B-mediated plant stress response mechanism in the relatively challenging wheat system.
Similar content being viewed by others

Introduction
Common wheat (Triticum aestivum L.) is the world’s second-largest food crop after rice, with the largest acreage and widest distribution. The increasing world population puts pressure on agriculture, making improved wheat production and quality one of the primary objectives of the breeding programs. Breeding for drought and salt tolerance are two cornerstone traits to improve productivity under projected climatic conditions. Phospholipase C (PLC) is one of the main lipid hydrolases in the plant cells that hydrolyze lipids to produce a series of secondary messenger molecules and lipid derivatives involved in signal transduction (Zheng et al. 2012; Pokotylo et al. 2014). In plants, PLCs can be broadly divided into two subgroups, phosphatidylinositol-specific (PI-PLC) and phosphatidylinositol-non-specific, based on substrate specificity (Hong et al. 2016). PI-PLC can hydrolyze phosphatidylinositol 4,5-diphosphate to fat-soluble diacylglycerol (DAG) and water-soluble inositol triphosphate (IP3), which are secondary messengers involved in cellular signal transduction (Pokotylo et al. 2014). These two signaling molecules trigger the release of Ca2+ ions from the cell, which activate related response genes to induce plant growth and development and response to biotic and abiotic stresses (Aggarwal et al. 2015; Rupwate and Rajasekharan 2012).
Previous studies have demonstrated that different PI-PLC genes express differently in various plants under abiotic stress. For instance, drought stress was shown to induce the expression of NtPI-PLC1 in tobacco and StPI-PLC1 and StPI-PLC2 in the potato (Wang et al. 2013; Kopka et al. 1998). On the other hand, OsPI-PLC1 showed significant upregulation under salt stress in rice (Li et al. 2017). OsPI-PLC3 exhibited induced expression under multiple stresses and a 22-fold higher expression under salt stress (Suzuki et al. 2007). OsPI-PLC4 showed enhanced expression under osmotic stress (Singh et al. 2013). In maize, ectopic expression of ZmPI-PLC1 showed drought tolerance (Wang et al. 2008). Similarly, in Arabidopsis, different PLCs exhibited induced expression under various stresses, for example, AtPI-PLC1 under hypertonic stress, AtPI-PLC3 and AtPI-PLC9 under heat stress (Zheng et al. 2012), AtPI-PLC6 under cold stress (Gaude et al. 2008), and AtPI-PLC7 and AtPI-PLC8 under salt stress, drought, and chilling injury (Peters et al. 2010). To sum up, PLCs played an important role in mitigating abiotic stresses in plants. Albeit the role of the PI-PLC gene being well studied in Arabidopsis and rice, the function of the wheat TaPI-PLC1 gene remains elusive.
Transcript profiling via RNA sequencing is a quantitative method that is increasingly used to compare transcriptional differences over the developmental time course and under various treatments within and between crop plants (Croucher et al. 2009; Xiao et al. 2013). Under biotic and abiotic stresses, plants exhibit significant alterations in their gene expression profiles and metabolism to resist the damage caused by these adversities (Okay et al. 2014; Smita et al. 2020). In this signaling cascade, plants first receive environmental cues and then respond via swiftly altering the gene expression patterns, inducing/suppressing the activity of various kinases, production and signaling of hormones and hormone-like molecules to timely respond to the stress, and minimizing the damage (Meijer and Munnik 2003; Smita et al. 2020). Therefore, transcriptional changes are the first step of a plant’s response to environmental stress, followed by physiological and phenotypic changes. In-depth analysis of gene expression networks and pathway enrichment under stress can help identify genes and regulatory pathways related to stress tolerance.
The effect of the TaPI-PLC1-2B gene on drought and salt tolerance in wheat and its mechanism of action is still unclear. In this study, an effort has been made to shed light on the mechanism of drought and salt tolerance offered by TaPI-PLC1-2B using a rice TaPI-PLC1-2B overexpressing line and comparing the transcript profile of it with the wild-type (WT) control under drought and salt stress. The regulatory network of genes induced by these stresses in the TaPI-PLC1-2B overexpression line was determined through functional enrichment of differentially expressed genes (DEGs) under drought and salt stress. It laid the foundation for applying the TaPI-PLC1-2B gene to improve drought and salt tolerance in wheat and other cereals.
Materials and methods
The generation of transgenic rice plants
The plasmid, pBI121-Ubi-TaPI-PLC1-2B (Supplementary Fig. S1), was constructed for use as the TaPI-PLC1-2B overexpression vector. For rice genetic transformation, the construct was used to transform Agrobacterium tumefaciens strain GV3101. Subsequently, the bacteria were used to co-culture with the WT rice cv. “Nipponbare.” Three primer combinations, M13F/R, TaPLC1-DF/R, and M13R/TaPLC1-DR, were used for the PCR-based identification of the transgenic rice plants (Supplementary Table S3–1).
Plant materials and growth conditions
Seeds of WT Oryza sativa cv. Nipponbare, and overexpressed lines 3 (L3) and 4 (L4) were the genetic material used in this study. The seeds were sterilized, placed on two layers of wet filter paper, and germinated in the dark at 28 °C in a growth chamber. When the buds were 2–3 cm, they were transplanted to the germination boxes containing 1/2 Hoagland nutrient solution for hydroculture at 28 °C day time and 26 °C night time temperatures and a 16 h photoperiod. After a week of growth, the seedlings were treated with 200 mM sodium chloride (NaCl) to impose salt stress or 20% polyethylene glycol (PEG6000) to simulate drought stress. The untreated seedlings were used as the control. Leaf samples for RNA extraction from the control and treatment groups were collected at 0, 6, 12, and 24 h on liquid nitrogen and stored at −80 °C for later use.
