
Abstract
Purpose
Heritable thoracic aortic aneurysms and dissections (hTAAD) are life-threatening complications of well-known syndromic diseases or underdiagnosed nonsyndromic heritable forms (nshTAAD). Both have an autosomal dominant transmission and are genetically heterogeneous. Our objective was to describe the relevance of molecular diagnosis in these patients and the contribution of each gene in nshTAAD.
Methods
Two hundred twenty-six consecutive nshTAAD probands, either young (<45 years) sporadic or familial cases were included. A next-generation sequencing capture panel comprising 23 known disease-causing genes was performed.
Results
Class 4 or 5 variants were identified in 18% of the nshTAAD probands, while class 3 variants were found in 10% of them. The yield in familial cases was greater than in sporadic cases. SMAD3 and FBN1 genes were the major disease-causing genes. Unexpectedly, no premature termination codon variant was identified in the FBN1 gene. Furthermore, we report for the first time that aortic dissection or surgery occurred significantly more often and earlier in probands with a class 4 or 5 pathogenic variant.
Conclusion
This study indicates that genetic screening using NGS is efficient in young and familial nshTAAD. The presence of a pathogenic variant has a possible predictive value, which needs to be further investigated because it may influence care.
Similar content being viewed by others


INTRODUCTION
Thoracic aortic aneurysms and dissections (TAAD) and their life-threatening complications are a common cause of premature deaths, accounting for over 10,000 and contributing to over 17,000 deaths annually in the United States.1,2 The etiologies underlying thoracic aortic enlargement are diverse and can range from degenerative or hypertension-associated aortic enlargement to more rare genetic disorders.3 The most well-known heritable diseases are the historic Marfan syndrome (MFS), the founding member of connective tissue disorders, and the more recently described Loeys–Dietz (LDS) and aneurysms–osteoarthritis (AOS) syndromes. However, even more recently recognized and still largely underdiagnosed are the nonsyndromic heritable forms of TAAD (nshTAAD). Transmitted as an autosomal dominant disease and presenting numerous sporadic cases, this clinical entity is highly heterogeneous at the genetic level with the involvement of rare variants in multiple genes. Genetic research progress in the past several years has enabled the identification of many nshTAAD genes. These results have unraveled four main types of TAAD-initiating pathogenic mechanisms: altered extracellular matrix proteins (pathogenic variants in FBN1, FBN2, LOX, BGN, MFAP5), altered TGF-β signaling proteins (TGFB2, TGFB3, TGFBR1, TGFBR2, SMAD2, SMAD3, SMAD4), altered smooth muscle cell (SMC) contraction (ACTA2, MYH11, MYLK, PRKG1, MAT2A, FLNA, LMOD1), and impaired SMC differentiation and proliferation (FOXE3). However, probably many more disease genes associated with nshTAAD are still unrecognized despite intensive research. This could be associated with the presence of variants in noncoding sequences of the known genes and thus located in noninvestigated regions or nonrecognized disease variants in intronic sequence reads. Furthermore, the last disease genes identified account for a very small number of probands. This could indicate that the genetic architecture underlying the disease is more complex than had been thought and that different inclusion criteria could be used to select nshTAAD probands for new research designs.
The past 20 years have completely altered the diagnosis of MFS and related disorders through the availability of capillary Sanger sequencers followed by the new next-generation sequencing (NGS) technologies and their platforms. Despite the availability of this exceptional technology and the dramatic drop in DNA sequencing costs, the cost of variant interpretation has not fallen as quickly. Thus, the low yield of positive results in highly heterogeneous genetic diseases and the rising load of inconclusive results question the cost-effectiveness of the strategy in diagnostic settings. Indeed, many publicly funded/institutional clinical laboratories still apply a combination of clinical and family criteria that probands need to meet to be investigated in order to raise the level of produced results that will inform clinical diagnosis and follow-up.
