
Abstract
Purpose
Congenital heart disease (CHD) affects up to 1% of live births. However, a genetic diagnosis is not made in most cases. The purpose of this study was to assess the outcomes of genome sequencing (GS) of a heterogeneous cohort of CHD patients.
Methods
Ninety-seven families with probands born with CHD requiring surgical correction were recruited for genome sequencing. At minimum, a proband–parents trio was sequenced per family. GS data were analyzed via a two-tiered method: application of a high-confidence gene screen (hcCHD), and comprehensive analysis. Identified variants were assessed for pathogenicity using the American College of Medical Genetics and Genomics–Association for Molecular Pathology (ACMG-AMP) guidelines.
Results
Clinically relevant genetic variants in known and emerging CHD genes were identified. The hcCHD screen identified a clinically actionable variant in 22% of families. Subsequent comprehensive analysis identified a clinically actionable variant in an additional 9% of families in genes with recent disease associations. Overall, this two-tiered approach provided a clinically relevant variant for 31% of families.
Conclusions
Interrogating GS data using our two-tiered method allowed identification of variants with high clinical utility in a third of our heterogeneous cohort. However, association of emerging genes with CHD etiology, and development of novel technologies for variant assessment and interpretation, will increase diagnostic yield during future reassessment of our GS data.
Similar content being viewed by others


Introduction
Congenital heart disease (CHD), structural abnormalities of the heart that arise during development, affects up to 1% of live births.1 Due to advancements in prenatal diagnosis and improvements to surgical intervention over the past 20 years, adults now comprise over two-thirds of the current CHD population.2 However, in most cases, the underlying cause is unknown, obstructing accurate predictions of familial recurrence and clinical reassessment associated with undiagnosed syndromes. Large-scale collaborative sequencing projects and family-based analyses have helped establish a genetic component to a portion of CHD,3 yet clinical implementation of these approaches has been limited by an assumption of low yield and an incongruity with clinical variant reporting practices. As the existing CHD population grows, achieving a successful clinical genetic diagnosis has become of paramount importance.
The focus of current clinical genetic diagnostic methods remains on identifying variants of largest effect in patients with a suspected genetic basis of disease.3 While these methods, such as targeted gene panels and chromosome microarrays, are able to identify specific types of genetic variation such as single-nucleotide variants (SNVs) and small insertions and deletions (indels) or copy-number variants (CNVs), a unified approach to interrogate all types of genetic variation has not yet been adopted into clinical practise. Additionally, in the research setting, a standard approach is not used to classify variants as disease-causal.
Genome sequencing (GS) combines all forms of genetic testing by allowing the detection of all genomic variation, and has been utilized for the genetic diagnosis of diverse conditions.4,5 We have previously shown that clinically actionable variants, according to the standards and guidelines defined by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG-AMP guidelines),6 can be efficiently identified from exome sequencing (ES) data by applying a two-tiered method that combines virtual gene panel analysis of sequencing data with comprehensive genomic analysis.7
The current study extends our analysis from coding to noncoding regions of the genome. We aim to assess the practicality of GS for providing an actionable genetic diagnosis for clinical geneticists and affected families by interrogating the genomes of 97 Australian families with heterogeneous, surgically corrected CHD.
Materials and methods
Study participants
Ethical approval for this study was obtained from the Sydney Children’s Hospital Network Human Research Ethics Committee (approval number HREC/16/SCHN/73). The cohort consisted of 97 families of which 36 were recruited at Princess Margaret Hospital for Children (PMH), Perth, Australia; 52 from the Kids Heart Research DNA Bank based at Children’s Hospital at Westmead (CHW), Sydney, Australia; and 9 via independent clinicians. Families from PMH and CHW were recruited at preadmission clinics prior to cardiac surgery. Informed, written consent was obtained from all participants. All surgically corrected cases lacking a preexisting genetic diagnosis or otherwise not offered genetic testing were selected for the study without enrichment for a particular mode of inheritance or CHD subtype. Heart defects in all affected individuals (n = 143) were confirmed by echocardiography. Principal component analysis confirmed the self-declared ethnicities of all individuals in the cohort (Supplementary Figure S1).
