
Abstract
Purpose
Neurodevelopmental disorders (NDDs) encompass a spectrum of genetically heterogeneous disorders with features that commonly include developmental delay, intellectual disability, and autism spectrum disorders. We sought to delineate the molecular and phenotypic spectrum of a novel neurodevelopmental disorder caused by variants in the GNAI1 gene.
Methods
Through large cohort trio-based exome sequencing and international data-sharing, we identified 24 unrelated individuals with NDD phenotypes and a variant in GNAI1, which encodes the inhibitory Gαi1 subunit of heterotrimeric G-proteins. We collected detailed genotype and phenotype information for each affected individual.
Results
We identified 16 unique variants in GNAI1 in 24 affected individuals; 23 occurred de novo and 1 was inherited from a mosaic parent. Most affected individuals have a severe neurodevelopmental disorder. Core features include global developmental delay, intellectual disability, hypotonia, and epilepsy.
Conclusion
This collaboration establishes GNAI1 variants as a cause of NDDs. GNAI1-related NDD is most often characterized by severe to profound delays, hypotonia, epilepsy that ranges from self-limiting to intractable, behavior problems, and variable mild dysmorphic features.
Similar content being viewed by others


INTRODUCTION
Neurodevelopmental disorders (NDDs) are heterogeneous disorders, often with a broad and overlapping range of features that commonly include developmental delay, intellectual disability, and autism spectrum disorder. This group of disorders also has an increased incidence of comorbidities such as epilepsy. Advances in genomic technologies have led to an exponential increase in the number of genes associated with NDDs. However, up to half of those affected do not have an identified genetic etiology,1 presenting challenges in understanding the long-term prognosis and accessing appropriate support. Phenotype-based genetic investigations of NDDs are hampered by the highly variable clinical manifestations, the phenotypic overlap with other closely related disorders, and the rarity of particular genetic subtypes.2 Instead, more recently, large-scale trio-based exome sequencing of clinically heterogeneous populations coupled with international data-sharing has proven a powerful strategy for discovering NDD-associated genes.1,2
G-protein subunits belong to a family of proteins that have previously been associated with NDDs.3,4 Heterotrimeric G proteins, composed of α, β, and γ subunits, transmit the signals of extracellular ligands bound to G-protein-coupled receptors (GPCRs) to intracellular signaling pathways.5 G-protein signaling has been implicated in a diverse range of biological functions including neuronal development and synaptic function.6 GNAI1 (MIM 139310) encodes the inhibitory Gαi1 subunit of heterotrimeric G-proteins. A recent study found a significant enrichment of de novo variants in GNAI1 in a diverse cohort of individuals with NDDs.2,7 Here, we describe 24 unrelated individuals with GNAI1 variants (23 de novo variants and 1 inherited from a mosaic parent) and delineate the associated phenotypic features, which include global developmental delay with intellectual disability, hypotonia, and seizures.
MATERIALS AND METHODS
Individuals with pathogenic and likely pathogenic variants in GNAI1 (NM_002069.5) were identified via the Deciphering Developmental Disorders (DDD) research study7 and international collaboration facilitated by GeneMatcher8 and MyGene2. Variants were classified using American College of Medical Genetics and Genomics (ACMG) guidelines.9 Individuals were identified via trio exome sequencing by the DDD study7 (individuals 11, 14, 16, 18–20, and 24), clinical trio exome sequencing at GeneDx as previously described10 (1–3, 5, 7–10, 15, 17, 22–23), trio exome sequencing via clinical practice (4, 6, 12), or by research-based trio exome sequencing (13, 21). The genetic details from 13 of these individuals was previously reported (2, 3, 7–11, 14, 16, 18–20, 24) with minimal clinical details;2,7 we obtained additional, detailed clinical information for these individuals for this study.
RESULTS
We identified 27 unrelated individuals (16 female) with rare variants in GNAI1 (GenBank: NM_002069.6). For three individuals with GNAI1 variants, parental samples were unavailable for testing (Table S1); clinical information for these individuals has not been included in this report. The remaining 24 variants would meet the criteria to be classified as likely pathogenic or pathogenic according to ACMG guidelines if GNAI1 were an established disease gene9 (Fig. 1, Table 1). Of these, 23 variants were de novo in the affected individual (somatic mosaic in Individual 2), while one variant was inherited from a mother with low-level mosaicism (individual 3, 6.0% alternate allele frequency). Where applicable, de novo status of variants and parental relationships were confirmed.