In the context described in the previous paragraphs, we set up a research project to inform our strategy for clinical diagnosis of nshTAAD in an economically constrained setting and also assess the missing heritability of nshTAAD when rigorous inclusion criteria are used to inform our research strategy for the identification of new disease genes. To address these aspects, we developed a research project built on a collection of at least 200 consecutive nshTAAD patients, either familial or occurring early in life (<45 years), recruited through the National French Reference Centre for Marfan syndrome and associated diseases in Paris as well as through the regional centers. In-depth clinical and family investigations were performed to exclude probands with syndromic forms of the disease and probands with well-known cardiovascular risk factors (mainly high blood pressure and bicuspid aortic valve). Our results show that a significant molecular event was identified in 18% of the probands. This yield was higher in familial cases than in sporadic cases of nshTAAD. SMAD3 and FBN1 genes were the two major disease-causing genes and unexpectedly, no premature termination codon (PTC) variant was identified in the FBN1 gene. Finally, we report for the first time that aortic dissection and surgery occurred significantly more often and earlier in patients who carried a pathogenic variant than in patients for whom no variant had been found.
MATERIALS AND METHODS
Patients
Two hundred twenty-six consecutive TAAD probands were recruited between 2012 and 2015 in the Programme Hospitalier de Recherche Clinique (PHRC) by Centre National Maladies Rares “Syndrome de Marfan et apparentés,” the French National Reference Centre located at Bichat Hospital (G.J., coordinator), and by seven affiliated competence centers located throughout the country (see URLs). The inclusion criteria were nonsyndromic TAAD, either sporadic in young patients (under the age of 45 years) or documented familial cases whatever the age. The exclusion criteria were syndromic TAAD, including MFS; vascular Ehlers–Danlos syndrome (vEDS); Loeys–Dietz syndrome (LDS); Noonan syndrome; and Turner syndrome. Patients presenting high blood pressure or bicuspid aortic valve (BAV) were also excluded. Patients were examined by several specialists: cardiologists, ophthalmologists, geneticists, rheumatologists, or pediatricians (depending on their age) with specific evaluation of clinical features to rule out syndromic diseases. Aortic diameter measurement was evaluated at the root level or at the level of the tubular portion of the ascending aorta. Aortic aneurysm was defined as a measure above mean +2 standard deviations (z-score > 2) as described in Campens et al.4 Systematic slit-lamp examination, cardiac ultrasonography, and radiological investigations were also performed. Family history was carefully detailed for each patient to investigate the recurrence of the disease in the family and systematic echocardiography was prescribed for all first-degree relatives. A proband was considered as a familial case if this pedigree investigation revealed other TAAD cases and/or several sudden deaths at a young age. Blood samples were obtained for all probands and relatives when possible. A specific informed consent was obtained for all patients in agreement with the requirements of French bioethics laws (institutional review board [IRB] approval from Comité de Protection des Personnes number 11 008, 13 January 2011).
DNA amplification and variant detection in known genes of nonsyndromic TAAD
Genomic DNA was isolated from peripheral blood leukocytes by standard procedures already reported.5 A custom capture array (NimbleGen SeqCap®, Roche, France) was designed and used to capture all coding exons, splice sites, and the flanking intronic sequences of 23 genes already known to be associated with heritable TAAD at the time: ACTA2 (NM_001613.3), COL3A1 (NM_000090.3), EFEMP2 (NM_016938.4), FBN1 (NM_000138.4), FBN2 (NM_001999.3), FLNA (NM_001110556.1), FOXE3 (NM_012186.2), LOX (NM_002317.6), MAT2A (NM_005911.5), MFAP5 (NM_003480.3), MYH11 (NM_001040113.1), MYLK (NM_053025.3), NOTCH1 (NM_017617.4), PRKG1 (NM_006258.3), SLC2A10 (NM_030777.3), SKI (NM_003036.3), SMAD2 (NM_001003652.3), SMAD3 (NM_005902.3), SMAD4 (NM_005359.5), TGFB2 (NM_001135599.2), TGFB3 (NM_003239.4), TGFBR1 (NM_004612.3), and TGFBR2 (NM_003242.5) (ref. 6). The total size of the target regions of the capture array was 132 kb. Briefly, oligonucleotide adapters