Cardiac phenotyping
Cardiac phenotypes of the 143 affected individuals were grouped into seven main categories (Table 1 and Supplementary Table S1): atrioventricular septal defect (AVSD) and variants (n = 8), septal defect (excluding AVSD; n = 35), septal defect with minor cardiac anomalies (n = 20), malformation of the outflow tract (n = 39), obstructive lesion (n = 11), functional single ventricle (n = 19), and other (n = 11). Any clinical phenotype that was not a heart defect was referred to as an extracardiac anomaly (ECA).
Sample preparation and GS library construction
Genomic DNA was extracted as described previously.8 DNA sample libraries were prepared using the Illumina TruSeq Nano DNA HT Library Prep Kit and genome sequenced (Illumina HiSeq X Ten, 150 base paired-end reads) at Genome.One, Garvan Institute of Medical Research, Sydney, Australia.
Calling and annotation of sequencing data
Quality control checks were performed on FASTQ files using FASTQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Sequence reads were mapped to the human reference genome hg38 using Burrows–Wheeler Aligner (BWA-mem v0.7.12) (ref. 9). Reads that mapped to regions with alternative contigs were remapped using the hg38 primary assembly that excludes the alternative contigs, to ensure reads mapped in these regions do not erroneously obtain mapping quality zero. SNVs and small insertions or deletions (indels) were called for each family using Platypus v0.8.1 (ref. 10) callVariants command with default options. The generated variant call format (VCF) files were annotated with various metrics using ANNOVAR v 2016Feb01 (ref. 11). Noncoding variants were called based on the Human Regulatory Features Dataset (GRCh38.p10) from the Ensemble Regulation 91 Database12 via ANNOVAR annotation. CNVs were called using Delly2 v0.7.7 with default parameters (unless otherwise stated) and the provided hg38 exclusion regions.13 Initial calls were generated individually for each sample with minimum length set to 50 bp and maximum length set to 1 Mb. To identify CNVs longer than 1 Mb, we calculated read depth across the genome in bin sizes of 100 kb. CNV calls of interest were validated by visualization with SVPV.14 We applied guidelines set by ACMG regarding the interpretation and reporting of CNVs15 to determine the pathogenicity of the called variants. For further detail, see Supplementary Methods.
Variant prioritization and analysis
Exonic variants were prioritized and quality-controlled using a previously described two-tiered approach with minor variations, comprising first a screen of strictly curated genes reproducibly shown to cause human CHD (hcCHD; Supplementary Table S2), then unbiased comprehensive analysis.7 CNVs overlapping genic regions (transcript and 10 kb upstream of transcript start site) were interrogated from genome data with respect to hcCHD and emerging CHD genes. Noncoding variants were prioritized using the hcCHD genes. ACMG-AMP guidelines were applied to all exonic and copy-number variants.6,15 Details are provided in the Supplementary Methods.
Results
Clinical cohort characteristics
The study cohort comprised 97 families with one or more individuals affected by CHD (60 sporadic cases, 37 familial cases; 143 affected individuals, 195 unaffected individuals). The characteristics of the cohort reflect an unbiased, diverse array of surgically corrected CHD cases in clinical practice (Table 1). Malformations of the outflow tract (39/143, 27%) and septal defects (35/143, 24%) were two of the most common cardiac lesions in the cohort. ECAs were observed in 53/143 (37%) affected individuals with developmental delay being the most common morbidity (22/53, 42%), while 70/143 (49%) affected individuals had no reported ECA (Supplementary Table S1).
We achieved 32× average coverage of genomic regions with 95% of sites covered >10× across all samples (n = 338) (Supplementary Figure S2). The hcCHD genes had an average coverage of 33× with 96% of exonic regions covered >10×, permitting unbiased identification of variants therein. Total numbers of variants identified within exonic and noncoding regions by GS in the cohort are presented in Supplementary Table S3.
hcCHD gene screen
To gauge the efficacy of interpreting patient genome data in a clinical setting, probands were screened for variants in a high-confidence list of genes associated with human CHD.7 This virtual gene panel (http://chdgene.victorchang.edu.au) was created with the aim of streamlining the identification of pathogenic variants per ACMG-AMP guidelines. Since the initial report of this list, 11 genes have been added based on supportive evidence from human cases in the literature, increasing the total to 101 genes (Supplementary Table S2).
