
Abstract
Purpose
Recent studies have identified suggestive prenatal features of RASopathies (e.g., increased nuchal translucency [NT], cystic hygroma [CH], hydrops, effusions, congenital heart diseases [CHD], polyhydramnios, renal anomalies). Our objective is to clarify indications for RASopathy prenatal testing. We compare genotype distributions between pre- and postnatal populations and propose genotype–phenotype correlations.
Methods
Three hundred fifty-two chromosomal microarray–negative cases sent for prenatal RASopathy testing between 2012 and 2019 were collected. For most, 11 RASopathy genes were tested. Postnatal cohorts (25 patients with available prenatal information and 108 institutional database genotypes) and the NSeuroNet database were used for genotypic comparisons.
Results
The overall diagnostic yield was 14% (50/352), with rates >20% for effusions, hydrops, and CHD. Diagnostic yield was significantly improved in presence of hypertrophic cardiomyopathy (HCM), persistent or associated CH, any suggestive finding combined with renal anomaly or polyhydramnios, or ≥2 ultrasound findings. Largest prenatal contributors of pathogenic variants were PTPN11 (30%), RIT1 (16%), RAF1 (14%), and HRAS (12%), which considerably differ from their prevalence in postnatal populations. HRAS, LZTR1, and RAF1 variants correlated with hydrops/effusions, and RIT1 with prenatal onset HCM.
Conclusion
After normal chromosomal microarray, RASopathies should be considered when any ultrasound finding of lymphatic dysplasia or suggestive CHD is found alone or in association.
Similar content being viewed by others
INTRODUCTION
RASopathies are a family of genetic disorders caused by dysregulated signal traffic through the Ras/mitogen-activated protein kinase (RAS-MAPK) signaling pathway,1,2,3,4 with more than 20 disease-associated genes identified thus far.3,4,5,6,7 As part of a common pathway, RASopathies share common clinical features involving craniofacial, cardiovascular, and lymphatic anomalies, as well as neurodevelopment and growth disorders.1,2 Other recurrent features include genitourinary, cutaneous, hematological and skeletal abnormalities, and cancer predispositions.1,2,8,9,10 Many genetic syndromes are part of the RASopathy spectrum, including Noonan syndrome (NS), Noonan syndrome with multiple lentigines (previously known as LEOPARD syndrome), Noonan-like syndrome with loose anagen hair, Costello syndrome (CS), cardiofaciocutaneous syndrome (CFCS), and other clinically related disorders.3,4,11,12 Collectively, RASopathies are frequent genetic conditions encountered in pre- and postnatal evaluations, occurring in 1:1,000 to 1:2,500 live births and probably more in the prenatal setting.1,3,8,13
Prenatal diagnosis of RASopathies can be challenging, mostly because of their variable expressivity, as well as their nonspecific prenatal presentation.8,14,15 In recent years, efforts have been made to clarify the prenatal phenotype of RASopathies and suggestive ultrasound (US) findings have been delineated, namely lymphatic dysplasia, such as increased nuchal translucency (NT), increased nuchal fold (NF), cystic hygroma (CH), hydrops fetalis (HF), ascites or thoracic effusions (TE), and other lymphatic anomalies; congenital heart disease (CHD), such as valvular dysplasia or hypertrophic cardiomyopathy (HCM); polyhydramnios and renal anomalies.14,15,16,17,18,19,20,21,22,23 Supporting US features have been identified, including macrosomia, mild ventriculomegaly, macrocephaly, and short long bones.19 In the presence of suggestive US finding(s), previous studies estimated that pathogenic variants in RASopathy genes could be detected in 6.7–21.7% of cases.15,16,18,22,24,25,26,27 Those studies, however, did not provide information regarding the prenatal findings considered as the most suggestive of RASopathies, either alone15 or in association.22 Moreover, they had heterogeneous cohorts in terms of (1) previous prenatal testing, as few studies excluded occurrence of aneuploidies and chromosomal structural rearrangements that have overlapping prenatal phenotypes with RASopathies;21,28,29 (2) patients tested, as many studies only included fetuses or only live children; and (3) RASopathy genes tested, with earlier studies testing for a limited number of genes.15,16,18,19,22,24,25,26,27
Prenatal diagnosis of RASopathies is important as it can improve parental counseling and allow families to make informed decisions with regard to pregnancy management, taking into account treatment options, living with a child with a genetic disorder, and availability of termination of pregnancy (TOP). It can enable providers to anticipate and screen for complications known to occur in pregnancies with RASopathies, like HCM, and prepare the medical team for management of neonatal complications.
The aim of this study was to establish which US findings are the most suggestive of RASopathies in an effort to better define the prenatal indications for RASopathy testing.