containing a barcode sequence as a sample index signature were ligated to the fragments. After the ligation was complete, DNA sample library was amplified by seven-cycle polymerase chain reaction (PCR) using a high-fidelity polymerase. This product was used to generate a library for further analysis, and the DNA adapter-ligated and indexed fragments from 24 libraries were pooled and hybridized to the customized oligonucleotide probes. After further amplification of the captured DNA sample, sequencing was carried out on a MiSeq (Illumina, USA), following the manufacturer’s standard cluster generation and sequencing protocols. Variant calling was performed through CLC Genomics Workbench v10.1.1 (Qiagen Bioinformatics, Denmark, see URLs). Variant annotation was done with an in-house Python script, which contains, in brief, the genomic position, information about the gene in question, the type of variant, allele frequency in different population databases, and the scores obtained from different in silico prediction tools. All variants of interest were systematically confirmed by bidirectional Sanger sequencing of the altered exon (Big Dye terminator kit, ABI 3100 Genetic Analyzer, Applied Biosystems, UK; primers are available on request). When possible, familial segregation of variants was investigated. Description of sequence variants was performed according to Human Genome Variant Society nomenclature.7 For a variant possibly affecting a splice site, the splicing effect at complementary DNA (cDNA) level was verified if patient blood sample on PaxGene® (Qiagen, Germany) could be obtained for cDNA generation in accordance with the manufacturer’s recommendations.
In silico variant analysis
Three consensual bioinformatics programs (Polyphen-2 [ref. 8], SIFT9 via Alamut Visual 2.10 [Interactive Biosoftware, France], and UMD-Predictor10) were used to predict pathogenicity of missense variants. Effect on splicing was analyzed through different tools including Human Splicing Finder,11 splicing prediction algorithm NNSPLICE,12 and MaxEntScan method.13 The UMD locus-specific databases and Human Gene Mutation Database (HGMD®) were queried for each identified variant (see URLs). The existence of each molecular event was looked for in the Genome Aggregation Database (gnomAD).14 However, the laboratory has been performing molecular diagnosis of MFS and TAAD for over 20 years and a population of over 5000 probands has been sequenced for the disease-causing genes. This has led to an in-house reference database of molecular events identified in each gene for which we have robust French population frequencies. Variants were classified according to recommendations of the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP).15
Statistical analysis
Statistical analyses were performed in an exploratory manner. All results for categorical variables were presented as percentages and numbers. Pearson correlation was used to compare percentages. Survival curves were estimated using the Kaplan–Meier method and compared by means of log-rank tests. P values below 0.05 were considered as statistically significant. All calculations were performed using R software.16
RESULTS
Description of the cohort and general molecular results
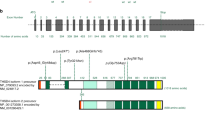
Two hundred twenty-six consecutive nshTAAD probands were included in the project. The mean age at diagnosis was 43.3 years (range: 12–76 years) with an approximate male/female sex ratio of 3.5/1. Women represented 22% (n = 50) of all the probands with a mean age of 45.6 years for women and 42.6 years for men. A family history of TAAD was found in 68.6% of the probands (n = 155 with 119 men) (familial cases) while the other part of the probands (n = 71 with 57 men) were isolated cases without any known family history of TAAD. The familial cases were on average older (mean age at diagnosis 46.5 years) than sporadic cases (mean age at diagnosis 36.3 years) as expected according to the inclusion criteria. A class 4 or 5 variant was identified in 18% of the probands (Table 1). This proportion was higher in familial cases (22%) compared with isolated cases (11%). No proband was found to carry more than one class 4 or 5 variant. Two thirds of class 4 or 5 variants were found in the SMAD3 and FBN1 genes (Fig. 1). The remaining third comprised variants identified in 9 other genes.

Gene distribution of pathogenic and likely pathogenic variants found in this study. The number of probands for each gene is indicated in parentheses.