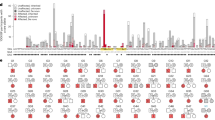
Clinically actionable variants were identified through strict application of the ACMG-AMP guidelines, resulting in the identification of pathogenic variants in 12 genes (ACTC1, BCOR, CFC1, CHD7, GATA4, GATA6, INVS, JAG1, MYH6, NODAL, NOTCH1, SMAD6) in 16/97 (16%) families (Figs. 1 and 2, Table 2, and Supplementary Figure S3).

Schematic summarizing the relationship between genomic loci harboring pathogenic or likely pathogenic variants and the corresponding congenital heart disease (CHD) lesion type in sporadic and familial cases. AVSD+ (yellow): atrioventricular septal defects and variants, FSV (orange): functional single ventricle, MOT (green): malformation of the outflow tracts, OL (pink): obstructive lesions, SD (pink): septal defects, SDMA (light blue): septal defects with minor abnormalities. Blue bars: familial cases; red bars: sporadic cases. aGenes with variants identified by comprehensive analysis.

Visual categorization of pathogenic and likely pathogenic variants identified in cohort by variant and lesion types. Count refers to the presence of a variant within an individual family. ACMG American College of Medical Genetics and Genomics, AVSD+ atrioventricular septal defects and variants, CNV copy-number variant, FSV functional single ventricle, MOT malformation of the outflow tracts, OL obstructive lesions, SD septal defects, SDMA septal defects with minor abnormalities.
In four additional families, five likely pathogenic variants were identified (Figs. 1 and 2, Table 2, and Supplementary Figure S3). A de novo missense variant in NF1, Leu1187Arg, was found in proband 3953 with tetralogy of Fallot (TOF). Heterozygous germline variants in NF1, a regulator of RAS signal transduction, are associated with neurofibromatosis type 1 (OMIM 162200); however, CHD has been recorded in up to 75/2322 (3.2%) cases including multiple records of variant carriers exhibiting TOF.16 We identified a heterozygous TLL1 missense variant, Thr860Ala, in proband 3695 with mitral and aortic valve anomalies inherited from the mother with patent foramen ovale (PFO). Heterozygous variants in TLL1, a procollagen protease, are associated with secundum atrial septal defect (ASD) (OMIM 613087) (ref. 17), and the more severe cardiac phenotypes observed in the proband have previously been seen in Tll1 knockout mice.18 Variants were identified in DOCK6 (homozygous Val1055Met) and ACVR2B (heterozygous Gly353Trp) in proband 150650086 with TOF and multiple ECAs. Recessive loss-of-function variants in DOCK6, an actin cytoskeletal remodeling protein, are associated with Adams–Oliver syndrome (OMIM 614219), characterized by limb and scalp defects, with 20% of cases presenting with CHD including TOF.19 An ACVR2B variant inherited from the asymptomatic mother is a potential disease-risk modifier in the proband. Heterozygous variants in ACVR2B, a gene encoding an activin receptor, have been associated with CHD in the context of heterotaxy (OMIM 613751) (ref. 20) potentially exacerbating the disease phenotype of the proband. A heterozygous variant in NOTCH1 was inherited by the proband 152900216 from the father, both of whom are affected by malformations of the outflow tract and limb abnormalities consistent with Adams–Oliver syndrome caused by NOTCH1 variants (OMIM 616028) (ref. 21).
All protein-coding variants in hcCHD genes identified in sporadic and familial CHD cases, and their ACMG-AMP classifications, are presented in Supplementary Tables S4 and S5, respectively.
As GS also allows for interrogation of CNVs, families were assessed for CNVs in genic regions of hcCHD genes. Variants ranging from 59 bp to 78.06 kb were identified across hcCHD genes. In 26/97 families (27%), CNVs overlapped hcCHD gene regions, comprising 20/60 (33%) sporadic and 6/37 (16%) familial cases (Supplementary Table S6). However, only one CNV was defined as pathogenic by ACMG guidelines.15 This 22.55-kb deletion in NOTCH1 identified in family 3173 results in loss of the first two protein-coding exons. This variant was shared by two siblings with septal defects and outflow tract malformations, and was inherited from the asymptomatic father, shown to be mosaic for the variant by polymerase chain reaction (PCR) and Sanger sequencing (Supplementary Figure S4A–C). Other CNVs, the majority of which were identified in intronic or intergenic regions of hcCHD genes, were classified as variants of uncertain significance (VUS) by the ACMG guidelines.