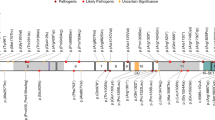
(a) Schematic showing the pathogenic and likely pathogenic variants identified in GNAI1, including one previously reported variant (*) (Kaplanis et al.2). Variants cluster within the first guanine nucleotide-binding domain (green box). Missense variants are represented as brown diamonds, coding deletion variants as blue circles, and the truncating frameshift variant as a yellow star. Each symbol represents one individual. (b) 3D structure of Gαi1. The left figure shows the structure of Gαi1 as part of the trimeric G-protein complex (PDB accession 6crk); Gαi1 is shown as a cyan ribbon, except for positions of novel variants which are colored as follows: missense, magenta; in-frame deletions, yellow; frameshifting insertion at Ile278, light green; bound GDP is shown as space-filling spheres, colored by atom type (white, carbon; blue, nitrogen; red, oxygen; orange, phosphorus); the molecular surface is shown for the βγ dimer, with the β1 and γ2 chains colored dark green and orange respectively. The right figure shows Gαi1 only, rotated around the vertical axis; Gly45 is obscured by the GDP ligand in this view. In both parts, labeling in bold font indicates residues making direct contact with GDP (Gly45, Thr48, Lys270, Ala326).
Among the 24 individuals with variants in GNAI1, there were 16 unique variants, with 7 recurrent variants identified in two or three individuals each. At three residues (Gly40, Thr48, Lys270), there were two different pathogenic variants resulting in different amino acid changes. Of the 16 variants, 12 were missense variants, 3 were small in-frame deletions, and 1 was a protein-truncating variant. The majority of missense and coding deletion variants (9/15; 60%) affect amino acids within the guanine nucleotide binding motifs of GNAI1 (Fig. 1). Variants predominantly cluster in the first GDP binding motif (also known as the Walker A motif or P-loop), where five variants at three sites (Gly40, Gly45, and Thr48) accounted for 10/24 individuals (42%), while variants at Arg270 and Ala326 affect residues in the fourth and fifth guanine nucleotide binding motifs, respectively.
We performed detailed phenotyping of the 24 affected individuals with de novo GNAI1 variants (Table 2, Table S1). Age at last medical review ranged from 3 years 10 months to 18 years (median age 11 years). All participants have global developmental delays ranging in severity from mild to profound. Speech is significantly affected with language delays reported in 21/23 (91%) individuals; 16 individuals are nonverbal (at ages 3–18 years), and only 1 individual has achieved fluent speech. Gross motor delays are also common. Delayed sitting was reported in 18/21 (86%) individuals, 5 of whom cannot yet sit independently (at ages 18 months–18 years). Delayed walking was reported in 19/23 (83%) individuals; 9 individuals remain nonambulatory (ages 18 months–18 years). Intellectual disability was reported in all individuals for whom data were available (20/20) and ranged from mild (3/20; 15%) to severe/profound (11/20; 55%).
Other prominent phenotypic features include hypotonia (20/23 individuals, 87%), and epilepsy (17/23 individuals, 74%). For many patients, hypotonia was severe and had a significant effect on daily functioning. Median age of seizure onset was 5 months (range 36 hours to 7 years), with seizures beginning in the first six months of life in 10/15 (67%) individuals for whom data were available. Seizure types were variable with the most common being absence seizures (n = 5), generalized tonic–clonic seizures (n = 5), and focal onset impaired awareness seizures (n = 4).
While behavioral anomalies were present in 17/19 individuals (89%), they were variable across the cohort. The most common behavioral features included aggression and temper tantrums (n = 7), autism (n = 7), hypersensitivity (n = 5), and hand stereotypies (n = 6). Other variable features included feeding difficulties (n = 9) and obesity (n = 7). Magnetic resonance image (MRI) abnormalities were reported for 10/20 (50%) individuals, with the most common finding being brain atrophy seen in four individuals.
Dysmorphic features were reported in 16/21 (76%) individuals (Fig. 2); however, the described physical features were variable. The most commonly reported features included tapered fingers (n = 9), a markedly long hallux (n = 5), and a short, upturned nose (n = 8). Other common facial features included a tented upper lip or open mouth appearance (n = 5), and a thin upper lip or prominent lower lip (n = 4).