Class 4 or 5 variants (likely pathogenic or pathogenic)
Forty-two of 226 probands carried a class 4 or 5 variant (18%). Thirteen variants were identified in the SMAD3 gene in 15 probands (Table 2, Supplementary Figure S1). Indeed, two SMAD3 variants were found twice in this cohort (c.401–6G>A and c.1117C>T). All of these 13 different variants are very rare molecular events because they are absent in the population database gnomAD. Furthermore, all variants but one had never been described in the UMD-SMAD3 locus-specific database or HGMD®. Six variants of 13 were PTC variants. Seven variants were missense variants and 6 of 7 were located in the MH2 domain of the SMAD3 protein. The missense variants in the SMAD3 gene were all predicted pathogenic by at least 2 of 3 of the missense prediction tools listed above. For the c.401–6G>A variant, an aberrant splice form with PTC was identified through transcript analysis. Because SMAD3 pathogenic variants were associated with early-onset osteoarthritis, Charcot–Marie–Tooth like neuropathy, and autoimmune features,17 patients carrying a SMAD3 variant were carefully reassessed in the search of these clinical signs. More than half of the probands or members of their family had a major joint involvement at an early age or autoimmune features. The diagnosis was then retrospectively repositioned as AOS18 in half of the SMAD3 gene variant carriers. For FBN1 gene, a class 4 or 5 variant was found in 12 probands (Table 2, Supplementary Figure S2). All but one were missense variants and no PTC variant was found. Two of the missense variants (p.[Arg485Cys] and p.[Ile2585Thr]) had already been reported in the UMD-FBN1 locus-specific database and HGMD®. Eight were located in a calcium-binding epidermal growth factor (cbEGF) domain of the protein: in five cases, it concerned either a cysteine involved in a disulfide bridge or a highly conserved residue involved in calcium binding. The last variant was a small deletion/insertion (indel) in the transforming growth factor β binding protein 7 (TGFBP7) domain. All variants were distributed throughout the gene and did not cluster in a specific region (Supplementary Figure S2). Clinical files of FBN1 variant carriers were systematically revisited to confirm the absence of skeletal or ocular manifestations. None were found and the systemic score calculated according to the Ghent criteria19 was under 4 for all of them. Finally, a small number of variants were found in either of the following genes: TGFBR1 and TGFB2 genes (three variants each); TGFBR2 and ACTA2 (two variants each); MYLK, FLNA, FBN2, LOX, and MFAP5 genes (one variant each) (Table 2).
Class 3 variants (variant of unknown significance [VUS])
Class 3 variants (n = 22) were found in 10% of all probands (Supplementary Table S1). This proportion was slightly higher in isolated cases (16%) versus familial cases (7%) (Table 1). Two SMAD3 variants were classified as class 3 variants; they were located in intron 4 and in the 5’UTR region of the gene, respectively. Splicing prediction tools were in favor of the pathogenicity but it could not be ascertained. Four VUS were found in the FBN1 gene: three missense variants were reported at very low frequency in gnomAD and warrant further familial investigations and the fourth one is intronic (c.5225–13T>G) and predicted to lead to altered splicing. It should be noted that none of these variants had ever been identified in our French reference collection.
Pathogenic variant as a possible predictor of severity
Patients with any pathogenic variant were studied together because the limited number of patients with a pathogenic variant in a given gene excluded any statistical analysis for each gene. They were compared with patients without any pathogenic variant (patients carrying a VUS were excluded). Log-rank analysis was performed; results are shown in Fig. 2. The presence of a pathogenic variant was associated with earlier aortic dissection or surgery for dilatation (p = 0.008). The percentage of aortic dissection was also higher in the patients with a pathogenic variant (42.9% [18/42] vs. 24.1% [39/162], p = 0.0156). Surgery for aortic dilatation tended to be performed at a younger age (40.3 years vs. 46.2 years) and more frequently (21.4% vs. 19.8%) in the group of pathogenic variant carriers but differences were not significant. Finally, the aortic dilatation could not be compared in both groups because a significant number of data were missing due to a large proportion of probands (111/226) who underwent surgery for aortic dilatation or dissection.

Kaplan–Meier analysis of event-free survival (aortic dissection or surgery for dilatation) in probands with variants classified as pathogenic or likely pathogenic (blue curve) versus those without any causative variant (red curve). Log-rank Chi-square: 6.87; p value: 0.008.