Gene expression analysis
Total RNA was isolated from 7-day-old seedlings, converted to cDNA (TIANGEN, Beijing, China), and used as a template in real-time quantitative PCR (qRT-PCR). The expression level of the TaPI-PLC1-2B genes in WT and TaPI-PLC1-2B overexpression transgenic lines, L3 and L4, was studied using the primers described in Wang et al. (2020). The TaPI-PLC1-2B expression level was normalized to the rice OsActin gene (LOC_Os03g61970).
Transcriptome sequencing and data processing
To determine the pathways and genes involved in the TaPI-PLC1-2B-mediated stress response, we performed transcript profiling of the seedling leaves from transgenic rice lines expressing the wheat TaPI-PLC1-2B gene and WT before (0 h) and after (24 h) drought and salt stress treatments. Leaf samples (collected at 0 and 24 h after treatment) were sent to Guangzhou Gene Denovo Biotechnology Co., Ltd. for high-throughput RNA sequencing using the Illumina Hiseq 2000 sequencing platform. Fastp software (https://github.com/OpenGene/fastp) was used to remove the low-quality sequences from the original data (raw data), and Hisat 2.1.0 software was used to compare the clean sequencing data with the rice reference genome to determine the genomic position of reads (Kim et al. 2015). Furthermore, Htseq-Count software was used to count the number of fragments (reads count) compared to each gene in each sample and calculate the number of sequences per million reads mapping to per kilobase length of a gene to yield the Fragments Per Kilobase per Million (FPKM) value.
Determination and analysis of the differentially expressed genes
DESeq2 (Love et al. 2014) software was used to analyze the read count with Q value <0.001 and |log2FC| >1 as a screening criterion for DEGs. Subsequently, Gene Ontology (Ashburner et al. 2000), gene functional annotation analysis, and Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa and Goto 2000) metabolic pathway enrichment analyses were performed on the obtained DEGs.
Localization of DEGs to rice chromosomes
Chromosomal and sub-chromosomal distribution of DEGs in the rice genome was studied by placing these genes on rice chromosomes via the Genome Data Viewer on the NCBI website (https://www.ncbi.nlm.nih.gov/genome). Some of the DEGs for which the conventional rice gene numbers (Os0Xg0XXXXXX) were unavailable, the gene sequences were blasted against the rice genomic DNA sequence using the BLAST Genomes option at the NCBI Blast server, putting the search settings to highly similar sequences (MegaBLAST). The genomic locations of the best matches (mostly perfect matches) were determined using the Genome Data Viewer. After determining the genomic locations of the DEGs, we determined if their locations overlap with genes/QTLs already identified to contribute to drought or salt stress tolerance. The information about known genes/QTLs for drought and salt stress was obtained from the recent reviews on this subject by Panda et al. (2021) and Qin et al. (2020).
In addition, to determine if a chromosome contributes more toward drought/salt stress tolerance, the biases of chromosome size and the number of genes (International Rice Genome Sequencing Project 2005) identified on each chromosome were eliminated via calculating the expected number of genes per Mb or based on the number of genes identified per chromosome following Yang et al. (2021) and comparing it with the observed number of DEGs.
Validation of differentially expressed genes
Ten genes (including Os03g0299700 and Os04g0397800) that exhibited significant differential expression between WT and L3 transgenic lines after PEG treatment were selected for qRT-PCR analysis to confirm the results of the RNA-Seq experiment. Gene-specific primers were designed for the qRT-PCR analysis using Oligo7 software (for primer sequences and PCR conditions, see Supplementary Table S3–2).
Determination of malondialdehyde and chlorophyll content
The content of malondialdehyde (MDA) was determined by the thiobarbituric acid method, specified in the MDA content detection kit (Solarbio BC0020, Beijing). The chlorophyll content was determined by spectrophotometer, following the instructions in the Plant Chlorophyll Content Assay Kit (Boxbio AKPL003M, Beijing).
Results
Heterologous expression of TaPI-PLC1-2B enhanced drought and salt tolerance in the rice
We obtained six independent TaPI-PLC1-2B overexpressing transgenic rice lines, L1–L6 (confirmed by PCR), and raised them to the T3 generation (Supplementary Fig. S2A). These overexpression rice lines were subjected to drought stress using 20% PEG and salt stress using 200 mM NaCl. All transgenic lines (GM) showed high TaPI-PLC1-2B expression relative to the WT. In particular, lines L3 and L4 showed higher TaPI-PLC1-2B expression levels than other lines and were selected for subsequent experiments (Supplementary Fig. S2B).
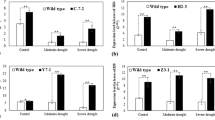
Before drought and salt treatment, the L3, L4, and WT seedlings were indistinguishable in morphology. Under 20% PEG simulated drought stress, transgenic L3 and L4 seedlings showed good tolerance, as most leaves maintained normal growth, except for tip necrosis in some leaves, whereas significant leaf wilting was observed after 2 h of treatment in control (WT) seedlings. With the increase in treatment time, leaf wilting and drooping increased significantly. On the contrary, most L3 and L4 seedlings exhibited normal growth after stress treatment, and no obvious curling, wilting, or lodging was observed (Fig. 1A).

A Phenotype, B the relative TaPI-PLC1-2B expression level, C chlorophyll content, and D MDA content. WT wild type, L3 TaPI-PLC1-2B overexpression transgenic line 3, L4 TaPI-PLC1-2B overexpression transgenic line 4. Each value is a mean ± SE, n = 3, a, b, c, d, e indicated significance at *p < 0.05 and **p < 0.01, and the statistics were calculated between L3/L4 and WT. Bar = 1 cm.
