
Abstract
Selection pressure from parasites is thought to be a major force shaping the extreme polymorphism of the major histocompatibility complex (MHC) genes, but the modes and consequences of selection remain unclear. Here, we analyse MHC class II and microsatellite diversity in 16 guppy (Poecilia reticulata) populations from two islands (Trinidad and Tobago) that have been separated for at least 10 ky. Within-population MHC diversity was high, but allele sharing was limited within islands and even lower between islands, suggesting relatively fast turnover of alleles. Allelic lineages strongly supported in phylogenetic analyses tended to be island-specific, suggesting rapid lineage sorting, and an expansion of an allelic lineage private to Tobago was observed. New alleles appear to be generated locally at a detectably high frequency. We did not detect a consistent signature of local adaptation, but FST outlier analysis suggested that balancing selection may be the more general process behind spatial variation in MHC allele frequencies in this system, particularly within Trinidad. We found no evidence for divergent allele advantage within populations, or for decreased genetic structuring of MHC supertypes compared to MHC alleles. The dynamic and complex nature of MHC evolution we observed in guppies, coupled with some evidence for balancing selection shaping MHC allele frequencies, are consistent with Red Queen processes of host-parasite coevolution.
Similar content being viewed by others

Introduction
Genes of the major histocompatibility complex, coding for proteins crucial for the vertebrate adaptive immune response, have several unique properties that have made them a research focus in evolutionary biology since their discovery half a century ago (Dausset 1958). These features include: extreme polymorphism; strong signatures of positive selection, indicating promotion of non-synonymous mutations; and apparently long persistence of allelic lineages, manifested as trans-species polymorphism (TSP; defined here as similarity between pairs of alleles from different species exceeding that between alleles within species; reviewed in Apanius et al. (1997); Radwan et al. (2020); Spurgin and Richardson (2010)). Despite decades of research, understanding of the mechanisms underlying these unique features remains incomplete (Radwan et al. 2020).
Because of their function in immune responses, selection generating and maintaining MHC diversity is presumed to arise from pathogens, although MHC-based mating preferences can also contribute (Bodmer 1972; Ejsmond and Radwan 2015; Hedrick 1992; Milinski 2006; Radwan et al. 2020; Spurgin and Richardson 2010; Takahata and Nei 1990; Winternitz et al. 2013). In its most basic form, the pathogen-mediated model is supported by numerous associations of MHC alleles with susceptibility or resistance to parasites (reviewed in Sommer 2005; Spurgin and Richardson 2010). Mechanisms proposed for how pathogen-mediated balancing selection may then prevent an MHC variant from becoming fixed in a population include heterozygote advantage (‘HA’ henceforth; Doherty and Zinkernagel 1975), fluctuating selection (FS; Hedrick 2002) resulting from temporal changes in parasite composition; and negative frequency-dependent selection (NFDS) underpinned by rare allele advantage resulting from host-parasite coevolution (a so-called ‘Red Queen’ process; Bodmer 1972; Borghans et al. 2004; Ejsmond and Radwan 2015). Empirical evidence exists for all these mechanisms (e.g. Oliver et al. 2009; Osborne et al. 2017; Phillips et al. 2018), which are not mutually exclusive, but their relative roles in generating the unique MHC features described above remain unclear (Radwan et al. 2020). This is further complicated by HA and NFDS being notoriously hard to disentangle in ecological MHC studies, as the rare variants, favoured under NFDS because selection on parasites to adapt to them is weaker compared to that on common alleles, occur almost exclusively in heterozygous states. Nevertheless, work using controlled crosses in the guppy-parasite system to disentangle these effects demonstrated that the advantage of alleles to which parasites have not had a chance to adapt (because they are rare or novel to a host population) can arise independently of HA, supporting the role of Red Queen processes (Phillips et al. 2018). However, such processes may cause rapid turnover of MHC alleles under some combinations of parameters, and thus may be incompatible with widespread TSP (Ejsmond and Radwan 2015).
Further evidence for the complexity of selection acting on the MHC comes from geographic structuring of MHC variation—pronounced structuring is not consistent with a dominant role of simple mechanisms of balancing selection, neither through HA nor NFDS. Balancing selection should reduce differentiation between populations compared to neutral expectations (Glémin et al. 2005; Schierup et al. 2000), but such reduction has been reported only in a minority of MHC population genetic studies (Bryja et al. 2007; Evans et al. 2010). MHC structuring similar to (Babik et al. 2008; Biedrzycka and Radwan 2008), or exceeding, that of neutral markers is more commonly reported (Aguilar and Garza 2006; Awadi et al. 2018; Cammen et al. 2011; Cortázar-Chinarro et al. 2017; Hansen et al. 2007), implying widespread local adaptation. Given that adaptation to local sets of pathogens is likely to select for different pools of MHC alleles (Eizaguirre et al. 2012), as long as species differ in the sets of pathogens they carry, local adaptation should act against long-term maintenance of allelic lineages in different species. This prediction is hard to reconcile with the prevalence of MHC TSP. Balancing selection on supertypes—groups of functionally similar alleles—has recently been proposed as a way to preserve lineages of functionally similar alleles despite rapid turnover of alleles within lineages (Lighten et al. 2017). However, this hypothesis, predicting weaker inter-population genetic differentiation at the level of supertypes compared to alleles, has not yet received convincing empirical support, and awaits firm theoretical underpinnings (Ejsmond et al. 2018). Alternatively, TSP-like patterns may arise from interspecific MHC introgression (Abi-Rached et al. 2011; Dudek et al. 2019), potentially driven by an advantage of immigrant alleles to which parasites have not had a chance to adapt (Phillips et al. 2018). Such introgression might be easier for more divergent foreign alleles that exhibit distinct antigen binding properties (divergent allele advantage; Arora et al. 2019; Pierini and Lenz 2018; Stefan et al. 2019; Wakeland et al. 1990). Finally, genetic structuring may not be a signature of diversifying selection arising from spatial differences in parasite composition, but rather a result of spatial asynchrony in balancing selection that could occur under NFDS or FS (Spurgin and Richardson 2010). Under this scenario NFDS or FS could lead to both strong differentiation of populations on the local scale, and the maintenance of allelic lineages in the long run, even in the absence of gene flow. Consistent with the last two scenarios (i.e. introgression and/or spatial asynchrony in balancing selection), lower differentiation between regions than between populations within regions has been reported for MHC, but without such a pattern being reflected in highly polymorphic neutral markers (Herdegen et al. 2014b; Sagonas et al. 2019). Such patterns could arise if NFDS or FS on its own stabilises isolated MHC gene pools in the long run, or because advantage of immigrant alleles (Phillips et al. 2018) helps to replenish lost MHC variation. Existing theory, however, argues against the former possibility, at least in the case of NFDS, which appears less effective than HA in long-term maintenance of allelic lineages (Ejsmond and Radwan 2015). The effectiveness of FS in this respect has not been, to our knowledge, modelled, but because its ability to maintain MHC polymorphism relies on overdominance of fitness calculated across generations (Hedrick 2002), it can be expected to be similar to HA in terms of the maintenance of TSP, but not necessarily in terms of decreasing population structuring if parasite composition fluctuates out of phase in different locations. Studying regions separated by impenetrable barriers that prevent gene flow, can be useful in distinguishing between these possibilities and ultimately, between the role of balancing selection vs introgression in maintaining TSP.