DISCUSSION
Through the study of 226 consecutively recruited nshTAAD probands, we identified a significant molecular event (class 4 or 5 variants) in 18%. Furthermore, class 3 variants were found in 10% of them. Our classification criteria rely on French population data accrued over 20 years and the class 3 variants reported in this study were never found in our French reference population. Only three approaches are available to meet a definite criterion to convert variants from class 3 to 4: functional studies in a relevant cell model, transcript analysis if the gene is expressed in accessible tissues, and familial segregation to reach a significant logarithm of the odds (LOD) score threshold. In the context of a diagnostic laboratory, only the last two studies are feasible. Both should be encouraged knowing that transcript analysis concerns only a limited number of variants while family studies may be performed extensively. Indeed, this approach is advisable not only for familial cases (in search of cosegregation of the variant and the TAAD phenotype) but also for sporadic cases (in search of a de novo event, a mosaic, or transmission from an undiagnosed affected parent). Nevertheless, in practice, it is more difficult to obtain samples from asymptomatic relatives of possible de novo cases. Furthermore, incomplete penetrance is frequent in TAAD20 and complicates the interpretation of segregation data in the trios. This could explain why in this study class 3 variants were more frequently identified in sporadic cases. Concerning class 4 and 5 variants, the discovery rate in our study was higher for familial cases (22%) enabling a definite diagnosis. However, because 11% of sporadic cases did indeed carry a pathogenic variant, a significant part of young sporadic cases can be explained by a genetic cause and this population should be included in the research of new causal genes of TAAD.
Our cohort is the largest sample of nonsyndromic TAAD described in the literature. The largest report to date among the six main reports21 has the closest number of probands but it included a significant percentage (30%) of probands with syndromic features and only seven genes were tested (Table 3). In other studies,21,22,23,24,25,26 the populations reported comprise carriers of syndromic forms as well as probands with cardiovascular risk factors such as hypertension, hyperlipidemia, and smoking. As an example, hypertension was reported in more than 60% of the probands reported by Poninska et al.22 and Ziganshin et al.26
The molecular results of this study are significant because SMAD3 and FBN1 genes were for the first time identified as major genes in nshTAAD. The high level of SMAD3 variant carriers can be explained by the fact that in approximately half of these cases, the presence of osteoarthritis had not been fully investigated in the family at large and was unappreciated by clinicians at first glance. This partial clinical assessment is detrimental for patients and this observation further supports the need to better disseminate in the medical community the full clinical description of AOS.17,18 This also illustrates the importance of a detailed clinical investigation in probands with TAAD. Contrary to the situation of SMAD3 molecular events, the large number of significant variants found in the FBN1 gene had never been delineated in nonsyndromic cases before. Therefore, clinical files of FBN1 variant carriers were systematically revisited to check for the presence of skeletal or ocular manifestations. None were found and therefore no diagnosis of MFS according to the revised Ghent criteria could be anticipated prior to genetic testing. This illustrates the large spectrum of phenotypes associated with variants in this gene.27 Unexpectedly, no PTC variant was identified in the FBN1 gene whereas in our experience, approximately one third of the molecular events found in this gene in the routine molecular screening of suspected MFS patients are PTCs.27 Thus, nshTAAD was associated with only missense FBN1 variants in this study, which suggests a genotype–phenotype correlation that needs to be further investigated. This is in keeping with reports from Campens et al.21 and Poninska et al.22 in which all FBN1 PTC variants are found in probands with syndromic forms of the disease. There is no evidence of a preferential location of the variants in the FBN1 gene and no difference as compared with variants found in Marfan syndrome patients. Interestingly, only a few missense variants involved a cysteine residue, while recent results show that there is a significant excess of loss of cysteine variants in severely affected MFS subjects.28 The significant number of SMAD3 variant carriers in our study is also in agreement with Campens et al.21 It should be noted that, besides SMAD3 and FBN1, half of the remaining variant carriers