Under drought stress, the expression pattern of TaPI-PLC1-2B was similar in transgenic lines, L3 and L4, exhibiting peak expression values, 3.6-fold of the initial value in L3 and 2.9-fold of the initial value in L4 plants at 12 h after drought treatment. The TaPI-PLC1-2B expression declined soon after reaching the maximum at 12 h (Fig. 1B). However, under salt stress, the TaPI-PLC1-2B expression in L3 and L4 plants exhibited a spike in expression (3-fold of the initial value in L3 and 4.4-fold of the initial value in L4 plants) at 6 h of treatment, followed by a significant decline in expression and later a slight increase at 24 h after treatment (Fig. 2B). The transgenic rice lines showed greater salt tolerance (Fig. 2A). However, there was no difference in chlorophyll and MDA content among WT, L3, and L4 before salt stress and drought stress treatment; however, after treatment, chlorophyll content decreased with the increase in the treatment time (Figs. 1C and 2C), while MDA content increased with the increase in treatment time (Figs. 1D and 2D). Although transgenic L3 and L4 showed the same trend as WT, the decline in chlorophyll content and increase in MDA have slowed down in transgenic lines relative to the WT. It suggested that the TaPI-PLC1-2B overexpression resisted salt and drought stress-induced physiological changes in the overexpression rice lines to a certain extent. Since L3 and L4 plants exhibited similar phenotypes and the TaPI-PLC1-2B expression patterns under drought and salt stress, only one of them, L3, was used in subsequent experiments.

A Phenotype, B the relative TaPI-PLC1-2B expression level, C chlorophyll content, and D MDA content. WT wild type, L3 TaPI-PLC1-2B overexpression transgenic line 3, L4 TaPI-PLC1-2B overexpression transgenic line 4. Each value is a mean ± SE, n = 3; a, b, c, d, e indicated significance at *p < 0.05 and **p < 0.01, and the statistics were calculated between L3/L4 and WT. Bar = 1 cm.
Statistical and comparative evaluation of transcriptome sequencing data
To determine how TaPI-PLC1-2B coordinates the rice response to drought and salt stress, we compared the expression profiles of six samples, namely WT-CK (untreated WT), WT-P (WT treated with drought stress for 24 h), WT-N (WT treated with salt stress for 24 h), GM-CK (untreated L3 transgenic line), GM-P (L3 treated with drought stress for 24 h), and GM-N (L3 treated with salt stress for 24 h). Each sample consisted of three biological replicates.
Collectively, the RNA-Seq analysis was performed on a total of 18 samples. The Q20 ratios of 18 samples ranged from 96.75 to 97.82% and Q30 ratios from 91.65 to 94.10%, indicating that the RNA-Seq data obtained in this study was reliable and could be used for further analysis. The sequence comparisons from all 18 samples with the rice reference genome returned a large number of hits (a minimum of 96.99% and a maximum of 97.86%; Supplementary Table S1), which met the needs of the later analysis. A total of 37,849 known genes (96.63% of the reference genome) and 1318 novel transcripts were identified. Principal component analysis (PCA) showed a clear separation between the WT and transgenic plant samples. The closer distance between replicates represented higher reproducibility (Fig. 3). The correlation heatmap analysis (Supplementary Fig. S4) also showed high reproducibility between biological replicates (higher the reproducibility among biological replicates, the closer the correlation index “R” value to 1). In addition, PCA showed significant differences in expression profiles between untreated and treated samples (Supplementary Fig. S3). Drought and salt stress amplified the difference in expression profiles between transgenic and WT (Supplementary Fig. S3), which was consistent with our phenotypic observations (Fig. 1A, B).

CK untreated, control, WT wild-type, GM transgenic overexpression lines, N NaCl treatment, P PEG treatment.
Analysis of differentially expressed genes
The DEGs between the combinations WT-P-vs-WT-CK, WT-N-vs-WT-CK, GM-N-vs-GM-CK, GM-P-vs-GM-CK, GM-P-vs-WT-P, GM-N-vs-WT-N, and GM-CK-vs-WT-CK were identified (FDR < 0.05 and |log2FC| > 1). A total of 16,794 and 15,421 DEGs were identified after PEG and NaCl treatments, respectively (Fig. 4). A total of 12,452 DEGs were identified in the WT-P-vs-WT-CK combination, among which 4905 were upregulated and 7547 were downregulated. In comparison, only 9692 DEGs, including 3603 upregulated and 6089 downregulated genes, were found in the GM-P-vs-GM-CK combination. Similarly, a total of 11,370 genes expressed differentially in the WT-N-vs-WT-CK combination, among which 4764 DEGs were upregulated, and 6606 DEGs were downregulated, whereas 9044 genes expressed differentially in the GM-N-vs-GM-CK combination, of which 3100 genes were upregulated, and 5944 genes were downregulated (Fig. 3).

A Under drought stress and B Under salt stress.
The results showed that under drought and salt stress, the number of upregulated genes in both WT and TaPI-PLC1-2B overexpression line (L3) was higher than that of downregulated genes, and the number of DEGs in the overexpression line before and after stress treatment was significantly lower than that of WT. When DEGs identified in combinations WT-P/N-vs-WT-CK and GM-P/N-vs-GM-CK were compared, 1759 DEGs were found exclusively in L3 after drought stress (Fig. 4A) and 2335 DEGs after salt stress (Fig. 4B).
GO annotation analysis of DEGs under stress
To explore the function of stress-responsive genes in TaPI-PLC1-2B-background, GO function enrichment analysis was performed on DEGs under each stress treatment. The results showed that 1759 DEGs were uniquely detected in L3 under drought stress, annotated to 48 GO terms belonging to three categories, biological processes, cell components, and molecular functions (Fig. 5A). Under the biological processes category, 563 genes were related to cellular process, 527 genes to metabolic process, and 416 genes to single-organism process. These are the most abundant of the three functional categories. Furthermore, 231 and 210 genes, respectively, fall under functional categories of biological regulation and regulation of the biological processes. Under the molecular functions category, 543 genes belonged to binding, 402 genes to catalytic activity, and 59 genes to transporter activity. Finally, the genes annotated to the cellular component category mainly fall under GO terms cell (516), cell part (507), organelle (360), and membrane (355).