MATERIALS AND METHODS
Objectives
This study objective is to clarify the indications for RASopathy prenatal testing by assessing as a primary outcome the diagnostic yields of suggestive US findings. Our hypothesis is that lymphatic dysplasia and suggestive CHD could motivate prenatal RASopathy testing, when isolated or in association. Secondly, we aim to compare genotype distributions between prenatal and postnatal cohorts, and evaluate prenatal genotype–phenotype correlations.
Patients
This retrospective study is an international multicenter collaboration with cases provided by Centre Hospitalier Universitaire Sainte-Justine, in Canada, and the Medical Genetics Unit of the Fondazione IRCCS-Casa Sollievo della Sofferenza, in Italy. The cohort consisted of 352 prenatal cases: 205 Canadian cases evaluated between July 2012 and July 2018, and 147 Italian cases evaluated between 2016 and 2019. The inclusion criteria were (1) fetuses referred to a prenatal genetics clinic because of US finding(s) suggestive of RASopathy, namely edema/lymphatic dysplasia: increased NT, increased NF, CH, HF, ascites or TE, including chylothorax, other lymphatic anomaly; and/or CHD, such as valvular dysplasia or HCM;14,16,19,23,24 (2) exclusion of a chromosomal anomaly explaining the US findings, by chromosomal microarray (CMA) (333/352) or karyotype (19/352); and (3) molecular RASopathy genetic testing during pregnancy or shortly after birth (for cases in which parents opted to postpone testing to neonatal period).
Data collection was based on chart review. Clinical information provided by the referring physician (“clinical indications for testing”) was collected for the 352 fetuses (50 cases with positive result), whereas detailed US findings, complete pregnancy longitudinal data (1st to 3rd trimester), as well as neonatal outcome, were available for 312 fetuses (47 with positive result). Data on clinical indications for testing were used to assess diagnostic yields. See Supplementary Materials and Methods 1 for details on phenotypic assessment and definitions.
A postnatal cohort of 25 children diagnosed molecularly with RASopathy conditions based on their postnatal clinical presentation, between 2012 and 2018, with available prenatal data, and selected from the same population as the prenatal cases, was added to obtain a wider spectrum of phenotypes and genotypes. Using this cohort and the prenatal cases, we attempted to determine if postnatal phenotype severity of RASopathy patients could correlate with the timing of diagnosis. The scoring system used to determine the postnatal phenotype severity is outlined in Table 3 and Table S1. Also, we accessed an institutional genotype database of 108 RASopathy patients analyzed postnatally between 2013 and 2020 at the Italian Medical Genetics Unit, and the European Network on Noonan syndrome and related disorders (NSEuroNet) database30 on gene-specific variant distribution in the general RASopathy population, to compare pre- and postnatal genotype frequencies.
Molecular analysis
Eleven percent (38/352) of the study cohort was tested for 9 genes, 43% (152/352) for 11 genes, 1% (4/352) for 13 genes, 1.5% (5/352) for 15 genes, 4% (13/352) for 16 genes, 34% (121/352) for 19 genes, and 5.5% (19/352) for 20 genes. The most commonly tested genes were BRAF (NM_001374258.1), HRAS (NM_005343.4), KRAS (NM_033360.4), MAP2K1 (NM_002755.3), MAP2K2 (NM_030662.3), NRAS (NM_002524.5), PTPN11 (NM_002834.4), RAF1 (NM_001354689.3), RIT1 (NM_006912.5), SHOC2 (exon 2) (NM_007373.4), and SOS1 (NM_005633.3). See Supplementary Materials and Methods 1 for details.
DNA was isolated from samples of amniotic fluid, chorionic villi sampling, or blood. For the Canadian cases, the samples were sent to private laboratories in the United States (mostly GeneDx) after written informed consent was obtained from the parents, and the diagnosis was made by molecular analysis using parallel sequencing or Sanger sequencing. For the Italian cases, molecular analyses were performed by parallel sequencing using either the HaloPlexHS target enrichment system or the SureSelectQXT target enrichment system (Agilent Technologies, http://www.genomics.agilent.com). The enriched libraries were sequenced by a MiSeq instrument (Illumina, https://www.illumina.com), and sequencing data were analyzed using an in-house implemented pipeline (see Motta et al.31 for further details). Variants validation and segregation analyses were performed by Sanger sequencing. Variants classification was determined using the American College of Medical Genetics and Genomics - Association for Molecular Pathology (ACMG-AMP) standards and guidelines for the interpretation of sequence variants.32
Statistical analysis
Chi-square test (χ2) one-sided was used for comparisons of categorical variables. Fisher’s exact test was used when the prerequisites for χ2 test could not be met. Wilcoxon–Mann–Whitney test was used for continuous variables, based on results of normality tests. A probability value less than 0.05 was considered significant.