Here, we analysed MHC class II and microsatellite variation in the guppy, P. reticulata, on the Caribbean islands of Trinidad and Tobago. Periodic fluctuations in sea level during the Pleistocene glacial cycles have isolated or connected these islands to each other and to the South American mainland. The connection between the islands probably existed during the last glacial maximum, with the sea barriers likely persisting since ca 10–15 ky ago (Alexander et al. 2006; Lambeck 2004). Previous work on this system has shown that positive selection acts on a number of codons (positively selected sites, PSS) in the MHC class II peptide binding region (Lighten et al. 2014), and that MHC alleles are associated with resistance to parasites (Fraser and Neff 2010; Phillips et al. 2018). Comparison of MHC class II variation between P. reticulata and its close relative P. obscura suggested extensive TSP. Despite 600 ky of divergence (Willing et al. 2010) between these two species, which inhabit allopatric areas separated by mountain ranges, they were reported to share as much as 31% of MHC alleles (Lighten et al. 2017). However, work in other parts of the guppy’s range has shown that mountains do not constitute impenetrable barriers to gene flow (Herdegen et al. 2014a), so it is not clear whether this extensive allele sharing is due to balancing selection or introgression. Studying MHC composition in the islands of Trinidad and Tobago, separated by the sea barrier gives an opportunity not only to study hierarchical spatial dynamics of allele frequencies in the absence of gene flow, but also to trace the origin of allelic lineages and their persistence vs replacement within the separated islands. The comparison within and between islands sets our empirical data apart from previous work on spatial patterns of MHC diversity in guppies, which have focused almost exclusively on Trinidad (Fraser et al. 2010; Lighten et al. 2017; but see Herdegen et al. 2014b). We first verified, using phylogenetic analysis of mtDNA sequences, whether or not the islands share mitochondrial lineages. Lack of such sharing would suggest impaired gene flow between the islands. We then compared patterns of population structure between and within islands at MHC and microsatellites. We took advantage of the fact that an earlier breeding experiment allowed us to phase MHC haplotypes containing more than one variant. This allowed us to use powerful population genetic tools available for codominant markers, and also to analyse similarity of MHC variants within haplotypes. Neither of these is usually possible in non-model species due to extensive gene duplication and conversion. The assignment of alleles to haplotypes also allowed us to test if divergence of duplicated alleles is favoured by selection, as could be expected if high individual MHC diversity enables response to a wider range of pathogens (Minias et al. 2018). We used the knowledge of MHC haplotypes and hierarchical structure of guppy populations at Trinidad and Tobago to investigate whether (i) MHC haplotypes are under balancing or diversifying selection, as inferred from their higher or lower inter-population differentiation compared to (presumably) neutral, highly polymorphic microsatellite markers, and (ii) whether balancing selection acts more strongly on MHC supertypes than on alleles. Furthermore, we test for divergent allele advantage by investigating (iii) whether MHC alleles are more divergent than expected within populations and within individual haplotypes. Finally, we ask (iv) whether allelic lineages are maintained in populations separated by impenetrable barriers, potentially leading to TSP in the long term, by testing for monophyly of MHC lineages on both islands.
Methods
Sampling and molecular methods
Guppies were collected form 16 rivers/streams across both Trinidad and Tobago in 2014 (locations and sample sizes in Table S1), within a stretch of ≤3 km of a given river sampled. Tail fin clips (2–4 mm2) were taken from 914 fish under light anaesthesia (0.02% methane tricaine sulfonate) and kept in 96% ethanol until DNA isolation. Total DNA was extracted from the tail fins using a MagJet magnetic beads-based nucleic acid purification kit (Thermofisher Scientific). Samples of 462 and 860 individuals, in both cases covering all 16 sampling locations, were used for microsatellite and MHC analyses, respectively. The large sample of MHC-typed fish was collected to maximise haplotyping power in an earlier study (Phillips et al. 2018). Of these, we microsatellite-genotyped a random subset of sufficient size for population genetics. Of these, a further random subset was taken for mtDNA phylogenetic analysis.
Microsatellite genotyping
Samples were genotyped at 15 previously described loci: AG9, AGAT11 (Olendorf et al. 2004); G75, G183, G239, G255, G289, G389 (Shen et al. 2007); Pret-27, Pret-48, Pret-77 (Watanabe et al. 2003); Pr39 (Becher et al. 2002); TACA033 (GenBank Acc. No. AY258896); TAGA033 (GenBank Acc. No. 258667); Pre26 (GenBank Acc. No. AY830946). Loci were amplified following the method of Kenta et al. (2008). PCR products were mixed with GeneScan 500 LIZ size standard and electrophoresed on ABI 3130xl machine. Genotyping was performed using GeneMapper v4.0 (ABI).
MHC genotyping
MHC sequences were amplified and genotyped following the protocol, optimised for guppy MHC II by Phillips et al. (2018); Appendix S1.3), which yielded 100% genotyping repeatability of fish tested in duplicate. Briefly, a 217 bp fragment of the major histocompatibility complex (MHC) class II beta second exon was amplified using forward primer Po_ii_2_01_F_new (GTTGTGTCTTTARCTCSHCTG) and reverse primer Po_eg2 in (ATCGGCTCACCTGATHTA). Primers were accompanied by 6-bp tags allowing for assigning reads to individuals/samples, as well as by the A and P1 adapters necessary for Ion Torrent sequencing. PCR products were divided into pools of ~200 amplicons each and sequenced on an Ion Torrent Personal Genome Machine (PGM; Life Technologies). AmpliSAS software (Sebastian et al. 2016; parameters as in Phillips et al. 2018) was used to infer individual MHC genotypes. MHC haplotypes were phased by genotyping the F1 progeny of captive-reared controlled crosses between pairs of sampled populations, as for the crosses of Phillips et al. (2018). As in Phillips et al. (2018), we inferred the guppy MHC class II to behave as a single locus, with discrete haplotypes that could be composed of multiple erstwhile alleles. Because of this, we performed all subsequent population genetics with the MHC treated as a single diploid marker, with each multi-variant haplotype treated as a single ‘allele’.
Ecological MHC studies routinely perform analyses with so-called MHC ‘supertypes’, clusters of alleles with similar physicochemical properties (e.g. Buczek et al. 2016; Lighten et al. 2017; Schwensow et al. 2007). These clusters are usually inferred statistically (Doytchinova and Flower 2005), but the concept comes from laboratory immunology (Sandberg et al. 1998; Sidney et al. 1996). We performed supertype clustering on a dataset that combined the guppy MHC alleles identified in our study with previously published guppy MHC alleles from Genbank (NCBI BLAST search ‘((poecilia reticulata [Organism]) AND mhc class II) NOT genome’, 14/02/2017). We used discriminant function analysis of principal components (DAPC), implemented in the R package adegenet (Jombart 2008; R Core Team 2016), with the 15 codons identified as under significant positive selection by Lighten et al. (2014), the five amino acid physicochemical descriptors of Sandberg et al. (1998), and the DAPC pipeline and parameters of Phillips et al. (2018).