concern genes involved in the TGF-β pathway genes (TGFBR2, TGFBR1, and TGFB2). Moreover, the detection rate of causal variants in the ACTA2 gene (<1% of all the probands; n = 2) contrasts with what has been reported in the first studies identifying ACTA2 variants in up to 16% of nonsyndromic hTAAD families.21,29,30 Indeed, pathogenic variants in the ACTA2 gene have been associated with TAAD in a context of coronary artery disease, stroke, and moyamoya disease.31 Our results are in line with more recent studies performed in similar patient populations.32 This illustrates the heterogeneity of recruitment among the studies in the literature. Thanks to NGS technologies and the large capture panel, we identified the molecular defects in 11 different genes. It should be noted that since this work, two new TAAD-causing genes have been identified: BGN encoding biglycan33 and HCN4 encoding hyperpolarization activated cyclic nucleotide gated potassium channel 4 (ref. 34). However, disease-causing variants in these genes are very rare and their study would probably not lead to major changes in our results. NGS is a powerful tool that supplies the irrefutable proof of a specific clinical disease that may lead to diagnosis repositioning. We provided some interesting examples of this. The woman for whom a pathogenic variant was found in the FLNA gene was diagnosed as nshTAAD at first. After this variant was found, she had a clinical examination focusing on features associated with pathogenic variants in the FLNA gene.35 It appeared she had a history of seizures in childhood and periventricular nodular heterotopia on a previous brain magnetic resonance image (MRI), which is consistent with the presence of the FLNA variant.
A class 3 variant (VUS) was reported in 10% of all probands. This is consistent with what is described in the literature.23 It is noteworthy that the “positive rate” in terms of molecular diagnosis is still weak and that in this field of pathologies, the study of many genes is necessary to inform clinical diagnosis. In other words, one of the limits of this work is the “missing heritability,” that is to say the probably numerous not yet recognized genes that could not be tested in this population.23,24 Indeed, the list of the known genes is incomplete and will evolve as scientific knowledge develops but it seems that no major genes are missing. The technology and the list of the tested genes have evolved throughout the study and will continue to evolve. The next challenge will be the reclassification of VUS,24 with the use of family segregation studies, transcript analyses, or based on updated variant frequencies in population databases.
The carriers of pathogenic variants have significantly more severe cardiovascular features (dissection/surgery for dilatation). This result supports the idea that surgery should be performed earlier in patients with a recognized pathogenic variant, which is in keeping with recent studies on specific genes and current guidelines.36,37
Conclusion
Thoracic aortic disease is complex and encompasses many disease states and presentations. This study broadens the spectrum of genetic background of thoracic aneurysms and dissections. A significant causal genetic component is identified either in sporadic (young patients under the age of 45 years without cardiovascular risk factors or BAV) and familial nshTAAD cases in 18% of the probands. An important part of the disease-causing variants concerned genes classically associated with syndromic forms of the disease, mainly SMAD3 and FBN1. When a genetic cause is identified, the probands have significantly more cardiovascular events (aortic dissection or surgery). The practical conclusions of our work are therefore twofold: (1) genetic screening using NGS is efficient in young (<45 years) sporadic or familial nshTAAD; and (2) the presence of a pathogenic variant has a possible predictive value that needs to be further investigated because it may influence care. Indeed, it could suggest a lower prophylactic surgical threshold in line with the present recommendations from the European Society of Cardiology.37
URLs
The URLs for data presented herein are as follows: National French Reference Centre for Marfan and associated diseases website: https://www.marfan.fr/ HGVS, Nomenclature for the description of sequence variants: http://www.hgvs.org/mutnomen/ CLC Genomics Workbench v10.1.1: https://www.qiagenbioinformatics.com/ PolyPhen-2: http://genetics.bwh.harvard.edu/pph2/ UMD-Predictor: http://umd-predictor.eu/ UMD locus-specific databases: http://www.umd.be/ Human Gene Mutation Database (HGMD®): http://www.hgmd.cf.ac.uk/ac/index.php gnomAD database: http://gnomad.broadinstitute.org/
References
Isselbacher EM, Lino Cardenas CL, Lindsay ME. Hereditary influence in thoracic aortic aneurysm and dissection. Circulation. 2016;133:2516–2528.