A Enrichment results of DEGs only in L3 under drought stress and B enrichment results of DEGs only in L3 under salt stress.
A total of 2130 genes uniquely differentially expressed in the L3 under salt stress were functionally annotated. These DEGs were mainly enriched in 45 GO terms in the three functional categories, biological processes, cell components, and molecular functions (Fig. 5B). The overall distribution was similar to that under drought stress, and the number of GO terms was also similar, but the number of genes involved in each GO term was different.
KEGG analysis of DEGs under stress
To understand the TaPI-PLC1-2B-mediated plant response to drought and salt stress, we conducted the KEGG pathway enrichment analysis of DEGs in the TaPI-PLC1-2B overexpression line. The results showed that DEGs were mainly enriched in metabolism, genetic information processing, environmental information processing, and organic system under drought and salt stress (Fig. 6 and Supplementary Table S2). The metabolic pathways under drought stress mainly involved galactose, tyrosine, fructose and mannose, pyruvate and phosphoinositol, propane, piperidine, and pyridine alkaloids, as well as phenylalanine, tyrosine, and tryptophan (Fig. 6A and Supplementary Table S2). In the metabolic pathway category “genetic information processing” mainly involved DNA replication (ko03030), mismatch repair (ko03430), homologous recombination (ko03440), and SNARE-related vesicular movement (ko04130). Similarly, the categories “organic system” and “environmental information processing” were significantly enriched in plant-pathogen interaction (ko04626), circadian rhythm (ko04712), mitogen-activated protein kinase (MAPK) signaling pathway (ko04016), and phosphatidylinositol signaling system (ko04070).

DEGs under drought stress (A) and salt stress (B). From outside to inside, the first circle represents the KO (KEGG ORTHOLOGY) pathway annotated, the second circle represents the number of genes annotated in this pathway, the third circle signifies the number of up- or downregulated genes, and the fourth circle represents rich factor (0–1).
Under salt stress, DEGs were primarily enriched in metabolic pathways involving pyrimidine, ether lipids, β-alanine, purine, thiamine, propane, piperidine, and pyridine alkaloid biosynthesis pathways. In the genetic information processing pathway, DEGs mainly showed enrichment in categories, DNA replication, mismatch repair, nucleotide excision repair (ko03420), basal excision repair (ko03410), and ribosome biosynthesis (ko03008). The circadian rhythms pathway was enriched in “plant-pathogen interaction,” and the environmental information processing was primarily enriched in plant hormone signal transduction (ko04075) and phosphatidylinositol signal system (Fig. 6B and Supplementary Table S2).
Localization of DEGs to rice chromosomes
The genomic distribution of 56 drought stress and 33 salt stress-responsive DEGs (consistently detected in all paired comparisons between genotypes and treatments) was studied on rice chromosomes. Eight genes, namely MSTRG.12253, MSTRG.1297, Os02g0237100, Os04g0249600, Os05g0369900, Os06g0200800, Os06g0705400, and Os07g0101000, were found common between the drought and salt stress pools and likely represented genes that express differentially due to the ectopic expression of the TaPI-PLC1-2B gene in rice (Fig. 7A, B). The greatest number of drought stress-responsive genes mapped to rice chromosomes 1 and 6 (eight each), followed by chromosomes 2 and 7 (seven each) and so on, and the least number of DEGs mapped to chromosomes 8 and 9 (one each) (Supplementary Table S4 and Fig. 7B). Similarly, the highest number of DEGs under salt stress mapped to chromosome 2 (six) followed by chromosomes 1, 6, and 7 (four each) to none on chromosome 11 (Supplementary Table S4 and Fig. 7A).

Chromosomal locations of salt (A) and drought (B) stress-responsive DEGs and known salt [OsSOS1 (SALT OVERLY SENSITIVE 1), oSSIT1 (SALT INTOLERANCE 1), and OsZFP245 (ZINC FINGER PROTEIN 245)] (A) and drought [DSM1 (DROUGHT-HYPERSENSITIVE MUTANT 1), AP37 (ASPARTIC PROTEASE 37), OsMIOX (MYO-INOSITOL OXYGENASE), CIPK12 (CALCINEURIN B-LIKE PROTEIN-INTERACTING PROTEIN KINASE 12), and OsCPK9 (CALCIUM-DEPENDENT PROTEIN KINASE 9)] (B) tolerance genes that map in the proximity of DEGs.
In addition, the expected number of DEGs was calculated based on the chromosome sizes and the even distribution of genes per Mb DNA sequence, and the number of genes predicted per chromosome (Supplementary Table S4). Comparing expected (based on chromosome size and gene numbers) and observed values for salt-responsive DEGs revealed that chromosomes 2, 5, 6, 7, and 8 carry more DEGs than the expected number of DEGs based on chromosome size and gene number (Supplementary Table S4). Similar comparisons for the drought-responsive DEGs revealed that chromosomes 1, 2, 4, 6, 7, and 11 carry more DEGs than expected (Supplementary Table S4). Also, many QTLs were mapped in the past for salt stress tolerance (Fan et al. 2021) and drought tolerance (Selamat and Nadarajah 2021) in rice. When studied, we observed a biased distribution of drought and salt stress tolerance QTLs; sometimes, it involved similar chromosomes for the drought and salt stress QTLs and sometimes different (Supplementary Table S4). We also studied genomic locations of known salt and drought tolerance genes in relation to the salt and drought-responsive DEGs to reflect on the genetic regulation of these traits (Fig. 7A, B).