Phylogenetic analyses
To assess between-island genetic differentiation in a wider phylogeographic context, we sequenced mtDNA from 45 individuals from 16 populations from Tobago and East and South Trinidad (for population names and locations see Table S1), randomly selected from samples genotyped for MHC. Additionally, to provide phylogenetic context to Trinidad-Tobago differentiation, we sequenced mtDNA in 19 samples from 5 populations from the Oropouche drainage. A 953 bp fragment of mitochondrial cytochrome b was amplified and sequenced using the primers and protocol described in Herdegen et al. 2014a, b. These sequences were pooled with a further 81 mitochondrial sequences of fish from 16 populations in Venezuela (Herdegen et al. 2014b; for all sampling sites see Table S1), and a maximum likelihood phylogenetic tree was constructed in MEGA 7 (Kumar et al. 2016) under a HKY substitution model.
Relationships of MHC alleles were visualised in a neighbour joining tree, constructed in MEGA 7 (Kumar et al. 2016) from a matrix of Jukes-Cantor nucleotide distances, with robustness tested using 1000 bootstrap replicates.
Population genetic analyses of microsatellite and MHC markers
All loci in each population were checked for deviations from Hardy–Weinberg expectations, and all pairs of loci, including the MHC, were tested for linkage disequilibrium in GENEPOP 4.3 (Rousset 2008). To control for familywise error rate from multiple testing, Holm-Bonferroni corrections (Holm 1979) were applied to the results of both analyses. Expected and observed heterozygosities were calculated using FSTAT v. 2.9.3 (Goudet 2001). The presence of null alleles in microsatellite markers was tested for, and their frequencies estimated, using the algorithm of Dempster et al. (1977) implemented in FreeNA (Chapuis and Estoup 2007). Per-population null allele frequencies at the MHC locus were estimated in GENEPOP 4.3 (Rousset 2008). Allelic richness (AR) or haplotype richness (HR), the per-population number of alleles or haplotypes, respectively, corrected for sample size, were estimated for each locus and population in FSTAT 2.9.3 (Goudet 2001). The relationship between population mean microsatellite AR and MHC HR was assessed with a linear model in R, with island as a fixed effect. The error distribution of the model did not deviate from normality. Model averaging of the subset with delta AIC less than 2 was performed in the R package MuMIn (Barton 2016).
For microsatellite markers and MHC, Nei’s GST was estimated in Arlequin v. 3.5, correcting for null allele frequencies (Excoffier and Lischer 2010), and visualised using metric multidimensional scaling (MDS) in R (Supplementary Material, Appendix S1). Analysis of molecular variance (AMOVA), also conducted in Arlequin, was performed to partition genetic variance between the two islands (Trinidad and Tobago), and among populations within islands, with the significance of the variance components assessed with 10,000 permutations. GST analysis and AMOVA were performed separately for microsatellite loci and for MHC. The correlations between the genetic distance (linearized pairwise GST values) and log-linear geographic distance between populations within each island were tested, also separately for microsatellite and MHC data, using Mantel’s test in IBD v1.53 (Bohonak 2002).
To test for patterns of genetic structure that might not conform to the pre-defined populations (i.e. sampled streams), such as several population comprising a single unit or one population consisting of multiple sub-units, we ran the HWE-optimising cluster analysis of STRUCTURE v2.3.4 (Pritchard et al. 2000) on microsatellite data. We used an admixture model with uncorrelated allele frequencies, and allowed for null alleles. The burn-in period and the number of MCMC steps were each set to one million. Twenty runs were performed for each K value (number of clusters) in the range 1-20.-20. In order to determine the most probable number of clusters we used two methods, as recommended by Janes et al. (2017). The method outlined by Evanno et al. (2005) is based on assessing the second-order rate of change of the probability of data (ΔK) against the range of K’s, and accepting the K corresponding to the maximum ΔK. The alternative method of Pritchard et al. (2000) recommends accepting the K value with the highest probability of the data. Both statistics were calculated and plotted in Structure Harvester (Earl and von Holdt 2012).
To check for signatures of diversifying or balancing selection on MHC alleles, the hierarchical genome scan method was used, following Excoffier et al. (2009). This method is based on simulating, for each marker, the range of between-population FST values expected under neutrality as a function of heterozygosity, and then identifying the markers that lie in the tails of the simulated distribution. Those are the FST outliers, i.e. loci putatively evolving under diversifying (higher than expected FST) or balancing (lower than expected FST) selection. The hierarchical approach allows assessment of and control for spatially stratified structure among the sampled populations and groups of populations. The analysis was performed in Arlequin, with 50,000 coalescent simulations to obtain confidence intervals under neutrality. To check for island-specific FST outliers, additional non-hierarchical analyses were performed for Trinidad and Tobago separately. For validation, the same three analyses were repeated in Bayescan (Foll and Gaggiotti 2008). If presumably neutral markers themselves are FST outliers, then the power to detect the outlier status of MHC could be reduced (Antao et al. 2008). We therefore replicated our analysis after excluding microsatellite markers that were themselves detected as outliers.
Functional divergence of MHC alleles
The patterns of within- and between-population functional MHC variation may indicate the character and scope of the prevailing selective pressures. For example, an excess of functional variation within populations compared to variation expected from allele frequencies in the region, may indicate selection pressures promoting/maintaining within-population diversity (Lighten et al. 2017). Similarly, reduced functional differentiation between populations could indicate that different populations are experiencing selection pressures that are spatially consistent. As a test of this, we used the Euclidean distances between pairs of MHC alleles in both amino-acid and physicochemical parameter space. For both analyses, we first reduced each MHC variant to the 15 amino acids at codon positions identified by Lighten et al. (2014); see also ‘MHC genotyping’ above) as being under positive selection (PSS). We then tested, using randomisations, if Euclidean distances based on PSS were significantly higher than expectations based on shuffling alleles between populations. We also explored whether patterns of divergence within populations could be influenced by variants born locally due to point mutations or recombination. To identify such candidate variants, we screened populations for private alleles that could be explained as substitutions or chimaeras from other alleles in the same population. If two such alleles were private, both were counted. Alleles that could be explained as either substitution or chimaera were classified as substitutions. We then tested for overrepresentation of local substitutions and chimaeras by pooling all alleles in the island and sampling without replacement for each population the set of alleles of the size equal to the number observed in the focal population. Each island’s P value was then calculated by comparing the observed value to this distribution.
To obtain the Euclidean physicochemical distances between alleles, we adapted the MHC supertyping pipeline described above (see section ‘MHC genotyping’), using each allele’s principal component values as its coordinates in the physicochemical parameter space. For each sampled population, we calculated the mean and standard deviation of Euclidean distance between 10,000 pairs of MHC variants drawn at random from the population. Random draws were made at the haplotype level, with a haplotype’s frequency being its probability of being drawn. If a drawn haplotype comprised multiple MHC variants, one variant was then selected at random. For each pair of populations, we similarly calculated the mean and SD of 10,000 between-population random draws. From these, we were able to derive overall values for within- and between-population Euclidean distance, and to derive these values separately for Trinidad and for Tobago. To derive null expectations against which to compare these values, we performed 500 randomisations in which we repeated the calculations after shuffling allele identities relative to the physicochemical descriptor lines (only 1000 draws were performed within each shuffle, due to computational limitations). Observed values falling significantly outside of their respective null distributions may indicate a significant selection pressure.
