
Abstract
Purpose
To evaluate the incidence of mosaicism in de novo neurofibromatosis 2 (NF2).
Methods
Patients fulfilling NF2 criteria, but with no known affected family member from a previous generation (n = 1055), were tested for NF2 variants in lymphocyte DNA and where available tumor DNA. The proportion of individuals with a proven or presumed mosaic NF2 variant was assessed and allele frequencies of identified variants evaluated using next-generation sequencing.
Results
The rate of proven/presumed mosaicism was 232/1055 (22.0%). However, nonmosaic heterozygous pathogenic variants were only identified in 387/1055 (36.7%). When variant detection rates in second generation nonmosaics were applied to de novo cases, we assessed the overall probable mosaicism rate to be 59.7%. This rate differed by age from 21.7% in those presenting with bilateral vestibular schwannoma <20 years to 80.7% in those aged ≥60 years. A mosaic variant was detected in all parents of affected children with a single-nucleotide pathogenic NF2 variant.
Conclusion
This study has identified a very high probable mosaicism rate in de novo NF2, probably making NF2 the condition with the highest expressed rate of mosaicism in de novo dominant disease that is nonlethal in heterozygote form. Risks to offspring are small and probably correlate with variant allele frequency detected in blood.
Similar content being viewed by others

INTRODUCTION
Neurofibromatosis 2 (NF2, MIM#101000) is characterized by the development of multiple benign nerve sheath schwannomas with the hallmark being bilateral vestibular schwannoma (VS).1,2,3,4,5 Affected individuals may also develop meningiomas and ependymomas with ocular features such as lens opacity and retinal hamartomas being frequent.1,3 The NF2 gene has a high de novo variant rate with 50–60% of NF2 patients having no affected parent.6,7 Mosaicism in the first affected family members of tumor prone kindreds has been recognized since at least 1992,8 and NF2 was one of the first conditions found to demonstrate this phenomenon.9 Since then a number of reports have assessed the frequency of mosaicism in NF2 to be ~25–33%.10,11,12
In addition to the high rate of mosaicism in de novo NF2, mosaicism may account for around 37% of de novo cases fulfilling schwannomatosis criteria without vestibular schwannomas.13,14 Newer sequencing techniques such as deep sequencing and next-generation sequencing (NGS) have improved the sensitivity of detection of mosaicism.15,16 We have reassessed the frequency of mosaicism in de novo NF2 in over 1000 first generation affected cases. More recently, we have been able to assess allele frequencies of germline NF2 variants using NGS in lymphocytes for the great majority of those previously identified as having two NF2 genetic “hits” in tumor, but where neither was identified on Sanger sequencing of lymphocyte DNA.
MATERIALS AND METHODS
Individuals meeting NF2 diagnostic criteria (Table 1) from the first generation (founders) had analysis of lymphocyte DNA and where possible DNA extracted from fresh NF2 tumor samples or paraffin-embedded tumor block sections.
Molecular analysis
All individuals underwent lymphocyte DNA analysis for NF2, with additional analysis in LZTR1 and SMARCB1 in cases with a unilateral VS or multiple schwannomas. From 1995 to 2012 genetic testing utilized Sanger sequencing of all exons and intron/exon boundaries. Since 2013 NGS has been employed to sequence NF2 with a read depth of 1000×. This includes the same regions as Sanger sequencing plus additional intronic sequence, which has led to an increase in the variant detection rate. NF2 pathogenic variant testing of lymphocyte DNA (and tumor when available) also employed multiple ligation-dependent probe amplification (MLPA) to assess copy-number variations (CNVs), mostly single or multiple exon deletions. In addition, loss of heterozygosity (LOH) was assessed on tumor specimens with intragenic and flanking polymorphic microsatellite markers. Similar analyses were performed for LZTR1 and SMARCB1. Individuals with de novo NF2 and learning problems also had chromosome analysis for ring 22 and those with unfound familial NF2 were tested for translocations. Mosaicism was considered confirmed when (1) a pathogenic variant was detectable in lymphocyte DNA at a substantially reduced level (starting below 30% mutant allele frequency and with the lowest levels often only detected after NGS guided by tumor analysis) or (2) an identical pathogenic variant was found in two tumors that were anatomically distinct. A third category of “presumed” mosaicism was described when an individual, fulfilling NF2 diagnostic criteria, but with only one tumor available for analysis, had both mutational events found in a single tumor, but not found in blood DNA. An NF2 diagnosis was refuted when molecular events were not consistent between two tumors or when NF2 testing did not identify a constitutional or mosaic variant and/or a pathogenic variant was found in another gene with an overlapping phenotype (e.g., LZTR1). Ethical approval was obtained from the North West 7–Greater Manchester Central Research Ethics Committee (reference 10/H1008/74).