Abdulameer H, Al Taii H, Al-Kindi SG, Milner R Epidemiology of fatal ruptured aortic aneurysms in the United States (1999-2016). J Vasc Surg. 2019;69:378–384.
Booher AM, Eagle KA. Diagnosis and management issues in thoracic aortic aneurysm. Am Heart J. 2011;162:38–46.e1.
Campens L, Demulier L, De Groote K, et al. Reference values for echocardiographic assessment of the diameter of the aortic root and ascending aorta spanning all age categories. Am J Cardiol. 2014;114:914–920.
Stheneur C, Collod-Béroud G, Faivre L, et al. Identification of the minimal combination of clinical features in probands for efficient mutation detection in the FBN1 gene. Eur J Hum Genet. 2009;17:1121–1128.
Milewicz DM, Regalado E Heritable Thoracic aortic disease overview. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews®. Seattle (WA): University of Washington, Seattle; 1993. http://www.ncbi.nlm.nih.gov/books/NBK1120/. Accessed 2018.
den Dunnen JT, Dalgleish R, Maglott DR, et al. HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat. 2016;37:564–569.
Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081.
Frédéric MY, Lalande M, Boileau C, et al. UMD-predictor, a new prediction tool for nucleotide substitution pathogenicity—application to four genes: FBN1, FBN2, TGFBR1, and TGFBR2. Hum Mutat. 2009;30:952–959.
Desmet F-O, Hamroun D, Lalande M, Collod-Béroud G, Claustres M, Béroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37:e67.
Adams MD, Celniker SE, Holt RA, et al. The genome sequence of Drosophila melanogaster. Science. 2000;287:2185–2195.
Yeo G, Burge CB. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol. 2004;11:377–394.
Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424.
R Development Core Team. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. 2014. http://www.R-project.org/.
Aubart M, Gobert D, Aubart-Cohen F, et al. Early-onset osteoarthritis, Charcot-Marie-Tooth like neuropathy, autoimmune features, multiple arterial aneurysms and dissections: an unrecognized and life threatening condition. PLoS One. 2014;9:e96387.
van de Laar IMBH, van der Linde D, Oei EHG, et al. Phenotypic spectrum of the SMAD3-related aneurysms-osteoarthritis syndrome. J Med Genet. 2012;49:47–57.
Faivre L, Collod-Beroud G, Adès L, et al. The new Ghent criteria for Marfan syndrome: what do they change? Clin Genet. 2012;81:433–442.
Albornoz G, Coady MA, Roberts M, et al. Familial thoracic aortic aneurysms and dissections--incidence, modes of inheritance, and phenotypic patterns. Ann Thorac Surg. 2006;82:1400–1405.
Campens L, Callewaert B, Muiño Mosquera L, et al. Gene panel sequencing in heritable thoracic aortic disorders and related entities—results of comprehensive testing in a cohort of 264 patients. Orphanet J Rare Dis. 2015;10:9.
Poninska JK, Bilinska ZT, Franaszczyk M, et al. Next-generation sequencing for diagnosis of thoracic aortic aneurysms and dissections: diagnostic yield, novel mutations and genotype phenotype correlations. J Transl Med. 2016;14:115.
Proost D, Vandeweyer G, Meester JAN, et al. Performant mutation identification using targeted next-generation sequencing of 14 thoracic aortic aneurysm genes. Hum Mutat. 2015;36:808–814.
Wooderchak-Donahue W, VanSant-Webb C, Tvrdik T, et al. Clinical utility of a next generation sequencing panel assay for Marfan and Marfan-like syndromes featuring aortopathy. Am J Med Genet A. 2015;167A:1747–1757.
Fang M, Yu C, Chen S, et al. Identification of novel clinically relevant variants in 70 Southern Chinese patients with thoracic aortic aneurysm and dissection by next-generation sequencing. Sci Rep. 2017;7:10035.
Ziganshin BA, Bailey AE, Coons C, et al. Routine genetic testing for thoracic aortic aneurysm and dissection in a clinical setting. Ann Thorac Surg. 2015;100:1604–1611.