We performed an analogous analysis of Euclidean distances between the pairs of variants comprising multi-variant haplotypes, at the island level rather than the population level. For the null distributions, we picked variants at random from each island’s genepool, without replacement. Random sampling was stratified to control for unequal sample sizes between sites—a population was first chosen at random, and a variant in that population was then chosen at random. The observed distributions of Euclidean distances between pairs of variants in multi-variant haplotypes were then compared against the simulated distribution using a Kolmogorov–Smirnov test. To assess whether alleles on the same haplotype may be evolving independently, we repeated the test described above for between-variant physicochemical Euclidean distances using nucleotide distances from the Jukes-Cantor matrix used for building the neighbour-joining phylogenetic tree.
As an additional exploration of functional population structuring at the MHC, we estimated population genetic structure using supertypes instead of alleles, and then used null simulations in which alleles were assigned to supertypes at random to test for biased patterns of structure (Ejsmond et al. 2018; Lighten et al. 2017). Strong stabilising selection on supertypes should produce a weaker population structure than null expectations, whereas divergent selection should produce stronger population structure.
Results
Phylogeography of mtDNA and MHC
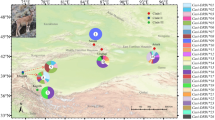
MtDNA haplotypes from Tobago clustered together on the phylogenetic tree, forming a weakly supported clade, while two mtDNA clades occurred in Trinidad—one predominated in the Oropouche drainage inhabited by P. obscura and the other in the Caroni drainage where P. reticulata occurs (Fig. 1c). The Tobago clade is ca. 0.4% divergent from the most closely related clades that occur in Venezuela and La Isla Margarita. If mtDNA is assumed to inform about population splits, the standard mtDNA divergence rate of 2%/my dates the divergence of the Tobago guppies as within the last ca. 200 ky.

a Geographic distribution of sampled populations. Pie diagrams show supertype frequencies in populations. Supertypes are color-coded as in B (dots on branch tips). b Neighbor Joining (NJ) tree showing relationships between MHC class II alleles was rooted with stickleback sequence. c NJ tree showing relationships between mtDNA cytochrome b sequences (945 bp) from P. obscura, P. reticulata and P. wingei, rooted with sequences from. Micropoecilia picta and P. latipinna. Both NJ were constructed from matrices of nucleotide distances; bootstrap supports >70% are marked with asterisks.
Relationships between all MHC alleles from Trinidad and Tobago are shown in Fig. 1b. There were only nine Trinidadian MHC sequences that were identical to those found at Tobago: five in Spring Site population, and the remaining four were each in a separate population. Apart from these shared alleles, lineages with high to moderate bootstrap support (>70%) are not shared among islands, except for a lineage containing five alleles from supertypes ST02 and ST07 (Fig. 1b; see below for supertyping details). The largest group of island-specific alleles, comprising 23 alleles that only occur on Tobago, corresponds to ST15 (yellow in Fig. 1b).
Variation at microsatellites and MHC
The number of microsatellite alleles per locus ranged from 6 to 77 (mean 28.9). At the MHC, 216 haplotypes were discovered, with 222 alleles. Haplotypes were composed of one (n = 187; frequency 0.865), two (n = 28; frequency 0.130) or three (n = 1; frequency 0.005) alleles, while individuals had from one to five alleles.
Population expected and observed heterozygosities are given in Table S2. After correcting for multiple comparisons, significant deviation from Hardy-Weinberg equilibrium was detected in 34/266 (13%) tests, including MHC in a single population (SC; Table S3). Evidence for the presence of null alleles in microsatellite loci, additionally supported by the presence of candidate null homozygotes (0.2 to 2.7% per locus/population), was found in all populations (Table S4) at frequencies ranging per locus/population from <0.01 to 0.31 (median = 0.03), which probably accounts for the deviations from Hardy-Weinberg equilibrium. For the MHC, null allele frequency estimates did not exceed 0.001 in any population. Linkage disequilibrium, after correcting for multiple comparisons, was only detected in 8/1736 tests (Table S5). This may result from drift or admixture rather than from physical linkage between loci, as any pair of loci was only ever in linkage disequilibrium in a single population. We thus did not exclude any loci from further analyses.
Sample size-corrected MHC haplotype richness was on average almost three times higher (10.6) than microsatellite allelic richness (mean across 15 loci = 3.4; Table S6). Both MHC HR and microsatellite AR were significantly higher in Trinidad (HRMHC = 12.95 ± (SE) 1.16, ARmsat = 3.67 ± 0.12) than in Tobago (HRMHC = 7.59 ± (SE) 1.64, ARmsat = 3.00 ± 0.12, t-test, MHC: t1,14 = 2.78, P = 0.015; msat: t1,14 = 3.80, P = 0.002). However, analysis of the set of models containing both the island and microsatellite AR to predict MHC HR retained two models within the best subset, one containing microsatellite AR only (relative weight = 0.66), and one containing both predictors but not their interaction (relative weight = 0.34), with only microsatellite AR remaining significant in model averaging (z = 2.475, P = 0.013; Table S7).
Population structure at MHC and microsatellites
For microsatellites, genetic differentiation between pairs of populations (GST) ranged from 0.02 to 0.79, with mean pairwise GST = 0.40 (Table S8). Mean GST values within islands were 0.41 and 0.33 for Trinidad and Tobago, respectively. The mean of between-island pairwise comparisons was 0.35. In AMOVA, differentiation among islands explained 26.7% of the total variance, while further 22.2% was explained by differences among populations from the same island (all P-values < 0.001). STRUCTURE analysis, interpreted with the Evanno’s method, suggested K = 2 as the most likely number of genetically differentiated clusters, with the clusters corresponding to the two islands (Fig. 2). Pritchard’s method based on the maximum probability of data indicated K = 9 as the most likely number (Fig. S1; see Supplementary Material, Appendix S2 for Structure Harvester results for both methods). Although the two methods gave different K values, no cluster crossed the between-island barrier, suggesting strong differentiation between Trinidad and Tobago. Mantel tests did not detect a significant isolation-by-distance pattern for either the MHC (Trinidad: r = 0.11, P = 0.24; Tobago: r = −0.14, P = 0.72) or microsatellites (Trinidad: r = −0.19, P = 0.80; Tobago: r = 0.16, P = 0.20).

Within-islands arrangement of populations on the graph corresponds to their geographic proximity.
At the MHC, differentiation between pairs of populations (GST) ranged from 0.06 to 0.47, with mean pairwise GST = 0.20 (Table S8). Mean GST value within Trinidad was 0.19, and within Tobago, 0.27. The average GST between populations from the two islands was 0.22.