Statistical analysis
Pairwise comparisons of statistical significance between groups were carried out using analysis of variance (ANOVA) or Kruskal–Wallis analysis.
RESULTS
In total there were 1055 de novo individuals and 359 with an affected parent who had molecular testing. Of the 1055 de novo patients fulfilling NF2 criteria only 387 (36.7%) had a nonmosaic, heterozygous pathogenic NF2 variant identified (Table 2). This included a total of 300 point variants (single base changes and small insertion/deletions), 85 CNVs, and 2 chromosome translocations, identified by a combination of Sanger sequencing, MLPA, and cytogenetic analysis.
A total of 232 patients (22.0%) were identified as proven/probable mosaic. Initially, Sanger sequencing identified 54 low allele frequency pathogenic single- or two-nucleotide variants and MLPA detected 16 low allele frequency CNVs. More recently, NGS testing identified a further ten pathogenic single-nucleotide variants at Sanger detectable allele fractions of over 10% (range 11.7–30%) using lymphocyte DNA alone, and four with the aid of tumor testing (range 11–30%). Ten patients were found to have ring 22 by cytogenetic analysis. As such, 471/1055 (44.6%; including 84 mosaic and 387 nonmosaic) pathogenic NF2 variants occurred in lymphocyte DNA at levels detectable by Sanger sequencing plus MLPA and 481/1055 (45.6%) on addition of cytogenetic analysis for ring 22, which causes a mosaic loss of the NF2 gene.
Detection of low allele frequency variants
Seven additional pathogenic variants were found on NGS testing below Sanger detection levels (<10%; range 1–8%) using lymphocyte DNA alone. A further case involving a clinically affected parent, who had mild NF2 features and an affected child, had the child’s pathogenic frameshift variant c.191–198del identified on NGS in lymphocyte DNA at only 0.2% allele fraction. Pathogenic variants were found in an additional 40 affected de novo individuals at allele fractions ≤10% (range 0.5–10%) using NGS, after tumor testing identified two pathogenic variant changes in tumor(s). In addition, 21 variants that were found to be identical in two separate tumors from the same individual were not identified in lymphocyte DNA using NGS. There were also seven CNVs that were found to be identical between two tumors that were not found in lymphocyte DNA by MLPA (we are not be able to detect a CNV at <10% allele fraction).
There were 47 patients in whom two pathogenic variants were confirmed in a single tumor, but testing lymphocyte DNA of 45 of these using NGS did not detect a variant (two lymphocyte DNA samples were exhausted after Sanger sequencing and therefore not assessable by NGS). Overall, in cases where only one tumor was assessable and Sanger sequencing was negative, NGS confirmed mosaicism in 30/75 (40%) of available lymphocyte DNA samples (allele fraction range 1.0–10%; median 2.9%), This was a little higher than the proportion of mosaic NF2 confirmed by NGS in cases with identical point variants identified in two tumors, 10/31 (32.3%) (allele fraction range 0.5–10%; median 1.5%).