Guidelines do not yet exist for clinical interpretation or efficient assessment of pathogenicity of noncoding variation. We observed rare, disease-segregating SNVs and indels within annotated regulatory regions of hcCHD genes in 29/97 families (30%), comprising 13/60 sporadic (22%) and 16/37 familial (43%) cases (Supplementary Table S7). Families 3438 and 3400, previously subjected to exome sequencing (ES) analysis, yet without a reportable genetic diagnosis,7 exhibited novel de novo variants in hcCHD gene regulatory regions (Supplementary Figure S3). Proband 3438, diagnosed with isolated, sporadic ASD, harbored an SNV ~450 bp upstream of the transcription start site of HAND1. This resides within the overlap of a CpG island, a DNase1 hypersensitivity site, and an H3K27me3 site, where transcription factor EZH2 is predicted to bind. Similarly, proband 3400, exhibiting right ventricular hypoplasia, pulmonic stenosis, and VSD, carried a unique intronic variant (NM_001105.4: c.790+371G>C) in ACVR1 overlapping a DNase1 hypersensitivity site. As pathogenic variants in both HAND1 and ACVR1 result in phenotypes similar to what we observe in our probands, it is conceivable that these noncoding variants lead to aberrant transcriptional regulation, which contributes to their disease phenotype. However, further functional studies are required to prove these associations.
Comprehensive analysis
To identify novel disease–gene relationships, unbiased analysis was performed on all families in the cohort by evaluating potential disease-causal variants across all protein-coding regions. Although all inheritance models were considered (Supplementary Tables S8 and S9), variant candidacy was given to those completely segregating with disease, present in all affected individuals per family (Supplementary Tables S10 and S11). Additional consideration was given to genes that, when knocked out in mice, result in abnormal heart phenotypes (Supplementary Table S12).
Comprehensive analysis identified all clinically reportable variants previously detected in the hcCHD gene screen. Additionally, 9/97 (9%) families were found to have pathogenic or likely pathogenic variants in emerging CHD genes.
In four families, pathogenic variants were identified, supported by human and knockout mouse evidence (Figs. 1 and 2, Table 2, and Supplementary Figure S3). A homozygous stopgain variant Trp186* in HAAO was found in proband 44 with hypoplastic left heart syndrome (HLHS) and ECA (OMIM 617660) (ref. 22). A homozygous variant in KIAA0586 leading to loss of the canonical splice acceptor of the C-terminal exon c.4268–1G>A was identified in proband 3651 with TOF and unknown ECA status. Recessive loss-of-function and splice-site variants in KIAA0586, a mediator of hedgehog signaling, have been associated with ciliopathies within which CHD is an uncommon feature (OMIM 616490) (ref. 23). An inherited heterozygous truncating variant Ser64* in SEMA3D was found in proband 2324 with TOF. CNVs or truncating variants in SEMA3D, encoding a secreted protein required for the migration of cardiac neural crest cells into the outflow tract, have been associated with nonsyndromic CHD including TOF.24,25 In proband 3397 with VSD, an inherited heterozygous missense variant Arg915His was identified in TEK, a tyrosine kinase involved in embryonic vascular development. Located in the first intracellular tyrosine kinase domain, this pathogenic variant has previously been identified in a family with perimembranous VSD (OMIM 600195), and was shown to cause ligand-independent receptor hyperphosphorylation.26
In four additional families, likely pathogenic variants were identified in genes with previous association with human CHD (Figs. 1 and 2, Table 2, and Supplementary Figure S3). Three de novo variants occurred in genes associated with syndromes where heart defects are inconsistently observed. An HNRNPK Ser420Leu variant was found in proband 1746 with ASD. Variants resulting in haploinsufficiency of HNRNPK, an RNA-binding protein, have previously been associated with a Kabuki-like syndrome (OMIM 616580), predominantly resulting in cardiac septal defects.27 Two KMT2C frameshift insertions were found in probands 939 and 3595. KMT2C encodes a lysine methyltransferase in which de novo truncating variants have been associated with Kleefstra syndrome-2 (OMIM 617768) manifesting in dominant intellectual disability,28 and less frequently, CHD.29 Accordingly, the Gln1880Alafs*9 and Glu2798Glyfs*11 variants in our cohort were identified in patients with both CHD and neurodevelopmental disorders (NDDs). The two siblings in family 2632, with HLHS and obstructive lesions, inherited a likely pathogenic Pro1109Leu variant in DCHS1, a cell adhesion molecule, previously implicated in isolated CHD (OMIM 607829) (ref. 30).