Aubart M, Benarroch L, Arnaud P, Collod-Béroud G, Jondeau G, Boileau C. Molecular Genetics of the Fibrillinopathies. In: Encyclopedia of Life Science (eLS). John Wiley & Sons, Ltd: Chichester; 2016:1–13.
Aubart M, Gazal S, Arnaud P, et al. Association of modifiers and other genetic factors explain Marfan syndrome clinical variability. Eur J Hum Genet. 2018;26:1759–1772.
Guo D-C, Pannu H, Tran-Fadulu V, et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007;39:1488–1493.
Morisaki H, Akutsu K, Ogino H, et al. Mutation of ACTA2 gene as an important cause of familial and nonfamilial nonsyndromatic thoracic aortic aneurysm and/or dissection (TAAD). Hum Mutat. 2009;30:1406–1411.
Guo D-C, Papke CL, Tran-Fadulu V, et al. Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and moyamoya disease, along with thoracic aortic disease. Am J Hum Genet. 2009;84:617–627.
Lerner-Ellis JP, Aldubayan SH, Hernandez AL, et al. The spectrum of FBN1, TGFβR1, TGFβR2 and ACTA2 variants in 594 individuals with suspected Marfan syndrome, Loeys-Dietz syndrome or thoracic aortic aneurysms and dissections (TAAD). Mol Genet Metab. 2014;112:171–176.
Meester JAN, Vandeweyer G, Pintelon I, et al. Loss-of-function mutations in the X-linked biglycan gene cause a severe syndromic form of thoracic aortic aneurysms and dissections. Genet Med. 2017;19:386–395.
Vermeer AMC, Lodder EM, Thomas D, et al. Dilation of the aorta ascendens forms part of the clinical spectrum of HCN4 mutations. J Am Coll Cardiol. 2016;67:2313–2315.
Reinstein E, Frentz S, Morgan T, et al. Vascular and connective tissue anomalies associated with X-linked periventricular heterotopia due to mutations in Filamin A. Eur J Hum Genet. 2013;21:494–502.
Jondeau G, Ropers J, Regalado E, et al. International registry of patients carrying TGFBR1 or TGFBR2 mutations: results of the MAC (Montalcino Aortic Consortium). Circ Cardiovasc Genet. 2016;9:548–558.
Erbel R, Aboyans V, Boileau C, et al. 2014 ESC guidelines on the diagnosis and treatment of aortic diseases: document covering acute and chronic aortic diseases of the thoracic and abdominal aorta of the adult. The Task Force for the Diagnosis and Treatment of Aortic Diseases of the European Society of Cardiology (ESC). Eur Heart J. 2014;35:2873–2926.
Acknowledgements
This work was supported by Programme Hospitalier de Recherche Clinique AOM10108 and CRC15014, Agence Nationale de la Recherche NONAGES ANR-14-CE15–0012–01, Fédération Française de Cardiologie, and Fondation Cœur et Recherche. The authors thank all colleagues who referred probands and families for molecular investigations. They also acknowledge the major contribution of Gwenaëlle Collod-Béroud for queries of the UMD locus-specific databases.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Arnaud, P., Hanna, N., Benarroch, L. et al. Genetic diversity and pathogenic variants as possible predictors of severity in a French sample of nonsyndromic heritable thoracic aortic aneurysms and dissections (nshTAAD). Genet Med 21, 2015–2024 (2019). https://doi.org/10.1038/s41436-019-0444-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-019-0444-y
Keywords
This article is cited by
-
A yeast based assay establishes the pathogenicity of novel missense ACTA2 variants associated with aortic aneurysms
European Journal of Human Genetics (2024)
-
Gendiagnostik bei kardiovaskulären Erkrankungen
Die Kardiologie (2023)
-
Identification of COL3A1 variants associated with sporadic thoracic aortic dissection: a case-control study
Frontiers of Medicine (2021)
-
Role of Clinical Genetic Testing in the Management of Aortopathies
Current Cardiology Reports (2021)
-
A new mutational hotspot in the SKI gene in the context of MFS/TAA molecular diagnosis
Human Genetics (2020)