In nine cases without proven mosaicism or a detectable variant in blood where two tumors were available for DNA analysis, mosaic NF2 could not be confirmed. In three of these cases it was not possible to identify a pathogenic variant (except LOH in one tumor in one case). In two cases it was possible to refute a diagnosis of NF2 as nonidentical genetic events were found in the two tumors. One of these two individuals had only bilateral vestibular schwannomas (BVS) as previously reported,17 and the other had presumed radiation-induced tumors from childhood lymphoma treatment.1 Two further cases with different point variants in each tumor, but with loss of the same allele identified by LOH analysis, were each found to harbor a germline pathogenic LZTR1 variant. These were both in a group of patients with unilateral VS and two or more schwannomas with no meningiomas or ependymoma disease. Conversely, there were 15 patients meeting schwannomatosis diagnostic criteria who were found to have identical pathogenic NF2 variants in two tumors and no pathogenic variants identified in SMARCB1 or LZTR1. These individuals were therefore reclassified as mosaic NF2. The final two individuals, with two tumors who could not be confirmed as mosaic NF2, had a point variant and LOH confirmed in one tumor and the point variant could not be confirmed in a second tumor. A large gene rearrangement could not be confirmed at a mosaic level as a cause of LOH in both tumors.
Nineteen individuals affected with NF2 and with a single tumor analyzed did not have a pathogenic NF2 variant identified in tumor, although four of these tumors did demonstrate LOH of chromosome 22. The 15/19 with no LOH is a high proportion compared with typical schwannomas and may indicate significant stromal contamination as the reason for failure to identify NF2 pathogenic variants. Overall, of those with at least one tumor analyzed and without a germline LZTR1 pathogenic variant, 134/160 (83.8%) had confirmed or presumed mosaic NF2 and 26/160 (16.3%) did not have NF2 mosaicism confirmed.
Frequent mosaic variants
The most frequent mosaic variants were the nonsense pathogenic variants c.784C>T, p.(Arg262Ter) (n = 14) and c.169C>T, p.(Arg57Ter) (n = 12). A number of other nonsense variants were seen at least four times (c.586C>T, p. (Arg196Ter) (n = 10); c.1021C>T, p. (Arg341Ter) (n = 6); c.1396C>T, p.(Arg466Ter) (n = 5); c.193C>T, p.(Gln65Ter) (n = 4). The c.784C>T, p.(Arg262Ter) variant was over-represented in mosaics 14/232 (6.3%) compared with de novo nonmosaic heterozygotes 9/385 (2.3%) (p = 0.025) and was seen only once in 359 inherited cases. The pathogenic missense variant c.655G>A, p.(Val219Met) was seen in four individuals with mosaic NF2 and a further variant at the same codon c.656T>A was seen in one mosaic individual, p.(Val219Glu), yet missense variants at this codon were absent from 744 NF2 affected patients with nonmosaic constitutional heterozygous pathogenic variants (p < 0.0001). The in-frame deletion c.357_359delCTT, p.(Phe119del) was seen in four individuals with mosaic NF2, yet was also not seen in 744 NF2 affected patients with nonmosaic constitutional heterozygous pathogenic variants. Indeed, in-frame deletions (n = 9/232) were highly significantly over-represented in mosaics (p < 0.0001) compared with nonmosaic heterozygotes (n = 2/744).
Overall likelihood of mosaicism
Assuming a 93% detection rate for a nonmosaic pathogenic variant using a combination of NGS and MLPA (146/157 second generation familial NF2 probands are detected in this way) the overall rate of mosaicism for de novo cases could be as high as 59.7% (Table 3). This assumes that 93% of those with a clinical diagnosis of NF2, but without an identified nonmosaic pathogenic variant, are probable mosaics. The highest rate of presumed mosaicism is found in the individuals with a unilateral VS and two or more nonvestibular schwannomas, but no meningiomas or ependymoma disease, at 79.5%. This is despite six cases (8.3%) having an LZTR1 pathogenic variant. This is reflected in the very low rate of only 8.3% for confirmed nonmosaic variants.
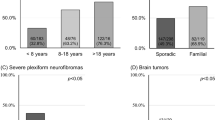
The rates of mosaicism in de novo cases correlate with increased age at diagnosis of NF2 (Table 4). The rates are increased from 21.7% probable mosaicism in those with BVS <20 years of age to over 80% in those diagnosed >60 years of age. The median age at diagnosis of mosaic NF2 was 35 (range: 1–76) compared with a median age at diagnosis of nonmosaic de novo heterozygotes of 21 (range 0.5–86) (p < 0.001). There were also significantly lower mean and median ages at onset in those with mosaicism confirmed in blood compared with those that were confirmed in tumor only, not confirmed in tumor (presumed), or had no tumor tested (Table 5).