(a) Individual 2; (b) individual 10; (c) individuals 11; (d) individual 16; (e) individual 18; (f) individual 19; (g) individual 20; (h) individual 22; (i) individual 23; (j) individual 24. Affected individuals have variable minor dysmorphic features and tend to have tapering fingers.
DISCUSSION
We describe a novel, severe neurodevelopmental disorder due to de novo variants in the GNAI1 gene, which encodes Gαi1, a member of the Gi/o inhibitory family of G-protein α-subunits. Heterotrimeric G-proteins act as a molecular switch. The GDP-bound Gα subunit binds the Gβγ dimer, maintaining the heterotrimeric protein in an inactive state. In response to an extracellular stimulant, bound GDP is replaced by GTP, resulting in a conformational change leading to the disassociation of the Gα subunit from the Gβγ dimer. Once separated, the Gα subunit and the Gβγ dimer are able to activate (or inhibit) downstream signaling pathways via modulation of cAMP levels. The intrinsic GTPase activity of the Gα subunit will eventually result in GTP hydrolysis, returning the protein to its GDP-bound inactive heterotrimeric state.5
Gαi1 is part of the Gi/o inhibitory family of α-subunits named for their ability to inhibit adenylyl cyclase activity. In the central nervous system, Gαi1 has been shown to mediate major signaling pathways Akt-mTORC1 and Erk–MAPK11,12 and control the gating of G protein-activated potassium channels.13 Other G-protein subunits have also been implicated in neurological disease including GNAQ (MIM 600998) associated with Sturge–Weber syndrome, GNAL (MIM 139312) associated with dystonia, GNAO1 (MIM 139311) in which loss-of-function variants are associated with developmental and epileptic encephalopathy while gain-of-function variants are associated with movement disorders,14 and GNB1 (MIM 139380) associated with developmental delay. Individuals with loss-of-function variants in GNB1, a β-subunit of heterotrimeric G-proteins, have a strikingly similar phenotype to individuals with de novo GNAI1 variants, including profound developmental delay commonly accompanied by hypotonia and seizures,4 suggesting there may be a common pathogenetic disease mechanism between GNB1 and GNAI1.
Gα proteins contain five highly conserved guanine nucleotide binding motifs that fold to form a single deep pocket for binding guanine nucleotides.10 Of the 16 pathogenic or likely pathogenic variants in GNAI1 reported in this study and one previously reported variant (p.Lys46Glu),2 nine variants at six sites (Gly40, Gly45, Lys46, Thr48, Lys270, and Ala326) are located in the guanine nucleotide binding pocket of GNAI1 (Fig. 1). Gly40, Gly45, Lys46, and Thr48 reside in the highly conserved first GDP binding motif. Gly40 lies at the mouth of the nucleotide binding pocket immediately N-terminal to a series of GDP-interacting residues, including Gly45 and Thr48. Notably, although Arg270 and Ala326 are distant in the linear sequence to Gly45 and Thr48, they lie in close spatial proximity on the opposite face of the GDP binding pocket.11 Gly45, Thr48, and Lys270 make direct contacts with the GDP ligand and therefore the Gly45Asp, Thr48Lys, Thr48Ile, Lys270Asn, and Lys270Arg substitutions are likely to disrupt these interactions. While Gly40 does not directly interact with the GDP ligand, structural modeling predicts that the Gly40Arg and Gly40Cys substitutions will have a significant destabilizing effect on the GDP binding pocket (increases in free energy compared with the native structure of ~9.4 kcal/mol and ~27.6 kcal/mol respectively; values >3 kcal/mol are generally regarded as strongly destabilizing15). As such, the pathogenic variants in guanine nucleotide binding motifs of Gαi1 are all predicted to have adverse effects on Gαi1 function through the disruption of Gαi1 ability to bind GDP and GTP and/or hydrolyze GTP.
GNAI1 is predicted to be intolerant to loss-of-function variants (pLI = 0.91; e/o = 0.12 [0.05–0.38]).16 Of the 16 variants in GNAI1 identified in 24 individuals, only one was a truncating variant: p.(Ile278Asnfs*20) frameshift identified in a single individual with profound developmental delay, axial hypotonia, and seizures; this variant is located within the last 50 bp of the penultimate exon and is predicted to escape nonsense-mediated decay. We identified one additional individual with an early truncating variant, but inheritance information was not available (Supplementary Table 1). We also previously identified large, heterozygous deletions encompassing GNAI1 (and additional genes) in two unrelated individuals with epilepsy.17 In one case, the deletion segregated in a large family, with at least five affected individuals in three generations; all had variable types of generalized epilepsy (ranging from mild to severe) and learning difficulties. Additional individuals with truncating variants need to be identified to determine if there are differences in phenotypes between individuals with missense and truncating variants in GNAI1.
