
Abstract
Purpose
Consanguineous couples are typically counseled based on familial pathogenic variants identified in affected children. The residual risk for additional autosomal recessive (AR) variants, however, remains largely understudied.
Methods
First, we surveyed pedigrees of 1,859 consanguineous families for evidence of more than one AR disease. Second, we mined our database of 1,773 molecularly tested consanguineous families to identify those with more than one AR disease. Finally, we surveyed 88 women from consanguineous unions who have undergone targeted prenatal testing for a familial AR variant and followed the pregnancy outcome (n = 144).
Results
We found suggestive evidence of more than one AR disease in 1.94% of consanguineous pedigrees surveyed. Of 1,773 molecularly characterized consanguineous families, 2.93% had evidence of at least two AR diseases (3.54% for first cousin or closer and 2.72% for second cousin or more distant). Furthermore, we found that in 2.78% of pregnancies negative for the familial variant, the pregnancy outcome was a child with a different AR disease.
Conclusion
Our results show that when counseling consanguineous couples for a familial AR variant, ~3% residual risk for additional AR variants should be discussed. This suggests that a broader testing strategy in consanguineous couples should be considered.
Similar content being viewed by others

INTRODUCTION
Consanguinity remains a common practice in many parts of the world and it is estimated that countries where 20% to over 50% of unions are between individuals who are third cousin or closer comprise one-seventh of the world population [1]. Regardless of the historical reasons that nurtured consanguinity, it remains a highly desirable option in many countries despite its documented health consequences. Its detrimental impact on several measures of health notwithstanding [2], it is well established that consanguinity increases the risk of rare autosomal recessive conditions given the higher probability of shared carrier status for rare alleles among consanguineous compared to randomly selected couples [3, 4]. Saudi Arabia is a country where more than half marriages are between consanguineous couples [5]. Consistently, large exome studies from Saudi Arabia have demonstrated a preponderance of autosomal recessive variants in the etiology of Mendelian diseases in a pattern strikingly different from outbred populations where de novo dominant variants contribute substantially more to the overall variant spectrum of diseases [3, 6,7,8].
Unlike de novo variants, which cannot be predicted a priori, recessive variants lend themselves readily to established preventive strategies such as carrier screening, prenatal diagnosis, and preimplantation genetic testing. This difference has also been the cornerstone of new initiatives aimed at expanding access to comprehensive carrier screening of severe pediatric onset diseases at the population level even in countries with very low consanguinity rates [9, 10]. At the level of individual couples who present for counseling regarding a familial recessive variant, it is the standard practice to offer reproductive options that are targeted to the familial variant in addition to the standard screening protocol in place, e.g., aneuploidy screen. The same practice is often employed when the couple is consanguineous because there are no clear guidelines that address this special scenario. Here, we aim to inform future guidelines by leveraging our large database of consanguineous couples to extract estimates of the residual risk for additional recessive diseases.
MATERIALS AND METHODS
Human subjects
Our database comprises consanguineous families that have been recruited over the period 2007–2020 because of family history of suspected Mendelian diseases. Informed consent was obtained from all families each of which was recruited under the relevant institutional review board (IRB)–approved research protocol for their respective disease (KFSHRC RAC #2070023, 2080006, 2090035, 2080033, 2140016, and 2121053). The consent permits us to construct detailed pedigrees and collect full clinical data including follow-up data on future pregnancies.
Residual risk estimation
We took three approaches to estimate the residual risk of a second (or more) recessive disease in consanguineous families:
Approach 1: We surveyed drawn pedigrees of consanguineous families in search of suggestive evidence of a second recessive disease. Given the extensive nature of our pedigrees, we opted to have a conservative approach by only considering evidence from the nuclear rather than the extended family. A pedigree was considered suggestive of more than one recessive disease if either of the following was observed: (1) one or more children with a known autosomal recessive disease other than the suspected recessive disease for which consultation was initiated, or (2) two or more children with a similar but undefined phenotype that is distinct from the suspected recessive disease for which consultation was initiated. We also opted to exclude seemingly dominant inheritance even though it may represent pseudodominance in consanguineous pedigrees to ensure a conservative estimate.