Offspring risk
Ten mosaic affected de novo cases had 12 affected children. All six de novo cases with a pathogenic single-nucleotide variant had a variant that was detectable in blood on NGS (at allele fractions of 0.2%, 4%, 15%, 20%, 22%, and 30%) and these parents had seven affected children (two from the case with a 15% allele frequency). For the 32 de novo cases with a mosaic CNV, only 2 that were detectable in the affected child were also present at a detectable level in blood from the parent, although an exon 1–16 deletion was found in the tumor of a third affected parent. One affected parent with an exon 7 deletion detected on MLPA in blood had two affected children. Although we do not have details on all children we have carried out a total of 75 predictive tests on children of mosaic cases with point variants detectable in blood and eight of these (10.7%) were positive. Of the 71 parents with presumed mosaic NF2 but without a pathogenic single-nucleotide variant detectable on NGS in blood, all 85 presymptomatic tests on children were negative.
Epidemiology
Within the boundaries of the North of England (population = 15.35 million) covered by the Manchester center there were 303 living NF2 patients as of January 2019 (prevalence = 1 in 50,660). Of the 303 patients, 207 (68.3%) were de novo cases and of these 60 had confirmed or probable mosaicism (29.0%) with 5/60 (8.3%) having ring 22 with a confirmed tumor diagnosis of NF2. Eighty further cases (38.6%) had no identified NF2 variant on DNA analysis in blood.
DISCUSSION
The present study has confirmed that mosaicism is extremely common in de novo NF2 patients. Our estimates of the frequency of mosaicism in de novo cases are now much higher than the previous 25–33% reported in the literature.11,12 This is based on the very high sensitivity of NF2 testing using a combination of high read depth NGS and MLPA in the second generation of families of 93%. Although the detection rate for both mutational “hits” in tumors is a little lower at 84%, this lower detection rate may be due to tumor contamination with macrophages, leading to a far lower tumor/Schwann cell content.18 Most of these tumors were assessed by Sanger sequencing and LOH analysis. Both of these techniques require greater than 10% tumor cell content to detect pathogenic variants and detection of LOH would usually require >50% tumor cell content. The overall predicted higher rate of mosaicism of 59.7% in de novo cases could be the result of the very high levels of ascertainment of milder later onset cases by the English highly specialized NF2 service, wherein four national centers care for all NF2 patients, and make ascertainment extremely high for the UK.2,14 Previous reports on rates of NF2 mosaicism are likely to be based on lower ascertainment levels with identified mosaic cases being more severely affected and more likely to have presented with classical bilateral VS. As previously described, detected levels of mosaicism are higher when patients present asymmetrically with a delay between diagnosis of the first and second VS or when they do not develop a second VS.12 Even for those presenting with bilateral VS, age at onset is a major factor with only 21.7% of those aged under 20 years at presentation predicted to be mosaic, compared with 80.7% of those presenting aged 60 years and over (Table 4).
Another confounding factor in identification of NF2 is the clinical diagnostic overlap between NF2 disease and SMARCB1-associated or LZTR1-associated schwannomatosis. In our cohort 15 individuals thought to have non-NF2 schwannomatosis were ultimately reclassified as having mosaic NF2 after identical pathogenic NF2 variants were identified in two separate tumors. This reclassification is not always possible due to a lack of available tumor material for genetic testing. Therefore, limitations of the study include incomplete screening for some patients, due to a lack of tumor material, or insufficient remaining DNA to allow retesting with NGS for many of the older tumors in which no pathogenic variants were detected by Sanger sequencing. In addition, some of the presumed mosaic cases may not have NF2 disease, but may have developed nonsyndromic tumors by chance.