In multidimensional scaling of microsatellite data, the first axis provided perfect discrimination between the two islands (Fig. 3). The second axis separated several of the Tobago populations while keeping the Trinidad populations tightly clustered, suggesting a stronger effect of drift on Tobagonian populations. MDS of MHC data produced a different pattern: the majority of sites on both islands formed a single central cluster on the first two axes, from which four populations (three Tobago, one Trinidad) were separated in three markedly different directions (Fig. 3).

Two-dimensional scaling of the matrix of pairwise GST values between all sampled populations based on MHC (left) and microsatellite (right) allele frequencies.
There were pronounced differences between the AMOVA breakdowns for the MHC and the microsatellites. At the MHC, 78.4% of the variance was at the within-population level, compared with 51.1% for microsatellites. The between-island level accounted for only 2.3% of MHC variance, compared with 26.7% for microsatellites (between population, within island = 19.3% and 22.2%, respectively).
In hierarchical FST outlier analysis, we found no evidence for MHC structuring departing from that in microsatellites. In contrast, non-hierarchical Bayescan analysis, detected significantly lower structuring in MHC (Supplementary File, Table. S9). However, several microsatellite loci also deviated from neutral expectations: TAGA033, AGAT11, TACA033, Pret77, Pre26, were in the 1st quantile in both analyses, and G239 and AG9 were in the 99th quantile. When we repeated the analysis in Arlequin excluding these outlying microsatellites, the results agreed with those form Bayescan, in that MHC was a significant FST outlier, showing lower structuring than expected under neutrality (Fig. 4). Similar analyses within each island separately revealed lower than expected MHC FST in Trinidad, but not in Tobago. Summaries of all FST outlier analyses are in the Supplementary Material, Table S9).

FST is plotted against expected heterozygosity (heterozygosity within populations/(1-FST)). Confidence intervals limits for FST estimated in relation to heterozygosity are dashed lines. Each molecular marker is a circle. Significant outlier loci at 5% and 1% level are shown in blue and red circles, respectively. The marker significant at 1% level (MHC) is labelled.
Average GST to other populations within the same island calculated based on supertype frequencies was higher than that expected based on haplotype frequencies for 5/9 populations in Trinidad, and lower than expected for 2/6 populations in Tobago (Fig. S2). When we repeated these analyses using DST, we obtained a qualitatively different result, with 2/9 populations remaining above expectations in Trinidad, but all Tobagonian populations showing lower supertype structure than expected (Fig. S2). Jack-knife removal of each supertype suggested a disproportionate influence of ST15 on the patterns observed in Tobago (Fig. S2, see also Supplementary Materials, Appendix S3 for scripts).
Divergent allele advantage
Alleles segregating within populations were not more divergent than expected by chance: Euclidean distances based on PSS were not significantly higher than expectations based on shuffling physicochemical descriptors among alleles within an island (Fig. S3). This result, however, could have been affected by locally born variants decreasing functional distances within populations. In Trinidad, both local substitution and local chimaeras were more common than expected, while in Tobago only local substitutions were more common (Table 1). The fractions of private alleles (binomial glm (Crawley 2013), z1,15 = −1.87, P = 0.061) and local substitutions (z1,15 = −1.85, P = 0.65) did not differ significantly between Trinidad and Tobago, whereas local chimaeras were marginally significantly less frequent in Trinidad (z1,15 = −1.96, P = 0.050).
Nucleotide distance between variants found on the same haplotype was significantly higher than expected from a distribution based on selecting pairs of MHC alleles at random in Trinidad (P = 0.006; Fig. S4), but not in Tobago. However, variants within haplotypes on Trinidad did not show significantly greater functional divergence than random variant pairs (P = 0.24; Fig. S4). The lone multi-variant haplotype on Tobago was not significantly different from random pairings measured in either nucleotide distance or Euclidean physicochemical distance or (P = 0.66 and 0.19, respectively; Fig. S4).
Discussion
Analyses of MHC variation in guppies in a hierarchical geographical context, including phylogenetic and population-genetic approaches, complemented by analyses of functional differentiation between MHC alleles, together provide several useful insights into the complex evolutionary processes shaping MHC variation.
Genetic variation within and between the islands
All analyses point to a strong genetic separation between Trinidad and Tobago. The islands are well separated in mtDNA and microsatellite data. According to geological data, the islands have been separated for approx. 10–15 ky, when sea levels rose by approx. 120 m to their present maximum depth of ~100 m (Alexander et al. 2006; Lambeck 2004; Mychajliw et al. 2020). The divergence of the guppy mtDNA between the islands suggests the separation is no older than 200 ky, assuming the standard mtDNA divergence rate of 2% per million years (Avise et al. 1998).
MHC haplotype richness was significantly higher for Trinidadian guppy populations than for Tobagonian ones. This likely reflects demographic history of the populations, as microsatellite allelic richness, lower on Tobago than on Trinidad, was a significant predictor of MHC haplotype richness. Tobago is much smaller than Trinidad (300 km2 vs. 4770 km2), its geomorphology offers only a relatively small number of suitable rivers for guppies (Mohammed et al. 2015), and, from personal observations, guppies occur at much lower densities in occupied rivers in Tobago than in occupied rivers in Trinidad. Higher effective size of the Trinidad guppy population is supported by the presence of several, divergent mtDNA haplotypes on this island. Haplotype diversity can also reflect colonization history, which could possibly involve several colonization waves, contributing to more mtDNA haplotypes coupled with higher MHC richness at Trinidad. The close grouping of most populations from Trinidad and Tobago in multidimensional scaling (Fig. 3), despite little MHC allele sharing between the islands, suggests high MHC polymorphism in those populations, while the relative separation of the remaining populations (DR, LD, Mag and Arp) may suggest a stronger influence of drift on these locations.
Importantly, we found a signal of local recruitment of new MHC alleles on both islands. This suggests that newly arisen alleles may remain ‘local’ for enough time for such a signal to be detected. However, whether this is due to a high allele ‘birth’ rate or a high probability of retention, both relative to gene flow between populations, remains an open question.
Selection on MHC inferred from population structuring
FST outlier analyses, showing reduced spatial structure in the MHC variation as compared to microsatellite, suggested that balancing selection may act on MHC II in the guppy, but the picture was not consistent among different analyses. FST in MHC (based on haplotype frequencies) was lower compared to expectations based on microsatellite markers in non-hierarchical Bayesian analyses, as well as on Trinidad when each island was analysed separately, but no such signal of balancing selection was found for Tobago, alone or in a hierarchical analysis. Stronger signatures of balancing selection on Trinidad than on Tobago may stem from a number of reasons, for example, stronger drift on Tobago implied by lower genetic diversity and suggested by MDS, stronger selection by parasites or higher connectivity between Trinidadian rivers, which may help equalize MHC allele frequencies by favouring rare immigrant alleles (Phillips et al. 2018). However, all analyses (except for separate analysis for Tobago) indicated 5–7 microsatellite loci as outliers (in both directions; Table S9), which suggests that some loci might be located in regions under selection, or that the population genetic model used to derive neutral expectations is oversimplified. When the seven significant outlier microsatellite loci were excluded, the results of hierarchical outlier analyses agreed with non-hierarchical Bayesian analyses in indicating balancing selection on MHC. While this result should be treated with caution, as after excluding significant microsatellite outliers, confidence intervals were based only on eight microsatellite loci, it is consistent with an earlier report of balancing selection inferred from population structuring in Trinidadian guppies by Fraser and Neff (2010). Nevertheless, based on our analyses it seems safe to conclude that diversifying selection for local adaptation, often inferred by the same method for other species (Aguilar and Garza 2006; Awadi et al. 2018; Cortázar-Chinarro et al. 2017; Hansen et al. 2007), is relatively weak in guppy populations in Trinidad and Tobago.