Approach 2: We identified all molecularly characterized consanguineous families in our cohort and calculated the percentage of those that were found to harbor two or more recessive pathogenic variants in the same nuclear family. This includes instances where the dual (or more) occurrence of these variants was observed in the same individual. To avoid artificial inflation of the estimate, the denominator was restricted to families in which the molecular analysis of all observed phenotypes has been completed. In other words, if a family with two or more phenotypes was only solved for one of these phenotypes, it did not count because it was considered “incomplete.”
Approach 3: We surveyed all consanguineous families in our cohort that underwent targeted prenatal diagnosis for a familial recessive variant in a previous child. By documenting the health outcome of these pregnancies, we estimated the percentage of those that tested negative for the known variant but were found to harbor a different autosomal recessive pathogenic variant.
Coefficient of relationship calculation
From our cohort of 1,773 molecularly characterized families, genome-wide genotyping based on Axiom SNP array was available for 1,233 families to calculate the coefficient of relationship. We used IBDelphi (http://www.insilicase.com/Guide/ibdelphi.aspx) to calculate the coefficient of relationship of the parents as a function of the proportion of identical by descent (IBD) regions in the parents’ genomes. When only children’s genotyping data were available, we deduced the parental coefficient of relationship by using PLINK to calculate the inbreeding coefficient of the children (coefficient of relationship = inbreeding coefficient × 2).
RESULTS
Prevalence of two or more recessive diseases in consanguineous pedigrees based on family history only
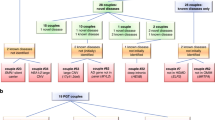
Our aim was to quantify the risk of multiple autosomal recessive diseases in consanguineous families. For our first approach, we surveyed the drawn pedigrees of 1,859 consanguineous families for evidence of more than one autosomal recessive disease. Pedigrees were scored based on the nuclear families and we were able to find 36 pedigrees where there was compelling evidence of more than one autosomal recessive disease (1.94%) (Fig. 1).

Approach 1 investigates the prevalence of recessive diseases in consanguineous pedigrees based on family history. Approach 2 investigates the prevalence of two or more recessive diseases in consanguineous pedigrees based on molecular analysis. Approach 3 considers the pregnancy outcomes in consanguineous couples who underwent targeted prenatal screening for a familial recessive variant. AR autosomal recessive.
Prevalence of two or more recessive diseases in consanguineous pedigrees based on molecular analysis
Our second approach was to survey our cohort of 1,773 molecularly characterized consanguineous families with at least one autosomal recessive disease and score for families with more than one molecularly characterized autosomal recessive disease. Our analysis showed 51 families with two autosomal recessive diseases and one with three autosomal recessive diseases (52/1,773 [2.93%]) (Table 1, Fig. 1 and Fig. S1). Please note the limited overlap between the cohorts used in approaches 1 and 2 (Fig. S2). This is due to lack of digital pedigrees for many families, the presence of multilocus phenotypes, and the fact that many families remain incompletely characterized molecularly.
To provide a higher resolution of the above estimated risk, we stratified our cohort based on the calculated coefficient of relationship into three categories:
Category 1: families with coefficient of relationship corresponding to first-degree cousins or higher (coefficient of relationship ≥12.5%)
Category 2: families with coefficient of relationship between first- and second-degree cousins (12.5% > coefficient of relationship ≥3.125%)
Category 3: families with coefficient of relationship lower than second-degree cousins (coefficient of relationship <3.125%).
Our analysis revealed that the risk of more than one autosomal recessive disease in the first category (families with coefficient of relationship corresponding to first-degree cousins or higher) was 3.65% (32 of 876 families). The calculated risk was lower in families with coefficient of relationship lower than first-degree cousins (2.72%).
Pregnancy outcomes of consanguineous couples who underwent targeted prenatal screening for a familial recessive variant
Finally, we assessed the pregnancy outcomes of 144 pregnancies from 88 consanguineous couples who underwent targeted prenatal diagnosis for a recessive variant that had been identified in a previously affected child/pregnancy. We found that of the 144 pregnancies, 81 resulted in healthy outcomes, 46 had a fetus affected with the same disease as the index, and 17 resulted in other outcomes. For the latter, these can be broken down as follows: two pregnancies with chromosomal abnormalities (trisomy 21), two cases of autosomal dominant diseases, nine cases that are still under molecular investigation (stillbirth, intrauterine fetal demise [IUFD], and congenital heart disease), three cases that are molecularly proven to be autosomal recessive, and one case showing an autosomal recessive mode of inheritance because the phenotype was also observed in cousins but is still under molecular investigation (Fig. 1 and Table S1). Therefore, our data show that 4 of the 144 pregnancies resulted in another autosomal recessive disease in the same family (2.78%).