The present study has also demonstrated that certain point variants appear to be extremely over-represented in mosaic cases. The high rate of the more severe19 nonsense pathogenic variants represent the known higher rate of de novo variant at CpG sites.20 However, the predominance of missense variants at codon 219 in five individuals and the in-frame deletion, c.357_359delCTT, p.(Phe119del) in four individuals with mosaic NF2, which were all absent from 744 NF2 affected patients with nonmosaic constitutional heterozygous pathogenic variants (p < 0.0001), is harder to explain. One possibility is that these variants are more likely to occur in embryogenesis than gametogenesis. Ordinarily these variants might be considered candidates for mild NF2, but their occurrence in the mosaic form suggests that they are more likely to cause severe NF2 in the heterozygous state.19,20
Patients with ring chromosome 22 have been included among the mosaic classification for NF2. Patients thus far reported with ring 22 have an intact NF2 gene within the ring, however, the ring is unstable and frequently lost in cell division.21,22 Thus patients with ring 22 are effectively mosaic for a whole NF2 gene deletion.21,22 Ring chromosomes are unstable not only in terms of frequent loss but also in being subject to bridge–breakage–fusion so that the ring may become much smaller (deleting the NF2 gene but not the entire chromosome) in some cells although this mechanism is yet to be proven as the mechanism of tumorigenesis. The present report shows that, in our highly ascertained region of Northern England, ring 22 constitutes 1.6% of all NF2 prevalent cases, 2.4% of de novo living cases, and 8.3% of confirmed mosaic cases. Also, 1 in 3 million people have ring 22 in the UK population with a confirmed NF2-associated tumor diagnosis. The penetrance of a clinical diagnosis of NF2 in ring 22 is not known but it is certainly possible that individuals with ring 22, who are typically institutionalized in adult years, may die with symptomatic tumors, but without a diagnosis of NF2.
The NGS era has revealed higher levels of mosaicism in disease than previously recognized,23 although most mosaicism may not be clinically recognized and can be found in unaffected parents of apparently de novo cases.23 Mosaicism has been reported in as high as 70–90% of cleavage and blastocyst stage embryos derived from in vitro fertilization, respectively.24
One of the highest rates for mosaicism is found for small supernumerary marker chromosomes that were found to be mosaic in around 50% of cases but the majority were not symptomatic.25 Mosaicism has been described in many tumor predisposition syndromes including familial adenomatous polyposis (FAP), neurofibromatosis 1, tuberous sclerosis (particularly for CNVs), and even BRCA1. However, none of these have been described at the frequencies seen in NF2. Some conditions are only seen in the mosaic state, such as Proteus syndrome, which is caused by mosaic AKT pathogenic variants.26 Overall, for conditions that are expressed clinically due to heterozygous point variants, NF2 appears to have by far the highest proportion of de novo mosaicism. One reason for this could be the known effects on mortality27 and genetic fitness6 that cause severely affected nonmosaic variant carriers to die early or choose not to have children, thus reducing the total number of affected nonmosaic individuals and increasing the proportion of mosaic disease.28
In conclusion, addition of high read depth NGS analysis to NF2 screening practices has improved our ability to detect mosaic NF2, indicating a higher level of mosaicism than previously thought and leaving only a small fraction with unidentified variants. These individuals may have more complex variants that remain undetected by standard analysis and may require alternative methods of detection.
References
Evans DG, King AT, Bowers NL, et al. Identifying the deficiencies of current diagnostic criteria for neurofibromatosis 2 using databases of 2777 individuals with molecular testing. Genet Med. 2018 Dec 7; https://doi.org/10.1038/s41436-018-0384-y [Epub ahead of print].
Baser ME, Friedman JM, Wallace AJ, Ramsden RT, Joe H, Evans DG. Evaluation of clinical diagnostic criteria for neurofibromatosis 2. Neurology. 2002;59:1759–1765.
Evans DG, Huson SM, Donnai D, et al. A clinical study of type 2 neurofibromatosis. Q J Med. 1992;84:603–618.
Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol. 1988;45:575–578.
Baser ME, Friedman JM, Joe H, et al. Empirical development of improved diagnostic criteria for neurofibromatosis 2. Genet Med. 2011;13:576–581.
Evans DG, Huson SM, Donnai D, et al. A genetic study of type 2 neurofibromatosis in the United Kingdom. I. Prevalence, mutation rate, fitness, and confirmation of maternal transmission effect on severity. J Med Genet. 1992;29:841–846.