GNAI1-related NDD is most often characterized by severe to profound delays, hypotonia, epilepsy that ranges from self-limiting to intractable, behavior problems, and variable mild dysmorphic features, though there is range of severity for all phenotypic features associated with pathogenic variants in GNAI1. Like many NDDs that have been recently described, GNAI1-related disorder may not be distinctly recognizable; many features overlap with other single-gene disorders including GNB1,4 PPP3CA,18 TANC2,19 and others. While many individuals have severe developmental delay accompanied by several additional comorbidities, there are also some more mildly affected individuals. In some cases, additional genetic variants may contribute to the phenotype; for example, individual 9 has a de novo, likely pathogenic variant in ACTA1, which can cause myopathy and may contribute to hypotonia or club feet in this case. Although we identified seven recurrent variants, our cohort is too small to determine whether there are clear genotype–phenotype correlations. The two individuals with p.(Lys270Arg) variants (individuals 19 and 20) have similar presentations, with similar ages at sitting and walking, development of some speech, and seizures. In contrast, for the two individuals with p.(Gln172del) variants, one is nonverbal, nonambulatory, and has intractable seizures (individual 13), while the other (individual 14) is ambulatory, has aggressive behaviors and has not had seizures. This lack of clear genotype–phenotype correlation will make it difficult to predict the severity of the disease progression in newly diagnosed individuals.
In summary, we report 24 individuals with de novo variants in GNAI1 and a neurodevelopmental disorder characterized by global developmental delay, intellectual disability, hypotonia, and seizures. While there is a spectrum of severity associated with pathogenic variants in GNAI1, most individuals are profoundly affected. Identification of additional cases as well systematic studies that implement uniform tools to evaluate phenotypes such as behavior and cognitive functioning will provide further insight into the full spectrum of neurological features associated with GNAI1 and potentially elucidate subtle genotype–phenotype correlations not apparent in this cohort.
URLs
gnomAD v2.1.1: https://gnomad.broadinstitute.org/. CADD: http://cadd.gs.washington.edu/. FoldX suite: http://foldxsuite.crg.eu/. GeneMatcher: https://www.genematcher.org/. MyGene2: https://mygene2.org/MyGene2/. Matchmaker Exchange: https://www.matchmakerexchange.org/. M-CAP Score: http://bejerano.stanford.edu/mcap/. OMIM: https://www.omim.org/.
Data availability
All methods and data are available on request.
References
Deciphering Developmental Disorders Study. Large-scale discovery of novel genetic causes of developmental disorders. Nature. 519, 223–228 (2015).
Kaplanis, J. et al. Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature. 586, 757–762 (2020).
Nakamura, K. et al. De novo mutations in GNAO1, encoding a Galphao subunit of heterotrimeric G proteins, cause epileptic encephalopathy. Am. J. Hum. Genet. 93, 496–505 (2013).
Petrovski, S. et al. Germline de novo mutations in GNB1 cause severe neurodevelopmental disability, hypotonia, and seizures. Am. J. Hum. Genet. 98, 1001–1010 (2016).
McCudden, C. R., Hains, M. D., Kimple, R. J., Siderovski, D. P. & Willard, F. S. G-protein signaling: back to the future. Cell. Mol. Life Sci. 62, 551–577 (2005).
Vassilatis, D. K. et al. The G protein-coupled receptor repertoires of human and mouse. Proc. Natl. Acad. Sci. U. S. A. 100, 4903–4908 (2003).
Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature. 542, 433–438 (2017).
Sobreira, N., Schiettecatte, F., Valle, D. & Hamosh, A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum. Mutat. 36, 928–930 (2015).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Retterer, K. et al. Clinical application of whole-exome sequencing across clinical indications. Genet. Med. 18, 696–704 (2016).
Cao, C. et al. Galpha(i1) and Galpha(i3) are required for epidermal growth factor-mediated activation of the Akt-mTORC1 pathway. Sci. Signal. 2, ra17 (2009).
Marshall, J. et al. Antidepression action of BDNF requires and is mimicked by Galphai1/3 expression in the hippocampus. Proc. Natl. Acad. Sci. U. S. A. 115, E3549–E3558 (2018).