DISCUSSION
The occurrence of multiple autosomal recessive conditions within consanguineous families has long been recognized. In the pre-exome era, this was considered a challenge in positional mapping especially when the phenotypes are overlapping such that affected members were lumped together even though their underlying variants are different [11]. We are not aware, however, of a systematic analysis of this phenomenon in a large cohort of consanguineous families. Thus, our finding that ~3% of consanguineous families have more than one autosomal recessive disease can be considered an important reference range given the large size of the studied cohort. This would be consistent with a prior study in which we specifically interrogated couples in our consanguineous population and found that 9.7% shared the carrier status for a recessive variant other than the one observed in their affected child [7]. Remarkably, a nearly identical estimate was arrived at by Mor-Shaked and colleagues who detected secondary shared carrier status for pathogenic and likely pathogenic variants leading to autosomal recessive disorders in 10 of 102 (9.8%) consanguineous couples they analyzed [12]. Unlike these previous reports that predicted the residual risk for additional autosomal recessive diseases based on the shared carrier status for variants among parents, our study is based on actual, observed outcomes in a large cohort of molecularly characterized consanguineous families including prospectively followed pregnancies.
Traditional genetic counseling informs couples of a background risk for major birth malformation of 3% and near doubling of this if the couple is consanguineous, which was borne out by a large population-based birth defect registry from our consanguineous population and others [13, 14]. What remains unclear, however, is how to translate this into actionable information in the era of genome sequencing. For example, can this be deconstructed into individual recessive diseases for which prevention can be offered? Since the previously mentioned estimate is based on a range of risks, it does not provide a personalized risk for the couple seeking counseling. Consanguineous couples continue to be offered targeted prenatal diagnosis for their familial recessive variants with ambiguous counseling about their residual risk for additional diseases. In view of our estimate of ~3% (with higher calculated risk if the couple are first cousins [3.65%]) it appears reasonable in an era where exome sequencing is commonplace to propose offering expanded carrier screening at least to those who choose it after they are informed of the significant residual risk. This is especially salient given that all the additional recessive variants encountered in our cohort are identifiable by exome sequencing. Therefore, we recommend that exome sequencing rather than targeted variant analysis should be offered to refine the residual risk by directly identifying other pathogenic variants.
We should emphasize that the ~3% residual risk is likely an underestimate. Many families with more than one phenotype did not contribute to this estimate because there was insufficient evidence of recurrence (approach 1) or because they have not yet been fully molecularly characterized (approaches 2 and 3). We have previously shown that the majority of Mendelian phenotypes in our highly consanguineous population are autosomal recessive as proven by molecular testing even when positive family history is lacking [15]. Thus, it is likely that at least some of those additional phenotypes are recessive. Indeed, despite the limited value of pedigree-only approach (approach 1), as revealed by our study, we opted to retain this part of our analysis to highlight its tendency to underestimate the residual risk for additional recessive diseases compared to the more reliable molecularly based estimates. This further supports our argument that broader molecular testing of consanguineous parents, e.g., exome sequencing is warranted regardless of the family history. We also note the value of approach 3 in providing real-world estimate of the residual risk for additional recessive diseases based on actual pregnancies despite the relatively small number of couples surveyed. The use of larger cohorts in the future should help refine this risk further.
In conclusion, we provide a minimum residual risk of ~3% above and beyond the recurrence risk of a previously documented recessive disease in consanguineous couples. Future guidelines should take this into account and consider a recommendation for exome/genome sequencing instead of targeted variant analysis for consanguineous couples who opt for preventive genetic tests.
Data availability
Data collected and analyzed for this paper are available upon request.
References
Bittles AH. A community genetics perspective on consanguineous marriage. Community Genet. 2008;11:324–330.
Clark DW, Okada Y, Moore K, Mason D, Pirastu N, Gandin I, et al. Associations of autozygosity with a broad range of human phenotypes. Nat Commun. 2019;10:1–17.
Alkuraya FS. Curt Stern Award address: a more perfect clinical genome—how consanguineous populations contribute to the medical annotation of the human genome. Am J Hum Genet. 2021;108:395–399.
Alkuraya FS. How the human genome transformed study of rare diseases. Nature. 2021;590:218–219.