RESULTS
Prevalence of ultrasound findings and diagnostic yields
Prevalence data are in Table 1. Increased NT was the most common indication for testing (230/312, 74% of fetuses), followed by CH (182/312, 58%). Increased NF, hydrops, effusions, CHD, polyhydramnios, and renal anomalies were significantly more prevalent in cases with a pathogenic or likely pathogenic variant (“positive cases” or “positive patients”) than in cases with a negative or inconclusive result (“negative cases”).
Diagnostic yields are in Table 2. A pathogenic or likely pathogenic variant was identified in 15% (47/312) of fetuses with longitudinal data available and in 14% (50/352) of fetuses with clinical indications for testing. These overall detection rates are consistent with results from previous studies that estimated a diagnostic yield between 4% and 22%.15,18,22,24,26,27
Of the 182 cases with CH, 30 (16%) had a positive result, with a rate similar to the literature (11–16%15,24,25,26,27). The detection rate of this US finding was significantly higher in cases with persistent CH into the second trimester (21%, 26/124) or when CH was associated with another suggestive US finding (28%, 26/94). Increased NT, hydrops, and CHD were frequently associated with CH.
Mean NT in positive cases (6.4 mm) was significantly higher than in negative cases (4.8 mm) (Table 1). The diagnostic yield of increased NT was significantly higher when NT was greater than 6 mm than when it was lower than 6 mm (20% [12/60] vs. 10% [17/165] respectively), but much lower when this finding was isolated (1%, 1/90) (Table 2). The diagnostic yield of increased NF (17/67, 25%) was significantly higher than the overall diagnostic yield (Table 2), and the mean NF was significantly larger in positive patients than in negative patients (9.6 mm vs. 6.3 mm respectively) (Table 1).
The diagnostic yields were significantly higher than the overall detection rate for TE (22/54, 41%), HF (20/60, 33%), and CHD (18/71, 25%). There were significantly higher diagnostic yields when HCM was present (9/13, 69%), when cardiac defect was combined with HCM (100%, 6/6), or when a suggestive US finding was associated with polyhydramnios or renal anomalies (39% [14/36] and 30% [14/46], respectively). Macrosomia, short long bones, and mild ventriculomegaly were often associated with a suggestive US finding, but numbers were too small to determine if their improvement of the test performance was statistically significant (Table 2).
Of the 251 cases with two or more US findings, 45 cases (18%) had a RASopathy diagnosis, which significantly exceeds the diagnostic yield of only one US finding (3%, 2/61) (Table 2). The most frequent isolated finding in RASopathy patients was CH (2/47, 4%). In cases of multiple findings, some combinations of US findings had significantly higher diagnostic yields (p value <0.02), namely CH, increased NT or increased NF in association with TE (49%, 19/39), HF (34%, 18/53), polyhydramnios (32%, 10/31), renal anomaly (30%, 12/40), or CHD (27%, 12/45). The majority of positive cases had at least three suggestive prenatal features (96% [45/47] had ≥2 findings at the end of the pregnancy), whereas the negative cases tended to get better with time as the US features resolved (Table 1).
Genotypic spectrum
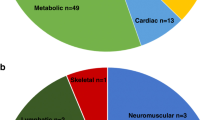
In the prenatal cohort, 65 variants were identified in 63 cases, namely 52 pathogenic or likely pathogenic variants in 50 cases (80% of all variants) and 13 variants of uncertain significance (VUS). Eighty-eight percent (46/52) of (likely) pathogenic variants were sporadic. One patient had two likely pathogenic variants in LZTR1 in compound heterozygous state, whereas another patient had a double diagnosis, neurofibromatosis type 1 (NF1) inherited from his affected father and NS (PTPN11). Two NS variants were inherited from a parent with a suggestive phenotype. One positive case had a maternal family history of NS. Distributions of genotypes in the prenatal and postnatal cohorts are in Fig. 1. In the prenatal cohort (Fig. 1a), variants were spread among 11 genes: BRAF, HRAS, KRAS, LZTR1, NRAS, PTPN11, RAF1, RIT1, SHOC2, SOS1, and SOS2. The largest contributors of positive cases were PTPN11 (30%, 15/50), RIT1 (16%, 8/50), RAF1 (14%, 7/50), HRAS (12%, 6/50), and LZTR1 (8%, 4/50). SOS1 contained the highest number of VUS, representing 46% of those identified (6/13).

(a) Gene-specific variant distribution in the prenatal cohort (50 positive cases). (b) Gene-specific variant distribution in the postnatal cohorts (133 cases: 25 postnatal positive cases and 108 postnatal genotypes). (c) Gene-specific variant distribution in both prenatal and postnatal cohorts (183 cases).
Genotype–phenotype correlations
Genotype–phenotype correlations are based on prenatal clinical features of 47 prenatal patients (Table S2) and 25 postnatal patients (Table S3). The distribution of genotypes according to prenatal features is in Fig. 2. Severe manifestations of lymphatic dysplasia like hydrops/effusions or cardiopathy were observed more frequently in patients with variants in RIT1, HRAS, or RAF1. PTPN11 was an important contributor to hydrops/thoracic effusions too, together with SOS1.