Divergent allele advantage
One reason for the balancing selection could be rare/novel allele advantage (Phillips et al. 2018), which may be enhanced if it is associated with high functional divergence of the advantageous alleles compared to locally abundant alleles. However, we have not found evidence for divergent allele advantage: alleles segregating within populations were no more divergent than expected by chance. This result is consistent with previous analyses showing that although novel MHC variants can confer significant advantages against local pathogen lineages, novel variants are not significantly more functionally divergent than random pairings of non-identical variants (Phillips et al. 2018, Appendix A6). However, it should be stressed that computationally inferred functional distance may not be able to capture more subtle differences that may still have significant effects on resistance to pathogens (e.g. Zernich et al. 2004). Similarly, we have not found higher than the expected functional divergence of MHC variants linked in the same haplotype, even though nucleotide divergence between the linked variants was higher than expected, indicating that loci within haplotypes evolve to some extent independently.
Overall, our results have not provided support for divergent allele advantage at either population or haplotype level. While genotypes containing more divergent alleles may bind a wider range of antigens (Pierini and Lenz 2018), in natural populations it may be as important to possess variants capable of responding to locally evolving parasites.
Selection on supertypes vs alleles
We did not find consistent evidence that balancing selection on MHC supertypes is stronger than on alleles, which has previously been suggested for guppies on Trinidad (Lighten et al. 2017). Rather, we found a more mixed pattern, with comparisons on Tobago tending towards a balancing selection-type effect, but populations on Trinidad tending towards a divergent selection-type effect (i.e. observed structure greater than expected). These patterns, though, come with two caveats. The first is that the interpretation is heavily influenced by the choice of population genetic statistic. Reduced supertype differentiation on Tobago compared to alleles was observed with DST, but not with GST. We stress that we are raising this as a cautionary note, and are not arguing for one method over the other, as both are useful and reflect different aspects of genetic differentiation (Jost 2008; Wang 2013). The second is that the pattern on Tobago is disproportionately affected by ST15, which occurs in all Tobagonian populations at high frequency. (Lighten et al. 2017) report of this balancing selection-type pattern was also disproportionately affected by a single, widespread supertype, although that effect was not a consequence solely of the supertype’s frequency (Ejsmond et al. 2018), and that supertype is not an analogue of our ST15. Thus (Lighten et al. 2017) method, though potentially informative, needs applying with care, given its extreme sensitivity. Overall, our analyses do not provide support for a special role of balancing selection on MHC supertypes, in agreement with no support for a special role of functional divergence as discussed above.
Maintenance of MHC allelic lineages across islands
For Venezuelan guppy populations, Herdegen et al. (2014) reported that, relative to microsatellites, regions differed less in MHC composition than populations within regions, which they ascribed to diversifying selection prevailing on local, short-term scales and balancing selection prevailing on a continental and longer-term scales. Our AMOVA results also ascribed little variance to the island level, but this appears to result from the fact that lower levels of population structure (i.e. within populations and between populations within islands) accounted for most MHC variation, leaving little variance to be explained by the highest (between-island) level. In fact, only nine alleles were shared between islands. Furthermore, MHC phylogeographic structure appears dynamic, casting doubts about the effectiveness of balancing selection in maintaining allelic lineages in the long term in the absence of gene flow. Most (31/36) of the reasonably well supported (>70%) monophyletic allelic lineages were confined to one island or the other, suggesting that lineage sorting has been progressing quickly since the islands became separated from each other. Still, as yet, there is no indication of reproductive isolation between the two species: fish from the two islands readily produce fully fertile hybrids in captivity (Phillips et al. 2018).
The dynamic nature of MHC evolution is well exemplified by the striking expansion of supertype ST15 on Tobago. One possible driver of this expansion could be differences between the parasite communities of Trinidad and Tobago. We are not aware of a published study that would allow such a comparison, though we note that the major guppy parasites Gyrodactylus turnbulli and G. bullatarudis are present on both islands. Alternatively, stochastic loss of entire supertypes from Tobago might have opened a space for compensatory expansion for the clade comprising ST15. Whatever the mechanism, this result is also not consistent with long-term stability of MHC supertypes. Likewise, the fact that the same clades on different islands preserved different supertypes (e.g. ST01 lost from Tobago), and lack of supertype monophyly, further count against this process. Based on our results, it seems highly improbable that Trinidadian P. reticulata and its allopatric sister species P. obscura, which are estimated to have diverged 600,000 years ago (Willing et al. 2010), share as much as 31% of alleles (Lighten et al. 2017) solely as a result of balancing selection. Future work should therefore investigate the role of introgression of MHC genes between these species.
Conclusions
Overall, our data indicate that selection on MHC class II in the guppy is balancing rather than diversifying, and that this selection is not associated with divergent allele advantage, or is consistently more pronounced if genotypes are classified according to supertypes. Thus, it appears that less functionally influential amino-acid substitutions are under selection in the guppy MHC. At the same time, the nature of processes driving MHC diversity are dynamic, characterised by apparently progressing loss of monophyly of MHC clades from different alleles, coupled with detectable signals of the local birth of new alleles and rapid expansion of some clades, as exemplified by a clade representing ST15. Taken together, our findings are consistent with Red Queen-like processes driving MHC evolution in the guppy, which are expected to be dynamic in nature on one hand, but to cause balancing selection favouring rare and novel alleles on the other hand.
Data archiving
Data and analysis scripts available from the Dryad Digital Repository: https://datadryad.org/stash/dataset/doi:10.5061/dryad.9kd51c5f4.