We extended CNV interrogation to include genes with knockout mouse evidence for a role in heart development. Excluding those identified in hcCHD genes, rare CNVs, segregating with disease, and overlapping regulatory or protein-coding regions of mouse heart genes were identified in 6/60 sporadic (10%) and 17/37 (46%) familial cases. A pathogenic 7.5-Mb deletion encompassing 75 genes at chr10q22.3-q23.2 was inherited by the proband of family 1456, with AVSD and ECA, from the affected mother (Supplementary Table S13 and Figure S5). The 10q22.3-q23.2 deletion (OMIM 612242), containing genes BMPR1A and LDB3, which are required for mouse heart development, has been previously reported in patients with cognitive and behavioral abnormalities often accompanied by CHD that includes VSD and AVSD.31 All other reported CNVs were classified as VUS (Supplementary Table S13).
Discussion
Genome sequencing presents the next step in the inevitable drive toward genotype-driven patient management. By application of a gene list–focused screening approach in combination with unbiased comprehensive analysis and strict application of ACMG-AMP variant interpretation guidelines, we have provided a clinically actionable genetic diagnosis for 30/97 (31%) families in our cohort (Figs. 1 and 3), providing an opportunity for tailored clinical guidance regarding family planning and health management, thereby ending what may otherwise be a prolonged period of uncertainty for families and clinicians.

Analysis of genome sequencing data from 97 families with congenital heart disease (CHD) by our two-tiered method identified clinically actionable variants in 30/97 (31%) families, thereby facilitating family counseling and further clinical investigation. Future reassessment of variants in 69% of families that did not receive an actionable diagnosis may reveal causal gene variants. hcCHD high-confidence gene screen.
Currently, there are no published CHD cohorts comparable with our study in terms of heterogeneity of the cohort, choice of sequencing technology, and comprehensive family-by-family analysis with stringent ACMG-AMP variant classification. A summary of findings from recent CHD cohort studies is presented in Supplementary Table S14. The overall diagnostic yield for our cohort does not stray from what has previously been reported,3 reflecting the limitations of the subset of gene variants that are currently clinically interpretable. However, due to the heterogeneity of our cohort, we are able to gauge diagnostic yields of various CHD subgroups present within our cohort. We observed higher diagnostic yields in certain CHD subgroups than others. In familial and CHD + ECA subgroups where the cause of disease is likely to be of genetic origin,3 we identified actionable variants in 49% and 43% of cases, respectively. In particular, the high yield for cases with ECA implies that these variants may occur in genes with pleiotropic roles in development, as has recently been suggested for de novo variants in CHD.32 The identification of a pathogenic or likely pathogenic variant in a known syndromic gene enables assessment of the proband and family for expected clinical features. This is especially important in cases where ECA cannot be determined during the neonatal period, as well as in cases with variants of incomplete penetrance or variable expressivity as observed in our own cohort and others.29,33
In contrast, for families with sporadic CHD in our cohort, where disease causality has been mostly attributed to multiple factors,3 our diagnostic yield was 20%. Five percent of our sporadic subcohort had actionable de novo variants, 7% had actionable recessive variants, and 8% of the sporadic subcohort inherited a pathogenic variant from one of the parents. The contribution of de novo and recessive variation toward causality of sporadic CHD has previously been observed,29,32 however, our results show that a proportion of pathogenic variants may also be inherited from a parent who does not exhibit overt disease phenotypes. This suggests that comprehensive phenotyping of parents and consideration of all modes of inheritance may expedite accurate genetic diagnosis and result in higher diagnostic yield in a sporadic cohort.