Gene-specific contributions to suggestive prenatal phenotypic features for the 72 patients with a positive result and available longitudinal data (47 prenatal and 25 postnatal positive cases). CH cystic hygroma, CHD congenital heart disease, HCM hypertrophic cardiomyopathy. Others: BRAF, CBL, MAP2K1, NRAS, RASA1, and SOS2.
Variants in PTPN11 were associated more often with a milder prenatal phenotype, as a little less than 50% of PTPN11 patients (13/28) had one (often nonspecific) or no US prenatal finding. More than 50% of PTPN11 patients (15/28) had a suggestive prenatal clinical presentation with wide variable expressivity, few PTPN11 variants being related to hydrops. SOS1 pathogenic variants were present in similar rates in the prenatal and postnatal cohorts (Fig. 1) and 60% (3/5) displayed suggestive prenatal US finding(s). Half of patients with a KRAS variant (2/4) did not have any prenatal features.
The majority of HRAS patients and all LZTR1 patients had ≥2 US findings. All patients with a variant in HRAS had a significant prenatal phenotype: 83% (5/6) had a severe lymphatic dysfunction, primarily severe HF (4/6). Seventy-five percent (3/4) of LZTR1 positive patients presented with a serious lymphatic dysplasia, namely important hydrops/effusions. Seven of eight patients with a variant in RIT1 had a multisystem prenatal phenotype: 25% (2/8) with important effusions/hydrops and 62.5% (5/8) with HCM. In contrast, no LZTR1 patient and only 29% (2/7) of RAF1 patients had an HCM identified prenatally. More than half of RAF1 patients (4/7) had important hydrops/effusions or CHD.
Regarding the postnatal phenotype severity (based on the scoring system by Baldassarre et al.,14 Table 3), RASopathy patients with severe postnatal phenotypes were significantly more likely to have been identified/diagnosed prenatally (78%, 7/9) than patients with mild or moderate phenotypes (27–29%, 3/11–4/14).
DISCUSSION
The collected data show that the US findings with the highest diagnostic yields for RASopathies (>20%) are, in decreasing order, as follows: HCM with or without cardiac defect; TE, HF, CH associated with another suggestive US finding; CHD; and persistent CH. RASopathies should be suspected when these prenatal findings are present alone or in association, as the diagnostic yield of a unique suggestive US finding is still 3–9% (2/61–13/142) (Table 2) and 11–19% in past studies,15,18,27 even though an association of at least two findings has a significantly higher diagnostic yield (18% in this study [45/251] and 28% in literature18), concurring with conclusions of other publications.15,17,18,19,24 The combinations of findings with the highest yields are CH, increased NT or increased NF combined with, in decreasing order, TE, HF, polyhydramnios, renal anomaly, or CHD.
The only suggestive US finding for which RASopathy prenatal testing may not be cost-efficient is isolated increased NT, as the diagnostic yield in the presence of this isolated finding is very low in this study (1%, 1/90) and in the literature (1.4–6.7%).15,25,27 RASopathy testing may be considered in cases of extreme isolated increased NT, such as NT above 6 mm, since many other publications observed a mean NT above 6 mm for RASopathy patients,16,22,25,26 like in this study. As for increased NF, this feature was never found alone, but always associated with CH, hydrops/effusion, or CHD in both positive and negative cases. Accordingly, we propose that RASopathy testing should be offered only if increased NF is combined with another highly suggestive US finding, like CHD.
As polyhydramnios and renal anomalies significantly improve the diagnostic yield for RASopathies, they should be considered meaningful associated US features. Macrosomia, mild ventriculomegaly, and short long bones were recurrently identified in positive cases, and should therefore be considered supporting findings of RASopathies, as suggested by Myers et al.19
When comparing diagnostic yields for RASopathies with the yield of chromosomal analyses for lymphatic dysplasia or CHD, which is estimated between 5% and 50%,28,29 RASopathies are certainly worth testing as a second/third-tier test, with diagnostic yields from 13% to 41% in this study and between 7% and 22% in the literature.15,16,18,22,24,25,26,27 However, the use of RASopathy testing as a reflex test after CMA has some practical implications. When amniocentesis is performed for prenatal diagnosis, if RASopathy testing is delayed after CMA, results may be obtained only after 24 gestational weeks, at which point they may not be available to contribute to reproductive decision-making, depending on the country’s legislation.