Alkuraya F. Impact of new genomic tools on the practice of Clin Genet in consanguineous populations: the Saudi experience. Clin Genet. 2013;84:203–208.
Monies D, Abouelhoda M, AlSayed M, Alhassnan Z, Alotaibi M, Kayyali H, et al. The landscape of genetic diseases in Saudi Arabia based on the first 1000 diagnostic panels and exomes. Hum Genet. 2017;136:921–939.
Monies D, Abouelhoda M, Assoum M, Moghrabi N, Rafiullah R, Almontashiri N, et al. Lessons learned from large-scale, first-tier clinical exome sequencing in a highly consanguineous population. Am J Hum Genet. 2019;104:1182–1201.
Martin HC, Jones WD, McIntyre R, Sanchez-Andrade G, Sanderson M, Stephenson JD, et al. Quantifying the contribution of recessive coding variation to developmental disorders. Science. 2018;362:1161–1164.
Kirk EP, Ong R, Boggs K, Hardy T, Righetti S, Kamien B, et al. Gene selection for the Australian reproductive genetic carrier screening project (“Mackenzie’s Mission”). Eur J Hum Genet. 2021;29:79–87.
Delatycki MB, Alkuraya F, Archibald A, Castellani C, Cornel M, Grody WW, et al. International perspectives on the implementation of reproductive carrier screening. Prenat Diagn. 2020;40:301–310.
Miano MG, Jacobson SG, Carothers A, Hanson I, Teague P, Lovell J, et al. Pitfalls in homozygosity mapping. Am J Hum Genet. 2000;67:1348–1351.
Mor-Shaked H, Rips J, Gershon Naamat S, Reich A, Elpeleg O, Meiner V, et al. Parental exome analysis identifies shared carrier status for a second recessive disorder in couples with an affected child. Eur J Hum Genet. 2021;29:455–462.
Majeed-Saidan MA, Ammari AN, AlHashem AM, Al Rakaf MS, Shoukri MM, Garne E, et al. Effect of consanguinity on birth defects in Saudi women: results from a nested case‐control study. Birth Defects Res A Clin Mol Teratol. 2015;103:100–104.
Bittles AH. Consanguineous marriages and congenital anomalies. Lancet. 2013;382:1316–1317.
Anazi S, Maddirevula S, Faqeih E, Alsedairy H, Alzahrani F, Shamseldin HE, et al. Clinical genomics expands the morbid genome of intellectual disability and offers a high diagnostic yield. Mol Psychiatry. 2017;22:615–624.
Acknowledgements
We thank the study families for their participation. We also thank Latifa Aljasser and our research coordinators (Mais Hashem, Firdous Abdulwahab, and Omar Abuyousef) for their help with data collection as well as Basel Alebdi for his help with data analysis. We would also like to thank Researchers Supporting Project number (RSP-2021/181), King Saud University, Riyadh, Saudi Arabia for providing funding for L.A.
Author information:
Conceptualization: F.S.A. Data curation: L.A., S.A., A.A., R.H. Resources: F.I., M.A. Investigation: L.A., S.A., A.A., R.H. Writing—original draft: L.A., F.S.A. Writing—review & editing: L.A., F.S.A.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics declaration
Informed consent was obtained from all families under the relevant IRB-approved (KFSHRC REC) research protocol for their respective disease. The consent permits us to construct detailed pedigrees and collect full clinical data including follow-up data on future pregnancies.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
AlAbdi, L., Alrashseed, S., Alsulaiman, A. et al. Residual risk for additional recessive diseases in consanguineous couples. Genet Med 23, 2448–2454 (2021). https://doi.org/10.1038/s41436-021-01289-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-021-01289-5
This article is cited by
-
Clinical application of next generation sequencing for Mendelian disease diagnosis in the Iranian population
npj Genomic Medicine (2024)
-
Cone dystrophy associated with autoimmune polyglandular syndrome type 1
Scientific Reports (2023)
-
Patterns and distribution of de novo mutations in multiplex Middle Eastern families
Journal of Human Genetics (2022)
-
An approach to recognising and identifying metabolic presentations in the paediatric Irish Traveller population
European Journal of Pediatrics (2022)
-
Prenatal exome sequencing and chromosomal microarray analysis in fetal structural anomalies in a highly consanguineous population reveals a propensity of ciliopathy genes causing multisystem phenotypes
Human Genetics (2022)