Validation of differentially expressed genes
The expression patterns of Os03g0299700, Os04g0397800, and eight other genes were studied using qRT-PCR in the PEG-treated WT and GM samples (Fig. 8B). The qRT-PCR results corresponded with the RNA-Seq FPKM values (Fig. 8A). Both techniques exhibited the expression level of the ten selected DEGs to be significantly higher in GM than the WT. The qRT-PCR results suggested that the transcriptome data were objective and reliable.

Expression patterns of differentially expressed genes in WT and L3 overexpression line under PEG treatment in RNA-Seq (A) and qRT-PCR (B).
Discussion
Challenged with various stresses, plants have developed a complex gene regulatory network to minimize damage. High-throughput sequencing methods allow obtaining the precise and in-depth transcriptional status of plants, which with the use of various biological databases could be functionally analyzed to shed light on the molecular mechanism of plant responses to various stresses.
In this study, we transformed the wheat gene TaPI-PLC1-2B into the rice cultivar “Nipponbare” to identify its role in drought and salt tolerance via its effect on the MDA and chlorophyll content, the transcription pattern of downstream genes under ambient conditions, and drought or salt stress. Furthermore, we studied the genomic distribution of DEGs under drought and salt stress in rice overexpression lines and their correspondence with known drought and salt tolerance QTLs/genes. This study contributes to our understanding of the TaPI-PLC1-2B-mediated drought and salt-tolerance plant responses and the genetic regulation of drought and salt-tolerance in rice.
The function of PI-PLCs in plant abiotic stress responses has received much attention in the last decade. Our previous study had shown that TaPLC1 was involved in heat and drought tolerance (Wang et al. 2021). In the present study, we observed that rice heterologously expressing TaPI-PLC1-2B also exhibits improved drought and salt tolerance. Under drought conditions, plants with higher chlorophyll content can make better use of light energy and hence generally exhibit greater drought tolerance. The MDA content represents the degree of damage to the plasma membrane. When a plant exhibits greater resistance to drought and salt, the chlorophyll content stays stable and the MDA content low (Luo et al. 2020; Sun et al. 2022). In line with these findings, our results (Figs. 1 and 2C, D) demonstrated that TaPI-PLC1-2B overexpression could improve drought and salt tolerance in monocotyledons via its effect on chlorophyll and MDA contents.
Interestingly, we found that the rice OsPI-PLC1’s expression was not different in the transgenic plants compared to the WT with or without drought stress, while under salt stress, OsPI-PLC1’s expression was higher in the transgenic rice (Supplementary Table S6). Li et al. (2017) showed that OsPLC1 improves salt tolerance in rice. Under the salt stress, OsPLC1 moves from the cytoplasm to the plasma membrane, where it increases cytosolic Ca2+, which controls sodium accumulation in the leaves, leading to improved salt tolerance (Li et al. 2017). Deng et al. (2019) observed that rice OsPI-PLC4 affected the changes in phosphatidic acid (PA), IP3, and Ca2+ content under osmotic stress and positively regulated the rice response to salt and drought.
In addition to monocotyledons, PI-PLC was studied in dicotyledons like Arabidopsis (Abd-El-Haliem et al. 2012; Darwish et al. 2009; Tasma et al. 2008). PLC was shown to mediate the differential expression of DREB1 and DREB2 in response to drought stress (Ruelland et al. 2013; Djafi et al. 2013). Furthermore, AtPI-PLC1 was shown to function downstream of the AP2/ERF transcription factors and induce the expression of downstream genes in response to drought and salt stress (Hsieh et al. 2013). Overexpression of AtPI-PLC3 was also shown to improve drought tolerance in plants, whereas AtPI-PLC4 was shown to negatively regulate salt tolerance by regulating salt stress-induced Ca2+ release and the transcription level of some salt stress-responsive genes (Zhang et al. 2018; Xia et al. 2017). Similarly, overexpression of BnPI-PLC2 in Brassica napus was shown to enhance drought tolerance and promote early flowering and maturation (Georges et al. 2009). AtPI-PLC9 in Arabidopsis was also shown to regulate the transcriptional level of some heat shock proteins by regulating the IP3 and Ca2+ content (Zheng et al. 2012). The heterologous expression of this gene in rice also improved its heat tolerance (Liu et al. 2020). In concert with earlier research, our previous work showed that allogeneic expression of TaPI-PLC1-2B can improve salt and drought tolerance of Arabidopsis (Wang et al. 2020).
In addition, the transcript sequencing of rice lines overexpressing TaPI-PLC1-2B before and after salt and drought stress was performed to understand the TaPI-PLC1-2B-mediated drought and salt responses in these lines. GO enrichment results showed that DEGs under salt and drought stress were significantly enriched in cell processes, metabolic processes, organelles, membranes, catalytic activity, and other functions. Meanwhile, KEGG pathway analysis showed that more genes differentially expressed under drought stress fall under categories like the regulation of plant and pathogen interactions, circadian rhythms, MAPK signaling pathway, and phosphatidylinositol signaling system, while under salt stress in categories like the regulation of plant and pathogen interactions, plant hormone signal transduction, and phosphatidylinositol signaling.