The large size and homogeneity of the present cohort enabled us to determine the most suggestive prenatal features of RASopathies and to clarify the most prevalent US findings in RASopathy patients, as opposed to past studies.16,18,22 It allowed us to compare the genotypic spectrum of positive cases between pre- and postnatal cohorts. However, for some subcategories of suggestive US findings, they were not enough patients to reach statistical significance, so the stratified analysis rendered few significant results. Moreover, there are inherent limitations to prenatal phenotyping, and it is possible that diagnoses were missed or variants were misclassified because of the difficulty of assessing some prenatal phenotypic features. Besides, not all patients had available data on third trimester US phenotypic features (43%, 134/312), as 69 pregnancies ended during the second trimester and 109 cases were sent back to their referring center for third trimester follow-up. As the information was collected retrospectively, this study is subjected to recall and information bias, especially for data concerning postnatal cases that were for some cases based on parents’ memory of prenatal events or for data concerning some negative cases that were lost to follow-up. We were limited in the selection of cases by the fact that referrals were made mostly by third parties (e.g., obstetricians) and that some parents were not included because they declined all investigations. Nevertheless, these limitations are minor, and we are confident that our cohort is representative of the targeted population. Some ascertainment bias could have influenced the results, such as the lack of systematic measurement of NT at the first trimester ultrasound and the fact that persistence of CH could have been underestimated because some pregnant women did not have access to a follow-up ultrasound earlier than 20 gestational weeks.
Concerning the genotype–phenotype correlations, variants in PTPN11, SOS1, and KRAS contributed to more than half of the cases without significant prenatal signs, and to a third of the hydrops/effusions, which reflects the wide phenotypic variability associated with RASopathies, even when the same gene is implicated1,8,18,19,22 (Fig. 2). In particular, some PTPN11 variants were more commonly observed in fetuses without a suggestive prenatal phenotype, like c.923A>G(p.Asn308Asp) and c.1403C>T(p.Thr468Met), which is congruent with the generally milder postnatal phenotype associated with p.Asn308Asp.33 Conversely, some other PTPN11 variants were consistently associated with severe lymphatic dysfunction. For example, two fetuses with PTPN11 variant c.218C>T(p.Thr73Ile) showed HF and other lymphatic anomalies, as in a recent study where it was associated with nonimmune HF.34 This variant is significantly more frequent in our prenatal cohort than in the RASopathy population (14% [2/14] vs. 2% [32/1,543] in NSEuroNet,30 p value 0.04). It is a particularly activating variant and one of the most common germline changes found in patients with NS and juvenile myelomonocytic leukemia (JMML),35,36 which could explain the complex prenatal presentation. Concurring with other studies,15,18 pathogenic variants in SOS1 were shown to have penetrance for RASopathy prenatal features. In regard to KRAS, the low occurrence of patients with suggestive prenatal features is in accordance with the atypical postnatal phenotypes of KRAS patients.33 A high proportion of HRAS, LZTR1, RAF1, and RIT1 patients had multisystemic prenatal presentation including severe lymphatic dysplasia and/or CHD/HCM. Variants in HRAS, LZTR1, and RAF1 seemed to be more associated with hydrops/effusions, whereas variants in RIT1 seemed to correlate with prenatal onset HCM, in accordance with the recurrent association of RIT1 variants with HCM early in life or prenatally20,33 (Fig. 2). Although patients with variants in LZTR1 or RAF1 have an increased prevalence of HCM in the postnatal setting,8,33 none of the LZTR1 cases and few RAF1 patients were identified having HCM prenatally, most probably because they develop HCM later in life.11,33,37 One liveborn patient with the RAF1 variant c.770C>T (p.Ser257Leu) had pulmonary hypertension, a genotype–phenotype correlation previously seen.37 Pathogenic variants linked to CFCS were rare in the prenatal cohort, as only one BRAF and no MAP2K1/MAP2K2 variant was identified. Similarly, SOS1 variants affecting Arg552, the hotspot residue associated with NS, were absent from the prenatal cohort. We think that pathogenic variants in these genes are rarely observed prenatally because their prenatal presentation generally does not include HCM or significant lymphatic dysplasia, or is nonspecific.19
Consistent with other publications,14,17 this study did not show any correlation between prenatal RASopathy US features and a complex postnatal clinical outcome, with the exception of severe prenatal HF and HCM, which were associated to a negative evolution, such as pregnancy loss (44% intrauterine fetal demise [IUFD], 4/9) or severe neonatal complications (Tables S2–S4). Severe postnatal phenotypes were significantly associated with a diagnosis in the prenatal setting (Table 3). Yet, the high percentage of TOP in the prenatal cohort (55%, 26/47) substantially reduced the population of prenatal positive cases available for this analysis and different results might be observed in a larger cohort.