Evans DG, Howard E, Giblin C, et al. Birth incidence and prevalence of tumor-prone syndromes: estimates from a UK family genetic register service. Am J Med Genet. 2010;152A:327–332.
Kovar H, Auinger A, Jug G, Muller T, Pillwein K. p53 mosaicism with an exon 8 germline mutation in the founder of a cancer-prone pedigree. Oncogene. 1992;7:2169–2173.
Bourn D, Carter SA, Evans DG, Goodship J, Coakham H, Strachan T. A mutation in the neurofibromatosis type 2 tumor-suppressor gene, giving rise to widely different clinical phenotypes in two unrelated individuals. Am J Hum Genet. 1994;55:69–73.
Evans DG, Wallace AJ, Wu CL, Trueman L, Ramsden RT, Strachan T. Somatic mosaicism: a common cause of classic disease in tumor-prone syndromes? Lessons from type 2 neurofibromatosis. Am J Hum Genet. 1998;63:727–736.
Kluwe L, Mautner V, Heinrich B, et al. Molecular study of frequency of mosaicism in neurofibromatosis 2 patients with bilateral vestibular schwannomas. J Med Genet. 2003;40:109–114.
Evans DG, Ramsden RT, Shenton A, et al. Mosaicism in neurofibromatosis type 2: an update of risk based on uni/bilaterality of vestibular schwannoma at presentation and sensitive mutation analysis including multiple ligation-dependent probe amplification. J Med Genet. 2007;44:424–428.
Kehrer-Sawatzki H, Kluwe L, Friedrich RE, et al. Phenotypic and genotypic overlap between mosaic NF2 and schwannomatosis in patients with multiple non-intradermal schwannomas. Hum Genet. 2018;137:543–552.
Evans DG, Bowers NL, Tobi S, et al. Schwannomatosis: a genetic and epidemiological study. J Neurol Neurosurg Psychiatry. 2018;89:1215–1219.
Contini E, Paganini I, Sestini R, et al. A systematic assessment of accuracy in detecting somatic mosaic variants by deep amplicon sequencing: application to NF2 gene. PLoS ONE. 2015;10:e0129099.
Louvrier C, Pasmant E, Briand-Suleau A, et al. Targeted next-generation sequencing for differential diagnosis of neurofibromatosis type 2, schwannomatosis, and meningiomatosis. Neuro Oncol. 2018;20:917–929.
Evans DG, Freeman S, Gokhale C, et al. Bilateral vestibular schwannomas in older patients: NF2 or chance? J Med Genet. 2015;52:422–424.
Lewis D, Roncaroli F, Agushi E, et al. Inflammation and vascular permeability correlate with growth in sporadic vestibular schwannoma. Neuro Oncol. 2019;21:314–325.
Hexter A, Jones A, Joe H, et al. Clinical and molecular predictors of mortality in neurofibromatosis 2: a UK national analysis of 1192 patients. J Med Genet. 2015;52:699–705.
Evans DG, Maher ER, Baser ME. Age related shift in the mutation spectra of germline and somatic NF2 mutations: hypothetical role of DNA repair mechanisms. J Med Genet 2005;42:630–632.
Tsilchorozidou T, Menko FH, Lalloo F, et al. Constitutional rearrangements of chromosome 22 as a cause of neurofibromatosis 2. J Med Genet. 2004;41:529–534.
Denayer E, Brems H, de Cock P, et al. Pathogenesis of vestibular schwannoma in ring chromosome 22. BMC Med Genet. 2009;10:97.
Gajecka M. Unrevealed mosaicism in the next-generation sequencing era. Mol Genet Genomics. 2016;291:513–530.
Taylor TH, Gitlin SA, Patrick JL, Crain JL, Wilson JM, Griffin DK. The origin, mechanisms, incidence and clinical consequences of chromosomal mosaicism in humans. Hum Reprod Update. 2014;20:571–581.
Liehr T, Klein E, Mrasek K, et al. Clinical impact of somatic mosaicism in cases with small supernumerary marker chromosomes. Cytogenet Genome Res. 2013;139:158–163.
Lindhurst MJ, Sapp JC, Teer JK, et al. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N Engl J Med. 2011;365:611–619.