Furthermore, we found that TaPI-PLC1-2B-mediated plant responses to environmental cues are channeled through the phosphatidylinositol signaling system and MAPK signaling pathway under drought stress via the upregulation of OsMAPK4, OsMKK5, OsCML9, and OsIPK1 genes (Supplementary Table S5). Studies have found that IP3 in plants does not directly regulate the release of intracellular calcium ions but rapidly produces IP6 by a series of phosphorylated kinases such as IPK2 and IPK1, which promotes the release of Ca2+ (Lemtiri-Chlieh et al. 2003; Michell 2008). IP6-mediated Ca2+ release is 100 times more efficient than IP3 (Lemtiri-Chlieh et al. 2000). Therefore, IP6 may be the dominant Ca2+ channel binding factor in plant cells, and no protein kinase C activated by DAG has been found in plant cells (Munnik and Testerink 2009). Studies have shown that PLC/DGK pathway is activated when plants are subjected to abiotic stress such as drought and salt, and DAG in plants can be rapidly phosphorylated by DGK to produce PA (Ruelland et al. 2002). PA was shown to activate MAPK, calcium-dependent protein kinase, ion channels, and NADPH oxidase cascade pathways and thus regulate plant responses to adversities (Härtel et al. 2000; Ruelland et al. 2002; Raho et al. 2011). Under salt stress, TaPI-PLC1-2B was related to the phosphatidylinositol signaling system and plant hormone signal transduction. The upregulated genes mainly included OsCML9, OsIAA10, OsbZIP62, and OsSAUR12 (Supplementary Table S5). In Arabidopsis, studies have shown that CML9 (Magnan et al. 2008), CML24 (Delk et al. 2005), CML37 (Scholz et al. 2015), CML38, and CML39 (Vanderbeld and Snedden 2007) transcripts are induced by salt or/and drought stress. OsCML16 (Jung et al. 2017), a direct target of OsERF48, appears to transduce OsERF48 actions to downstream target genes that confer the acquired root phenotype and drought tolerance in rice. Overexpression of OsbZIP62-VP64 (OsbZIP62V) enhanced the drought tolerance and oxidative stress tolerance of the transgenic rice (Yang et al. 2019). This is consistent with our results and provides a reference for further exploring the mechanism of TaPI-PLC1-2B in plants. In Arabidopsis, the SAUR12 gene was 4-fold induced in response to brassinosteroids application (Mourik et al. 2017). Similarly, the knockdown of OsIAA10 enhanced the rice’s resistance to Rice Dwarf Virus (Jin et al. 2016). It is different from what was observed in the present study; hence, we will further study the functions of OsIAA10 and OsSAUR12 in salt and drought tolerance.
In addition, the chromosomal localization of salt and drought-responsive DEGs showed a clear biased distribution of these genes on specific rice chromosomes (Supplementary Table S4 and Fig. 8A, B). The biased distribution pattern of DEGs somewhat coincided with the distribution of QTLs for drought and salt tolerance mapped in earlier studies (Supplementary Table S4) (Fan et al. 2021; Selamat and Nadarajah 2021). The chromosomal distribution of DEGs reconfirmed the complex regulation of these traits (Fan et al. 2021; Selamat and Nadarajah 2021), where some of the DEGs coincided with the known salt (OsSSIT1, OsZFP245, and OsSOS1) and drought (CIPK12, DSM1, OsCPK9, AP37, and OsMIOX) tolerance genes (Fig. 8A, B) (Panda et al. 2021; Qin et al. 2020). This mapping effort might help the future QTL cloning activities for drought and salt tolerance and breeding for these traits via developing markers for these genes to be used in the foreground or background selections in the conventional marker-assisted or genomic selection schemes.
Conclusion
In this study, high-throughput transcriptome sequencing was used to sequence and analyze WT and transgenic rice seedlings (overexpressing the wheat TaPI-PLC-2B gene) under salt and drought stress, and the components of the TaPI-PLC-2B-mediated stress responses were revealed. Precisely, the DEGs and significantly enriched signaling pathways related to the TaPI-PLC-2B-mediated stress responses were identified, providing data to launch further studies to decipher the molecular mechanism of TaPI-PLC-2B-mediated stress responses. In addition, the genomic location of DEGs deciphered in this study could be used to develop molecular markers to breed for these quantitative traits. Furthermore, this study provided ample evidence of salt and drought tolerance conferred by this gene in cereals to undertake a similar TaPI-PLC-2B overexpression study in technically challenging (difficult to transform) common wheat.
References
Abd-El-Haliem A, Meijer H, Tameling W, Vossen JH, Joosten M (2012) Defense activation triggers differential expression of phospholipase-C (PLC) genes and elevated temperature induces phosphatidic acid (PA) accumulation in tomato. Plant Signal Behav 7(9):1073–1078
Aggarwal S, Shukla V, Bhati KK, Kaur M, Sharma S, Singh A et al. (2015) Hormonal regulation and expression profiles of wheat genes involved during phytic acid biosynthesis pathway. Plants 4(2):298–319
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM et al. (2000) Gene ontology: tool for the unification of biology. Nat Genet 25(1):25
Croucher NJ, Fookes MC, Perkins TT, Turner DJ, Marguerat S, Keane T et al. (2009) A simple method for directional transcriptome sequencing using Illumina technology. Nucleic Acids Res 37(22):148–148
Darwish E, Testerink C, Khalil M, El-Shihy O, Munnik T (2009) Phospholipid signaling responses in salt-stressed rice leaves. Plant Cell Physiol 50(5):986–997