The overrepresentation of highly activation variants in our prenatal cohort concurs with the accepted model that strongly activating RASopathy gene alterations are not or poorly compatible with embryonal/fetal development and are almost exclusively observed as somatic (cancer) events or else associated with severe phenotypes.2 For example, one pathogenic variant in HRAS, c.182A>G (p.Gln61Arg), was never encountered as a germline substitution, being previously described in the somatic state in Schimmelpenning syndrome or cancer. This variant lead to severe HF and early spontaneous fetal demise, most probably because the germline state or very high level of mosaicism of this deleterious variant was not viable. Moreover, we observed that the most frequent HRAS pathogenic variant in CS (p.Gly12Ser) (78% (521/669) in NSeuroNET30), a mildly activating substitution, was found to be absent from our prenatal cohort, while other rare highly activating HRAS pathogenic variants, such as p.Gly12Asp and p.Gly13Asp, were relatively frequent in our prenatal cohort (each variant was found in two fetuses vs. prevalence of 0.7% [5/669] and 1.3% [9/669] in NSeuroNET,30 p value <0.00001). CS patients with these variants have a high prevalence of HCM, arrhythmia, and fetal hydrops.38,39 Deleterious RAS-MAPK variants are probably underdiagnosed in the prenatal setting because of their nonspecific presentation and early demise. In the prenatal cohort, 88% of variants (46/52) were sporadic, which is more than the literature reported de novo rate (25–70%).13,33 These differences may be the consequence of the lack of consideration of lethal prenatal presentations or TOP in the calculation of de novo cases in RASopathy reviews.22
Our study shows that the genotypic spectrum of prenatal cases is wider and different from previous publications15,18,22 (Table S5). Notably, LZTR1, HRAS, and RIT1 variants were overrepresented in our study (8–16%) compared with the literature (0–7%), and PTPN11 represented only 30% of cases versus 67–81% in other publications. This is mainly due to the fact that those studies used smaller molecular panels; did not test for LZTR1, even though variants in this gene are a common cause of NS;11,33 and poorly tested for RIT1, whereas RIT1 patients frequently display important prenatal features20. We systematically assessed HF, which increased HRAS cases.19 The lower-than-expected frequency of PTPN11 variants can be attributed to the underrepresentation of variants with milder effect in our prenatal cohort as these variants usually have little penetrance for prenatal signs.
We recommend that a large prenatal molecular panel should be systematically offered in the prenatal setting when a RASopathy is suspected, including not only the ten most commonly tested genes—BRAF, HRAS, KRAS, MAP2K1, MAP2K2, NRAS, PTPN11, RAF1, SHOC2, and SOS1—but also at least CBL, LZTR1, MRAS, RIT1, and SOS2. Even though no pathogenic variant was identified in MAP2K1, MAP2K2, and CBL in this study, characteristic prenatal phenotypes were reported previously for these genes.16,18 We particularly recommend the inclusion of MRAS, based on the generally severe postnatal phenotype of MRAS patients, namely the invariant occurrence of HCM at an early age.6,40 Considering the peculiar genotypic spectrum characterizing prenatal RASopathies, and possible occurrence of previously unreported pathogenic variants, we recommend full sequencing of RASopathy genes rather than targeted genotyping, including full sequencing of SHOC2, since a pathogenic variant outside of exon 2 was detected in this study (c.807_808delinsTT) in a fetus with early-onset HCM.31
In conclusion, after a normal CMA, RASopathy prenatal testing should be offered when any prenatal US finding suggestive of lymphatic dysplasia (CH, increased NT or NF, hydrops, effusions, other lymphatic anomalies) or suggestive CHD (like valvular dysplasia and/or HCM) is found alone or in association, except for isolated increased NT below 6 mm or isolated increased NF. Some US features should be considered in the prenatal evaluation because they increase the likelihood of RASopathy diagnosis, such as associated findings of polyhydramnios and renal anomaly. Our data support the view that a subset of RASopathy genes and variants are more frequently associated with complex prenatal features such as hydrops/effusions or serious cardiopathy.
Data availability
Data and materials can be supplied individually, upon request.
References
Tajan, M., Paccoud, R., Branka, S., Edouard, T. & Yart, A. The RASopathy family: consequences of germline activation of the RAS/MAPK pathway. Endocr. Rev. 39, 676–700 (2018).
Tartaglia, M. & Gelb, B. D. Disorders of dysregulated signal traffic through the RAS-MAPK pathway: phenotypic spectrum and molecular mechanisms. Ann. N. Y. Acad. Sci. 1214, 99–121 (2010).
Tidyman, W. E. & Rauen, K. A. Expansion of the RASopathies. Curr. Genet. Med. Rep. 4, 57–64 (2016).
Aoki, Y., Niihori, T., Inoue, S. & Matsubara, Y. Recent advances in RASopathies. J. Hum. Genet. 61, 33–39 (2016).
Capri, Y. et al. Activating mutations of RRAS2 are a rare cause of Noonan syndrome. Am J. Hum. Genet. 104, 1223–1232 (2019).