Baser ME, Friedman JM, Aeschliman D, et al. Predictors of the risk of mortality in neurofibromatosis 2. Am J Hum Genet. 2002;71:715–723.
Smith MJ, Urquhart JE, Harkness EF, et al. The contribution of whole gene deletions and large rearrangements to the mutation spectrum in inherited tumor predisposing syndromes. Hum Mutat. 2016;37:250–256.
Acknowledgements
The authors wish to acknowledge NHS England for their support of the National NF2 program. D.G.E., E.F.H., and M.J.S. are supported by the all Manchester National Institute for Health Research (NIHR) Biomedical Research Centre (IS-BRC-1215–20007).
English Specialist NF2 Research Group members
Cambridge and Central: Patrick Axon, Juliette Gair, James Tysome, Neil Donnelly, Lucy Raymond, Anke Hensiek, Rajesh Jena, Robert Macfarlane, Richard Mannion, James Nicholson, Brinda Muthusamy, Amy Taylor, Richard Price, Gabriella Rands, Nicola Gamazo, Zebunnisa Vanat, Daniel Scoffings, Sarah Jefferies, Richard Knight, Tamara Lamb, Yu Chuen Tam, Karen Foweraker, Fiona Harris, Paul Sanghera, Sara Meade, Richard Irving, Peter Monksfield, Saba Sharif, Nicola Ragge, Melanie Murrell, Julian Barwell, Martin English, Gary Doherty, Rikin Trivedi, Ilse Patterson
London: Shazia K. Afridi, Rosalie E. Ferner, Rupert Obholzer, Victoria Williams, Chris Hammond, Karine Lascelles, Chris Skilbeck, Shakeel Saeed, Adam Shaw, Angela Swampillai, Suki Thomson, Nick Thomas, Eleni Maratos, Sinan Barazi, Rebecca Mullin, Susie Henley, Sally Trump, Vanessa Everett, Terry Nunn, Charles Nduka
Manchester and North: D. Gareth Evans, Raji Anup, Chris Duff, Simon R. Freeman, Emma Stapleton, Nicola Jarvis, Ian Kamaly-Asl, Andrew T. King, Mark Kellett, John-Paul Kilday, Simon K. Lloyd, Connor Malluci, Deborah Mawman, Catherine McBain, Roger Laitt, Martin O'Driscoll, Martin McCabe, Mary Perry, Scott A. Rutherford, Kirsty Henshaw, Stavros M. Stivaros, Owen Thomas, Grace Vassallo, Charlotte L. Hammerbeck-Ward, Omar N. Pathmanaban, Jincy Kurian
Oxford and South West: Claire Hobbs, Kate Browne, Bruce Castle, Rosie Crabtree, Lucy Cogswell, Louise Dalton, Caroline Dodridge, Beatrice Emmanouil, Henk Giele, Dorothy Halliday, Jane Halliday, C. Oliver Hanemann, Wendy Howard, Richard Kerr, Elle Mace, Sam MacKeith, Allyson Parry, Peter Pretorius, James Ramsden, Carolyn Redman, Srilakshmi Sharma, Ros Taylor, Helen Tomkins, Shaun Wilson, Rachael Woolrich
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Evans, D.G., Hartley, C.L., Smith, P.T. et al. Incidence of mosaicism in 1055 de novo NF2 cases: much higher than previous estimates with high utility of next-generation sequencing. Genet Med 22, 53–59 (2020). https://doi.org/10.1038/s41436-019-0598-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-019-0598-7
Keywords
This article is cited by
-
Human embryonic genetic mosaicism and its effects on development and disease
Nature Reviews Genetics (2024)
-
Gene Therapy for Neurofibromatosis Type 2-Related Schwannomatosis: Recent Progress, Challenges, and Future Directions
Oncology and Therapy (2024)
-
The genetic landscape and possible therapeutics of neurofibromatosis type 2
Cancer Cell International (2023)
-
Correlation between genotype and phenotype with special attention to hearing in 14 Japanese cases of NF2-related schwannomatosis
Scientific Reports (2023)
-
Management of Central and Peripheral Nervous System Tumors in Patients with Neurofibromatosis
Current Oncology Reports (2023)