Change history
09 February 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41437-021-00407-y
References
Abi-Rached L, Jobin MJ, Kulkarni S, McWhinnie A, Dalva K, Gragert L et al. (2011) The shaping of modern human immune systems by multiregional admixture with archaic humans. Science 334(6052):89–94
Aguilar A, Garza JC (2006) A comparison of variability and population structure for major histocompatibility complex and microsatellite loci in California coastal steelhead (Oncorhynchus mykiss Walbaum). Mol Ecol 15(4):923–937
Alexander HJ, Taylor JS, Wu SS-T, Breden F (2006) Parallel evolution and vicariance in the guppy (Poecilia reticulata) over multiple spatial and temporal scales. Evolution 60(11):2352–2369
Antao T, Lopes A, Lopes RJ, Beja-Pereira A, Luikart G (2008) LOSITAN: a workbench to detect molecular adaptation based on a Fst-outlier method. BMC Bioinforma 9(1):323
Apanius V, Penn D, Slev PR, Ruff LR, Potts WK (1997) The nature of selection on the major histocompatibility complex. Crit Rev Immunol 17(2):179–224
Arora J, Pierini F, McLaren PJ, Carrington M, Fellay J, Lenz TL (2019) HLA heterozygote advantage against HIV-1 is driven by quantitative and qualitative differences in HLA allele-specific peptide presentation. Mol Biol Evol. 37(3):639–650
Avise JC, Walker D, Johns GC (1998) Speciation durations and Pleistocene effects on vertebrate phylogeography. Proc R Soc B-Biol Sci 265(1407):1707–1712
Awadi A, Ben Slimen H, Smith S, Knauer F, Makni M, Suchentrunk F (2018) Positive selection and climatic effects on MHC class II gene diversity in hares (Lepus capensis) from a steep ecological gradient. Sci Rep. 8(1):11514
Babik W, Pabijan M, Radwan J (2008) Contrasting patterns of variation in MHC loci in the Alpine newt. Mol Ecol 17(10):2339–2355
Barton K (2016) MuMIn: Multi-Model Inference. R package version 1.15.6. https://CRAN.R-project.org/package=MuMIn
Becher SA, Russell ST, Magurran AE (2002) Isolation and characterization of polymorphic microsatellites in the Trinidadian guppy (Poecilia reticulata). Mol Ecol Notes 2:456–458
Biedrzycka A, Radwan J (2008) Population fragmentation and major histocompatibility complex variation in the spotted suslik, Spermophilus suslicus. Mol Ecol 17(22):4801–4811
Bodmer W (1972) Evolutionary significance of the HL-A system. Nature 237:139–145
Bohonak AJ (2002) IBD (Isolation by Distance): a program for analyses of isolation by distance. J Heredity 93(2):153–154
Borghans JAM, Beltman JB, De Boer RJ (2004) MHC polymorphism under host-pathogen coevolution. Immunogenetics 55(11):732–739
Bryja J, Charbonnel N, Berthier K, Galan M, Cosson JF (2007) Density-related changes in selection pattern for major histocompatibility complex genes in fluctuating populations of voles. Mol Ecol 16(23):5084–5097
Buczek M, Okarma H, Demiaszkiewicz AW, Radwan J (2016) MHC, parasites and antler development in red deer: no support for the Hamilton & Zuk hypothesis. J Evol Biol 29(3):617–632
Cammen K, Hoffman JI, Knapp LA, Harwood J, Amos W (2011) Geographic variation of the major histocompatibility complex in Eastern Atlantic grey seals (Halichoerus grypus). Mol Ecol 20(4):740–752
Chapuis MP, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evolution 24(3):621–631
Cortázar-Chinarro M, Lattenkamp EZ, Meyer-Lucht Y, Luquet E, Laurila A, Höglund J (2017) Drift, selection, or migration? Processes affecting genetic differentiation and variation along a latitudinal gradient in an amphibian. BMC Evolut Biol 17(1):189
Crawley MJ (2013) The R Book, 2nd edn. Wiley, Chichester
Dausset J (1958) Iso-leuco-anticorps. Acta Haematologica 20(1-4):156–166
Dempster AP, Laird NM, Rubin DB (1977) Maximum Likelihood from Incomplete Data Via the EM Algorithm. J R Stat Soc: Ser B (Methodol) 39(1):1–22
Doherty PC, Zinkernagel RM (1975) Enhanced immunological surveillance in mice heterozygous at H-2 gene complex. Nature 256(5512):50–52
Doytchinova IA, Flower DR (2005) In silico identification of supertypes for class II MHCs. J Immunol 174(11):7085–7095
Dudek K, Gaczorek TS, Zieliński P, Babik W (2019) Massive introgression of major histocompatibility complex (MHC) genes in newt hybrid zones. Mol Ecol 28(21):4798–4810
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Eizaguirre C, Lenz TL, Kalbe M, Milinski M (2012) Divergent selection on locally adapted major histocompatibility complex immune genes experimentally proven in the field. Ecol Lett 15(7):723–731
Ejsmond MJ, Phillips KP, Babik W, Radwan J (2018) The role of MHC supertypes in promoting trans-species polymorphism remains an open question. Nat Commun 9(1):4362
Ejsmond MJ, Radwan J (2015) Red Queen processes drive positive selection on Major Histocompatibility Complex (MHC) genes. PLoS Comput Biol 11(11):e1004627–e1004627
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14(8):2611–2620
Evans ML, Neff BD, Heath DD (2010) MHC genetic structure and divergence across populations of Chinook salmon (Oncorhynchus tshawytscha). Heredity (Edinb) 104(5):449–459
Excoffier L, Hofer T, Foll M (2009) Detecting loci under selection in a hierarchically structured population. Heredity 103:285–298
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567
Foll M, Gaggiotti O (2008) A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a bayesian perspective. Genetics 180(2):977
Fraser A, Neff B (2010) Parasite mediated homogenizing selectionat the MHC in guppies. Genetica 138:273–278
Fraser A, Ramnarine I, Neff B (2010) Selection at the MHC class IIB locus across guppy (Poecilia reticulata) populations. Heredity 104:155–167
Glémin S, Gaude T, Guillemin M-L, Lourmas M, Olivieri I, Mignot A (2005) Balancing selection in the wild: testing population genetics theory of self-incompatibility in the rare species Brassica insularis. Genetics 17:279–289
Goudet J (2001) Available from http://www.unil.ch/izea/softwares/fstat.html
Hansen MM, Skaala O, Jensen LF, Bekkevold D, Mensberg KLD (2007) Gene flow, effective population size and selection at major histocompatibility complex genes: brown trout in the Hardanger Fjord, Norway. Mol Ecol 16(7):1413–1425
Hedrick PW (1992) Female choice and variation in the major histocompatibility complex. Genetics 132:575–581
Hedrick PW (2002) Pathogen resistance and genetic variation at MHC loci. Evolution 56(10):1902–1908
Herdegen M, A. HJ, Babik W, Mavárez J, Breden F, Radwan J (2014a) Population structure of guppies in north-eastern Venezuela, the area of putative incipient speciation. BMC Evolut Biol 14:28
Herdegen M, Babik W, Radwan J (2014b) Selective pressures on MHC class II genes in the guppy (Poecilia reticulata) as inferred by hierarchical analysis of population structure. J Evolut Biol 27(11):2347–2359
Holm S (1979) A simple sequentially rejective multiple test procedure. Scand J Stat 6(2):65–70
Janes JK, Miller JM, Dupuis JR, Malenfant RM, Gorrell JC, Cullingham CI et al. (2017) The K = 2 conundrum. Mol Ecol 26(14):3594–3602
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24(11):1403–1405