Higgins, E. M. et al. Elucidation of MRAS-mediated Noonan syndrome with cardiac hypertrophy. JCI Insight 2, e91225 (2017).
Motta, M. et al. Enhanced MAPK1 function causes a neurodevelopmental disorder within the RASopathy clinical spectrum. Am J. Hum. Genet. 107, 499–513 (2020).
Tartaglia, M., Gelb, B. D. & Zenker, M. Noonan syndrome and clinically related disorders. Best Pract. Res. Clin. Endocrinol. Metab. 25, 161–179 (2011).
Dunnett-Kane, V., Burkitt-Wright, E., Blackhall, F. H., Malliri, A., Evans, D. G. & Lindsay, C. R. Germline and sporadic cancers driven by the RAS pathway: parallels and contrasts. Ann. Oncol. 31, 873–883 (2020).
Kratz, C. P. et al. Cancer spectrum and frequency among children with Noonan, Costello, and cardiofaciocutaneous syndromes. Br. J. Cancer 112, 1392–1397 (2015).
Gripp, K. W. et al. The sixth international RASopathies symposium: precision medicine-From promise to practice. Am. J. Med. Genet. A 182, 597–606 (2020).
D’Amours, G., Brunel-Guitton, C., Delrue, M. A., Dubois, J., Laberge, S. & Soucy, J. F. Prenatal pleural effusions and chylothorax: an unusual presentation for CM-AVM syndrome due to RASA1. Am. J. Med. Genet. A. 182, 2454–2460 (2020).
van der Burgt, I. Noonan syndrome. Orphanet J. Rare Dis. 2, 4 (2007).
Baldassarre, G. et al. Prenatal features of Noonan syndrome: prevalence and prognostic value. Prenat. Diagn. 31, 949–954 (2011).
Leach, N. T., Wilson Mathews, D. R., Rosenblum, L. S., Zhou, Z., Zhu, H. & Heim, R. A. Comparative assessment of gene-specific variant distribution in prenatal and postnatal cohorts tested for Noonan syndrome and related conditions. Genet. Med. 21, 417–425 (2019).
Croonen, E. A. et al. Prenatal diagnostic testing of the Noonan syndrome genes in fetuses with abnormal ultrasound findings. Eur J. Hum. Genet. 21, 936–942 (2013).
Gaudineau, A. et al. Postnatal phenotype according to prenatal ultrasound features of Noonan syndrome: a retrospective study of 28 cases. Prenat. Diagn. 33, 238–241 (2013).
Hakami, F., Dillon, M. W., Lebo, M. & Mason-Suares, H. Retrospective study of prenatal ultrasound findings in newborns with a Noonan spectrum disorder. Prenat. Diagn. 36, 418–423 (2016).
Myers, A. et al. Perinatal features of the RASopathies: Noonan syndrome, cardiofaciocutaneous syndrome and Costello syndrome. Am. J. Med. Genet. A. 164A, 2814–2821 (2014).
Yaoita, M. et al. Spectrum of mutations and genotype–phenotype analysis in Noonan syndrome patients with RIT1 mutations. Hum. Genet. 135, 209–222 (2016).
Bakker, M., Pajkrt, E. & Bilardo, C. M. Increased nuchal translucency with normal karyotype and anomaly scan: what next? Best. Pract. Res. Clin. Obstet. Gynaecol. 28, 355–366 (2014).
Stuurman, K. E. et al. Prenatal ultrasound findings of rasopathies in a cohort of 424 fetuses: update on genetic testing in the NGS era. J. Med. Genet. 56, 654–661 (2019).
Noonan Syndrome Guideline Development Group. Management of Noonan syndrome – a clinical guideline. University of Manchester, DYSCERNE. https://rasopathiesnet.org/wp-content/uploads/2014/01/265_Noonan_Guidelines.pdf (2010).
Houweling, A. C., de Mooij, Y. M., van der Burgt, I., Yntema, H. G., Lachmeijer, A. M. & Go, A. T. Prenatal detection of Noonan syndrome by mutation analysis of the PTPN11 and the KRAS genes. Prenat. Diagn. 30, 284–286 (2010).
Pergament, E., Alamillo, C., Sak, K. & Fiddler, M. Genetic assessment following increased nuchal translucency and normal karyotype. Prenat. Diagn. 31, 307–310 (2011).
Ali, M. M., Chasen, S. T. & Norton, M. E. Testing for Noonan syndrome after increased nuchal translucency. Prenat. Diagn. 37, 750–753 (2017).
Lee, K. A. et al. PTPN11 analysis for the prenatal diagnosis of Noonan syndrome in fetuses with abnormal ultrasound findings. Clin. Genet. 75, 190–194 (2009).
Chen, Y. N., Chen, C. P., Lin, C. J. & Chen, S. W. Prenatal ultrasound evaluation and outcome of pregnancy with fetal cystic hygromas and lymphangiomas. J. Med. Ultrasound 25, 12–15 (2017).