Delk NA, Johnson KA, Chowdhury NI, Braam J (2005) CML24, regulated in expression by diverse stimuli, encodes a potential Ca2+ sensor that functions in responses to abscisic acid, daylength, and ion stress. Plant Physiol 139(1):240–253
Deng XJ, Yuan S, Cao HS, Lam SM, Shui GH, Hong YY et al. (2019) Phosphatidylinositol-hydrolyzing phospholipase C4 modulates rice response to salt and drought. Plant Cell Environ 42(2):536–548
Djafi N, Vergnolle C, Cantrel C, Wietrzyñski W, Delage E, Cochet F et al. (2013) The Arabidopsis DREB2 genetic pathway is constitutively repressed by basal phosphoinositide-dependent phospholipase C coupled to diacylglycerol kinase. Front Plant Sci 4:307
Fan X, Jiang H, Meng L, Chen J (2021) Gene mapping, cloning and association analysis for salt tolerance in rice. Int J Mol Sci 22(21):11674
Gaude N, Nakamura Y, Scheible W, Ohta H, Dörmann P (2008) Phospholipase C5 (NPC5) is involved in galactolipid accumulation during phosphate limitation in leaves of Arabidopsis. Plant J 56(1):28–39
Georges F, Das S, Ray H, Bock C, Nokhrina K, Kolla V et al. (2009) Over-expression of Brassica napus phosphatidylinositol-phospholipase C2 in canola induces significant changes in gene expression and phytohormone distribution patterns, enhances drought tolerance and promotes early flowering and maturation. Plant Cell Environ 32(12):1664–1681
Härtel H, Dörmann P, Benning C (2000) DGD1-independent biosynthesis of extraplastidic galactolipids after phosphate deprivation in Arabidopsis. Proc Natl Acad Sci USA 97(19):10649–10654
Hong Y, Zhao J, Guo L, Kim S, Deng X, Wang G et al. (2016) Plant phospholipases D and C and their diverse functions in stress responses. Prog Lipid Res 62:55–74
Hsieh E, Cheng M, Lin T (2013) Functional characterization of an abiotic stress-inducible transcription factor AtERF53 in Arabidopsis thaliana. Plant Mol Biol 82(3):223–237
International Rice Genome Sequencing Project (2005) The map-based sequence of the rice genome. Nature 436:793–800
Jin L, Qin Q, Wang Y, Pu Y, Liu L, Wen X et al. (2016) Rice Dwarf Virus P2 protein hijacks auxin signaling by directly targeting the rice OsIAA10 protein, enhancing viral infection and disease development. PLoS Pathog 12(9):e1005847
Jung H, Chung PJ, Park SH, Redillas M, Kim YS, Suh JW et al. (2017) Overexpression of OsERF48 causes regulation of OsCML16, a calmodulin-like protein gene that enhances root growth and drought tolerance. Plant Biotechnol J 15(10):1295–1308
Kanehisa M, Goto S (2000) KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 28(1):27–30
Kim D, Langmead B, Salzberg S (2015) HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12(4):357–360
Kopka J, Pical C, Gray J (1998) Molecular and enzymatic characterization of three phosphoinositide-specific phospholipase C isoformsfrom potato. Plant Physiol 116:239–250
Lemtiri-Chlieh F, Macrobbie E, Brearley C (2000) Inositol hexakisphosphate is a physiological signal regulating the K+-inward rectifying conductance in guard cells. Proc Natl Acad Sci USA 97(15):8687–8692
Lemtiri-Chlieh F, MacRobbie E, Webb A, Manison N, Brownlee C, Skepper J et al. (2003) Inositol hexakisphosphate mobilizes an endomembrane store of calcium in guard cells. Proc Natl Acad Sci USA 100(17):10091–10095
Li L, Wang F, Yan P, Jing W, Zhang C, Kudla J et al. (2017) A phosphoinositide-specific phospholipase C pathway elicits stress-induced Ca2+ signals and confers salt tolerance to rice. N Phytol 214(3):1172–1187
Liu Y, Liu X, Wang X, Gao K, Qi W, Ren H et al. (2020) Heterologous expression of heat stress-responsive AtPLC9 confers heat tolerance in transgenic rice. BMC Plant Biol 20(1):514
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15(12):550
Luo XY, Li C, He X, Zhang XL, Zhu LF (2020) ABA signaling is negatively regulated by GbWRKY1 through JAZ1 and ABI1 to affect salt and drought tolerance. Plant Cell Rep 39(2):181–194
Magnan F, Ranty B, Charpenteau M, Sotta B, Galaud JP, Aldon D (2008) Mutations in AtCML9, a calmodulin-like protein from Arabidopsis thaliana, alter plant responses to abiotic stress and abscisic acid. Plant J 56:575–589
Meijer H, Munnik T (2003) Phospholipid-based signaling in plants. Annu Rev Plant Biol 54:265–306
Michell R (2008) Inositol derivatives: evolution and functions. Nat Rev Mol Cell Biol 9(2):151–161
Mourik H, Dijk A, Stortenbeker N, Angenent GC, Bemer M (2017) Divergent regulation of Arabidopsis SAUR genes: a focus on the SAUR10-clade. BMC Plant Biol 17(1):245
Munnik T, Testerink C (2009) Plant phospholipid signaling: “in a nutshell”. J Lipid Res 50(Suppl):S260–S265
Okay S, Derelli E, Unver T (2014) Transcriptome-wide identification of bread wheat WRKY transcription factors in response to drought stress. Mol Genet Genomics 289(5):765–781
Panda D, Mishra SS, Behera PK (2021) Drought tolerance in rice: focus on recent mechanisms and approaches. Rice Sci 28(2):119–132
Peters C, Li M, Narasimhan R, Roth M, Welti R, Wang X (2010) Nonspecific phospholipase C NPC4 promotes responses to abscisic acid and tolerance to hyperosmotic stress in Arabidopsis. Plant Cell 22(8):2642–2659
Pokotylo I, Kolesnikov Y, Kravets V, Zachowski A, Ruelland E (2014) Plant phosphoinositide-dependent phospholipases C: variations around a canonical theme. Biochimie 96:144–157
Qin H, Li Y, Huang R (2020) Advances and challenges in the breeding of salt-tolerant rice. Int J Mol Sci 21(21):8385