Peleg, S., Varon, D., Ivanina, T., Dessauer, C. W. & Dascal, N. G(alpha)(i) controls the gating of the G protein-activated K(+) channel, GIRK. Neuron. 33, 87–99 (2002).
Feng, H. et al. Movement disorder in GNAO1 encephalopathy associated with gain-of-function mutations. Neurology. 89, 762–770 (2017).
Tokuriki, N., Stricher, F., Schymkowitz, J., Serrano, L. & Tawfik, D. S. The stability effects of protein mutations appear to be universally distributed. J. Mol. Biol. 369, 1318–1332 (2007).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 536, 285–291 (2016).
Mefford, H. C. et al. Rare copy number variants are an important cause of epileptic encephalopathies. Ann. Neurol. 70, 974–985 (2011).
Myers, C. T. et al. De novo mutations in PPP3CA cause severe neurodevelopmental disease with seizures. Am. J. Hum. Genet. 101, 516–524 (2017).
Guo, H. et al. Disruptive mutations in TANC2 define a neurodevelopmental syndrome associated with psychiatric disorders. Nat. Commun. 10, 4679 (2019).
Acknowledgements
We thank all affected individuals and their families for participating in this study. A.M.M. received support from the American Epilepsy Society. H.C.M. received support from the National Institute of Neurological Disorders and Stroke (R01 NS069605). F.L. and A.G. received funding from European Union and Région Normandie in the context of Recherche Innovation Normandie (RIN 2018). FL and AG received fundings from European Union and Région Normandie in the context of Recherche Innovation Normandie (RIN 2018) and the European Regional Development Fund (ERDF). Sequencing and analysis were provided by the Broad Institute of MIT and Harvard Center for Mendelian Genomics (Broad CMG) and was funded by the National Human Genome Research Institute, the National Eye Institute, and the National Heart, Lung and Blood Institute grants UM1 HG008900, R01 HG009141 and by the Chan Zuckerberg Initiative to the Rare Genomes Project. The DDD study presents independent research commissioned by the Health Innovation Challenge Fund (grant number HICF-1009–003). This study makes use of DECIPHER (http://decipher.sanger.ac.uk), which is funded by Wellcome. See Nature PMID: 25533962 or www.ddduk.org/access.html for full acknowledgement.
Author information
Authors and Affiliations
Contributions
Conceptualization: L.R., A.M.M., J.F.G., V.C.V., H.C.M. Data curation: A.M.M., J.F.G., R.H.v.J., I.M.d.L., J.J.v.d.S., G.N.W., H.D., E.M.G., L.Z., C.Br., J.M., M.S., A.Ac., A.Al., M.M., A.Go., C.Bu., R.C.C., T.M., C.M., S.M., C.P., P.C.V., A.F.B., S.J., C.T., G.N., M.T., L.B., M.Z., S.A.S.V., B.M., L.S., B.E.H., K.S., D.D., D.J.H., I.M.W., R.E.S., K.G.M., J.J., L.R., W.B.D., F.L., A.Gr., T.P., S.A.-S., K.P., C.K., T.L.H., D.G.M., M.C.O., G.E.V., E.E., V.C.V., H.C.M. Formal analysis: L.R., A.M.M., J.F.G., V.C.V., H.C.M. Writing—original draft: A.M.M., J.F.G., H.C.M. Writing—review & editing: R.H.v.J., A.M.M., J.F.G., H.C.M.
Corresponding author
Ethics declarations
Competing interests
I.M.W., R.E.S., K.G.M., J.J., and L.R. are employees of GeneDx, Inc.
Ethics declaration
This study was approved by local institutional review boards of the participating centers (University of Washington and UK Ethics Research Committee). Informed consent was obtained from all individuals or was provided by a parent or legal guardian in the case of minors or individuals with intellectual disability. Their permission for inclusion in this case series, including photographs, was obtained locally.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Muir, A.M., Gardner, J.F., van Jaarsveld, R.H. et al. Variants in GNAI1 cause a syndrome associated with variable features including developmental delay, seizures, and hypotonia. Genet Med 23, 881–887 (2021). https://doi.org/10.1038/s41436-020-01076-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-01076-8
This article is cited by
-
Consensus reporting guidelines to address gaps in descriptions of ultra-rare genetic conditions
npj Genomic Medicine (2024)
-
Novel de novo pathogenic variant in the GNAI1 gene as a cause of severe disorders of intellectual development
Journal of Human Genetics (2022)