Jost L (2008) G(ST) and its relatives do not measure differentiation. Mol Ecol 17(18):4015–4026
Kenta T, Gratten J, Haigh NS, Hinten GN, Slate J, Butlin RK et al. (2008) Multiplex SNP-SCALE: a cost-effective medium-throughput single nucleotide polymorphism genotyping method. Mol Ecol Resour 8(6):1230–1238
Kumar S, Stecher G, K. T (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evolution 33:1870–1874
Lambeck K (2004) Sea-level change through the last glacial cycle: geophysical, glaciological and palaeogeographic consequences. Comptes Rendus Geosci 336(7):677–689
Lighten J, Papadopulos AST, Mohammed RS, Ward BJ, G. Paterson I, Baillie L et al. (2017) Evolutionary genetics of immunological supertypes reveals two faces of the Red Queen. Nat Commun 8(1):1294
Lighten J, Van Oosterhout C, Paterson IG, McMullan M, Bentzen P (2014) Ultra-deep Illumina sequencing accurately identifies MHC class IIb alleles and provides evidence for copy number variation in the guppy (Poecilia reticulata). Mol Ecol Resour. 14(4):753–767
Milinski M (2006) The major histocompatibility complex, sexual selection, and mate choice. Annu Rev Ecol Evolution Syst 37:159–186
Minias P, Pikus E, Dunn PO, Whittingham LA (2018) Evolution of copy number at the MHC varies across the avian tree of life. Genome Biol Evolution 11(1):17–28
Mohammed RS, Phillips K, Khan K, Bhagan A (2015) A survey of freshwater fish distribution in Tobago, West Indies. Living World, J Trinidad Tobago Field Naturalists’ Club 2015:44–51
Mychajliw AM, Mohammed RS, Rice KA, Farrell AB, Rincón AD, McAfee R et al. (2020) The biogeography of “breas”: Contextualizing the taphonomy, ecology, and diversity of Trinidad’s asphaltic fossil record. Quat Sci Rev 232:106179
Olendorf R, Reudi B, Hughes KA (2004) Primers for 12 polymorphic microsatellite DNA loci from the guppy (Poecilia reticulata). Mol Ecol Notes 4:668–671
Oliver MK, Telfer S, Piertney SB (2009) Major histocompatibility complex (MHC) heterozygote superiority to natural multi-parasite infections in the water vole (Arvicola terrestris). Proc R Soc B-Biol Sci 276(1659):1119–1128
Osborne MJ, Pilger TJ, Lusk JD, Turner TF (2017) Spatio-temporal variation in parasite communities maintains diversity at the major histocompatibility complex class IIβ in the endangered Rio Grande silvery minnow. Mol Ecol 26(2):471–489
Phillips KP, Cable J, Mohammed RS, Herdegen-Radwan M, Raubic J, Przesmycka KJ et al. (2018) Immunogenetic novelty confers a selective advantage in host–pathogen coevolution. Proc Natl Acad Sci USA 115(7):1552–1557
Pierini F, Lenz TL (2018) Divergent allele advantage at human MHC Genes: signatures of past and ongoing selection. Mol Biol Evolution 35(9):2145–2158
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
R Core Team (2016) R Foundation for Statistical Computing: Vienna.
Radwan J, Babik W, Kaufman J, Lenz TL, Winternitz J (2020) Advances in the evolutionary understanding of MHC polymorphism. Trends Genet. 36(4):298–311
Rousset F (2008) GENEPOP ‘ 007: a complete re-implementation of the GENEPOP software for Windows and Linux. Mol Ecol Resour 8(1):103–106
Sagonas K, Runemark A, Antoniou A, Lymberakis P, Pafilis P, Valakos ED et al. (2019) Selection, drift, and introgression shape MHC polymorphism in lizards. Heredity 122(4):468–484
Sandberg M, Eriksson L, Jonsson J, Sjostrom M, Wold S (1998) New chemical descriptors relevant for the design of biologically active peptides. A multivariate characterization of 87 amino acids. J Medicinal Chem 41(14):2481–2491
Schierup MH, Vekemans X, Charlesworth D (2000) The effect of hitchhiking on genes linked to a balanced polymorphism in a subdivided population. Genet Resour 76:63–73
Schwensow N, Fietz J, Dausmann KH, Sommer S (2007) Neutral versus adaptive genetic variation in parasite resistance: importance of major histocompatibility complex supertypes in a free-ranging primate. Heredity 99(3):265–277
Sebastian A, Herdegen M, Migalska M, Radwan J (2016) AMPLISAS: a web server for multilocus genotyping using next-generation amplicon sequencing data. Mol Ecol Resour 16(2):498–510
Shen X, Guanpin Y, Meijie L (2007) Development of 51 genomic microsatellite DNA markers of guppy (Poecilia reticulata) and their application in closely related species. Mol Ecol Notes 7:302–306
Sidney J, Grey HM, Kubo RT, Sette A (1996) Practical, biochemical and evolutionary implications of the discovery of HLA class I supermotifs. Immunol Today 17(6):261–266
Sommer S (2005) The importance of immune gene variability (MHC) in evolutionary ecology and conservation. Front Zool 12:16
Spurgin LG, Richardson DS (2010) How pathogens drive genetic diversity: MHC, mechanisms and misunderstandings. Proc R Soc B Biol Sci 277(1684):979–988
Stefan T, Matthews L, Prada JM, Mair C, Reeve R, Stear MJ (2019) Divergent allele advantage provides a quantitative model for maintaining alleles with a wide range of intrinsic merits. Genetics 212(2):553–564
Takahata N, Nei M (1990) Allelic genealogy under overdominant and frequency-dependent selection and polymorphism of Major Histocompatibility Complex loci. Genetics 124(4):967–978
Wakeland EK, Boehme S, She JX, Lu CC, Mcindoe RA, Cheng I et al. (1990) Ancestral polymorphisms of Mhc class-II genes—divergent allele advantage. Immunologic Res 9(2):115–122
Wang J (2013) On the measurements of genetic differentiation among populations. Genet Res 94(5):275–289
Watanabe T, Yoshida M, Nakajima M, Taniguchi N (2003) Isolation and characterization of 43 microsatellite DNA markers for guppy (Poecilia reticulata). Mol Ecol Notes 3(4):487–490
Willing EM, Bentzen P, van Oosterhout C, Hoffmann M, Cable J, Breden F et al. (2010) Genome-wide single nucleotide polymorphisms reveal population history and adaptive divergence in wild guppies. Mol Ecol 19(5):968–984
Winternitz JC, Minchey SG, Garamszegi LZ, Huang S, Stephens PR, Altizer S (2013) Sexual selection explains more functional variation in the mammalian major histocompatibility complex than parasitism. Proc R Soc B Biol Sci 280(1769):2013.1605
Zernich D, Purcell AW, Macdonald WA, Kjer-Nielsen L, Ely LK, Laham N et al. (2004) Natural HLA class I polymorphism controls the pathway of antigen presentation and susceptibility to viral evasion. J Exp Med 200(1):13–24
Acknowledgements
We thank Associate Editor and three anonymous reviewers for their comments of the previous version of our manuscript. Most mtDNA sequences have been produced by SMTB2018 students. This project was funded by a Polish National Science Centre grant UMO-2013/10/M/NZ8/00253. KPP is supported by Science Foundation Ireland.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Associate editor Paul Sunnucks
Supplementary information
Rights and permissions
About this article

Cite this article
Herdegen-Radwan, M., Phillips, K.P., Babik, W. et al. Balancing selection versus allele and supertype turnover in MHC class II genes in guppies. Heredity 126, 548–560 (2021). https://doi.org/10.1038/s41437-020-00369-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-020-00369-7