Raho N, Ramirez L, Lanteri ML, Gonorazky G, Lamattina L, Have AR et al. (2011) Phosphatidic acid production in chitosan-elicited tomato cells, via both phospholipase D and phospholipase C/diacylglycerol kinase, requires nitric oxide. J Plant Physiol 168(6):534–539
Ruelland E, Cantrel C, Gawer M, Kader JC, Zachowski A (2002) Activation of phospholipase C and D is an early response to a cold exposure in Arabidopsis suspension cells. Plant Physiol 130:999–1007
Ruelland E, Djafi N, Zachowski A (2013) The phosphoinositide dependent-phospholipase C pathway differentially controls the basal expression of DREB1 and DREB2 genes. Plant Signal Behav 8(10):e26895
Rupwate SD, Rajasekharan R (2012) Plant phosphoinositide-specific phospholipase C: an insight. Plant Signal Behav 7(10):1281–1283
Scholz SS, Reichelt M, Vadassery J, Mithofer A (2015) Calmodulin-like protein CML37 is a positive regulator of ABA during drought stress in Arabidopsis. Plant Signal Behav 10(6):e1011951
Selamat N, Nadarajah KK (2021) Meta-analysis of quantitative traits loci (QTL) identified in drought response in rice (Oryza sativa L.). Plants 10(4):716
Singh A, Kanwar P, Pandey A, Tyagi A, Sopory S, Kapoor S et al. (2013) Comprehensive genomic analysis and expression profiling of phospholipase C gene family during abiotic stresses and development in rice. PLoS One 8(4):e62494
Smita S, Katiyar A, Lenka SK, Dalal M, Kumar A, Mahtha SK et al. (2020) Gene network modules associated with abiotic stress response in tolerant rice genotypes identified by transcriptome meta-analysis. Funct Integr Genom 20(1):29–49
Sun HM, Xie YZ, Yang WB, Lv Q, Chen LP, Li JT et al. (2022) Membrane-bound transcription factor TaNTL1 positively regulates drought stress tolerance in transgenic Arabidopsis. Plant Physiol Biochem 182:182–193
Suzuki M, Tanaka K, Kuwano M, Yoshida K (2007) Expression pattern of inositol phosphate-related enzymes in rice (Oryza sativa L.): implications for the phytic acid biosynthetic pathway. Gene 405(1-2):55–64
Tasma IM, Brendel V, Whitham SA, Bhattacharyya MK (2008) Expression and evolution of the phosphoinositide-specific phospholipase C gene family in Arabidopsis thaliana. Plant Physiol Bioch 46(7):627–637
Vanderbeld B, Snedden WA (2007) Developmental and stimulus-induced expression patterns of Arabidopsis calmodulin-like genes CML37, CML38 and CML39. Plant Mol Biol 64(6):683–697
Wang CR, Yang AF, Yue GD, Gao Q, Yin HY, Zhang JR (2008) Enhanced expression of phospholipase C 1 (ZmPLC1) improves drought tolerance in transgenic maize. Planta 227(5):1127–1140
Wang L, Zhu X, Liu J, Chu X, Jiao J, Liang Y (2013) Involvement of phospholipases C and D in the defence responses of riboflavin-treated tobacco cells. Protoplasma 250(2):441–449
Wang XG, Liu Y, Li L, Gao X, Dong J, Zhang JC et al. (2020) Genome-wide identification and expression profile analysis of the phospholipase C gene family in wheat (Triticum aestivum L.). Plants 9(7):885
Wang XG, Yao XL, Zhao AH, Yang MM, Zhao WC, LeTourneau MK et al. (2021) Phosphoinositide-specific phospholipase C gene involved in heat and drought tolerance in wheat (Triticum aestivum L.). Genes Genomics 43(10):1167–1177
Xia K, Wang B, Zhang J, Li Y, Yang H, Ren D (2017) Arabidopsis phosphoinositide-specific phospholipase C 4 negatively regulates seedling salt tolerance. Plant Cell Environ 40(8):1317–1331
Xiao J, Jin X, Jia X, Wang H, Gao A, Zhao W et al. (2013) Transcriptome-based discovery of pathways and genes related to resistance against Fusarium head blight in wheat landrace Wangshuibai. BMC Genomics 14:197
Yang MM, Wang XG, Dong J, Zhao WC, Alam T, Thomashow LS et al. (2021) Proteomics reveals the changes that contribute to Fusarium head blight resistance in wheat. Phytopathology 111(2):386–397
Yang S, Xu K, Chen S, Li T, Xia H, Chen L et al. (2019) A stress-responsive bZIP transcription factor OsbZIP62 improves drought and oxidative tolerance in rice. BMC Plant Biol 19(1):260
Zhang Q, Wijk R, Zarza X, Shahbaz M, Hooren M, Guardia A et al. (2018) Knock-down of Arabidopsis PLC5 reduces primary root growth and secondary root formation while overexpression improves drought tolerance and causes stunted root hair growth. Plant Cell Physiol 59(10):2004–2019
Zheng SZ, Liu YL, Li B, Shang Z, Zhou RG, Sun DY (2012) Phosphoinositide-specific phospholipase C9 is involved in the thermotolerance of Arabidopsis. Plant J 69(4):689–700
Acknowledgements
This work was supported by grants from National Natural Science Foundation of China (31801443), Fundamental Research Funds for the Central Universities (Z109021623), Chinese postdoctoral science foundation (2016M602871), and NIFA Hatch/Multi-State Grant (S009).
Author information
Authors and Affiliations
Contributions
Conceptualization: AZ, MY. Methodology: AZ, CC. Validation: AZ, CC, FL, CL, MY. Formal analysis: AZ, SN. Investigation: AZ, CC. Resources: XG, JD, MY. Writing—original draft preparation: AZ, MY, SR. Writing—review and editing: MY, SR, SW. Supervision: SR, XG, JD, MY. funding acquisition: MY.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Associate editor: Yuan-Ming Zhang.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhao, A., Cui, C., Li, F. et al. Heterologous expression of the TaPI-PLC1-2B gene enhanced drought and salt tolerance in transgenic rice seedlings. Heredity 129, 336–345 (2022). https://doi.org/10.1038/s41437-022-00566-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-022-00566-6