Grande, M. et al. Genomic microarray in fetuses with increased nuchal translucency and normal karyotype: a systematic review and meta-analysis. Ultrasound Obstet. Gynecol. 46, 650–658 (2015).
European Network on Noonan syndrome and related disorders. University Hospital Magdeburg, Genetics IoH. https://nseuronet.com/php/ (2020).
Motta, M. et al. Clinical and functional characterization of a novel RASopathy-causing SHOC2 mutation associated with prenatal-onset hypertrophic cardiomyopathy. Hum. Mutat. 40, 1046–1056 (2019).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. (2015).
Allanson, J. E. & Roberts, A. E. Noonan syndrome. In GeneReviews (eds Adam, M. P. et al.) (Seattle, University of Washington, 1993).
Schönfeld, M. et al. Rapid detection by hydrops panel of Noonan syndrome with PTPN11 mutation (p.Thr73Ile) and persistent thrombocytopenia. Mol. Genet. Genomic Med. 8, e1174 (2020).
Strullu, M. et al. Juvenile myelomonocytic leukaemia and Noonan syndrome. J. Med. Genet. 51, 689–697 (2014).
Tartaglia, M. et al. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat. Genet. 34, 148–150 (2003).
Thompson, D. et al. RAF1 variants causing biventricular hypertrophic cardiomyopathy in two preterm infants: further phenotypic delineation and review of literature. Clin. Dysmorphol. 26, 195–199 (2017).
Lorenz, S., Petersen, C., Kordaß, U., Seidel, H., Zenker, M. & Kutsche, K. Two cases with severe lethal course of Costello syndrome associated with HRAS p.G12C and p.G12D. Eur J. Med. Genet. 55, 615–619 (2012).
Bertola, D. et al. Phenotypic spectrum of Costello syndrome individuals harboring the rare HRAS mutation p.Gly13Asp. Am. J. Med. Genet. A. 173, 1309–1318 (2017).
Motta, M. et al. Activating MRAS mutations cause Noonan syndrome associated with hypertrophic cardiomyopathy. Hum. Mol. Genet. 29, 1772–1783 (2019).
Acknowledgements
This study was supported, in part, by funding from European Joint Programme on Rare Diseases: EJP-RD (NSEuronet, to M.T.), Associazione Italiana per la Ricerca sul Cancro: AIRC (IG 21614, to M.T.), and the Italian Ministry of Health (Ricerca Corrente 2019 and 2020, to M.T. and A.D.L.).
Author information
Authors and Affiliations
Contributions
Conceptualization: A.S., M.-A.D., A.D.L. Formal analysis: A.S., N.D.G., V.P., P.D., T. Mazza, A.-M.L., M.-A.D., A.D.L. Funding acquisition: M.T., A.D.L. Investigation: A.S., N.D.G., V.P., M.T., M.-A.D., A.D.L. Methodology: A.S., M.-A.D., A.D.L. Project administration: A.S., M.T., M.-A.D., A.D.L. Resources: V.P., P.D., S.C., V.D., E.A., A.M., F.S., G.T., D.M., C.D.M., M.R., I.D., A.Z., E.G., V.G.N., G.M., P.V., F.P., F.C.R., T. Mattina, G.D., L.P., T. Mazza, A.G., A.P., M.-A.D., A.D.L. Supervision: A.S., M.T., M.-A.D., A.D.L. Visualization: A.S. Writing—original draft: A.S., N.D.G., M.T., M.-A.D., A.D.L. Writing—review & editing: A.S., N.D.G., A.-M.L., M.T., M.-A.D., A.D.L.
Corresponding authors
Ethics declarations
Ethics declaration
The study was performed in accordance with the principles set out in the Declaration of Helsinki. The CHU Sainte-Justine Research Ethics and the Fondazione IRCCS Ethics Committees approved the study based on chart review. Because patients and clinical data were de-identified and the study was considered to have minimal risks, the need for consent was waived by the Research Ethics committee.
Competing interests
A.-M.L. received an honorarium for a conference given by Sobi (Swedish Orphan Biovitrum AB [publ] reg. number 556038–9321). The other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Scott, A., Di Giosaffatte, N., Pinna, V. et al. When to test fetuses for RASopathies? Proposition from a systematic analysis of 352 multicenter cases and a postnatal cohort. Genet Med 23, 1116–1124 (2021). https://doi.org/10.1038/s41436-020-01093-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-01093-7
This article is cited by
-
Defining the variant-phenotype correlation in patients affected by Noonan syndrome with the RAF1:c.770C>T p.(Ser257Leu) variant
European Journal of Human Genetics (2024)
-
RASopathies due to de novo pathogenic variants: clinical features, genetic findings and outcomes in nine neonates born with congenital heart defects
BMC Medical Genomics (2022)