Interestingly, we were able to identify a genetic component in 27% of patients in our subgroup of patients with isolated CHD. Our findings suggest that this particular patient group, who is not routinely offered clinical genetic testing due to suspected heterogeneous etiology,3,29 may receive greater benefit from genetic testing than previously anticipated. Additionally, a higher diagnostic yield was achieved for probands with functional single ventricle and outflow tract malformations (47% and 37%, respectively) than other lesion types in our cohort (Figs. 1 and 2). Cardiac septal defects, the second most common surgically actionable lesion type within our cohort after outflow tract malformations, had the lowest diagnostic yield at 20%, suggesting that disease causality may not be restricted to the contribution of a single gene variant, as previously highlighted.29
Our methodology also allowed us to evaluate the different types of genetic variation that underlie CHD causality. SNVs comprised the largest portion of actionable variants at 56% (19/34), followed by indels at 32% (11/34) and CNVs at 6% (2/34) (Fig. 2). The two pathogenic CNVs that we identified, a deletion in NOTCH1 and a multigene deletion on chromosome 10, account for 2% of the cohort, a lower than expected yield.3 However, we did not recruit patients who had received a genetic diagnosis by conventional diagnostic testing, which may have led to a depletion of pathogenic CNVs in our cohort. Regardless, our findings exemplify the ability of GS to identify a comprehensive range of clinically actionable gene variants. As conventional methods for CNV detection are not able to resolve smaller CNVs such as the 22-kb NOTCH1 deletion,3 the ability to detect such CNVs from GS data, which are more likely to disrupt single genes and mimic a monogenic cause of CHD, is an important technological advantage.
The use of our hcCHD gene screen allowed for the identification of rapid, clinically translatable findings in 22% of our cohort. However, the use of a virtual gene panel does not facilitate the expansion of current knowledge regarding causes of human CHD and phenotypic manifestation of disease genotypes. Here, comprehensive analysis of families allowed for the implication of prospective genes in human CHD. For example, the identification of two frameshift variants in the chromatin modifier KMT2C in two unrelated probands with CHD and NDD presents KMT2C, predominantly associated with NDD,28 as a promising syndromic CHD gene candidate.
Our dynamically curated gene panel evolves with insight from comprehensive CHD cohort analyses such as this. Variants that may otherwise be classified as VUS can be functionally validated or be reassessed in light of recurrent human disease–gene associations, a process that can be expedited by data-sharing between laboratories. Furthermore, clinical reassessment of families with VUS in syndromic genes may reveal expected ECA phenotypes, allowing reclassification of variants. We expect diagnostic yield by this approach to increase with time, as has proved fruitful for previous studies of clinical genome reanalysis,34 thereby creating an efficient link between the research and clinical sides of the patient genome.
For the purposes of our study, we adopted the ACMG-AMP guidelines to ensure that our findings, which are obtained in a research setting, have clinical relevance. Care was taken in the interpretation and utilization of ACMG-AMP guidelines to avoid misclassification of variants. Although we have been transparent in classifications of pathogenicity of our variants, they may be subject to alternative interpretation, thus highlighting the need for further refinement of the guidelines with respect to CHD.35,36
Despite the advantages offered by GS coupled with our comprehensive family analysis, 69% of the families in our cohort did not receive a genetic diagnosis (Fig. 3). Firstly, this relates to the etiology of CHD. To provide a clinically actionable genetic diagnosis for each family, our analysis presumes a Mendelian origin for CHD where the vast majority of actionable variants are found in exonic regions of protein-coding genes that have recurrent associations with CHD. However, evidence suggests a more complex etiology in a majority of CHD cases.3 Complex interactions between multiple gene variants or environmental factors and gene variants, contribution of noncoding and epigenetic variation, as well as causal variation in genes with no current association with CHD may underlie the majority of the unresolved cases. Secondly, the limitation lies in our inability to interpret the clinical implications of such complex interactions. As GS is able to identify all genomic variants, it allows a comprehensive overview of potential multigenic contributions to disease, the majority of which are classified as VUS (Supplementary Table S15 and Figure S6). Although variants of uncertain significance are not clinically actionable, they may yet contribute to phenotypic presentation. Burden analysis of these cardiac gene variants to gauge their contribution to a complex model of disease may be beneficial for patient prognosis; however, currently such an analysis is impeded by our cohort size and the absence of equivalently sequenced and analyzed controls.
By utilizing GS, we were able to examine noncoding regions of the genome. Although we identified noncoding variants in disease-related genes, their role in CHD causality could not be determined. Promisingly, emerging evidence is shedding light on the role of noncoding variation in disease.37 Tools and techniques are currently being developed to interpret the wealth of noncoding data produced by studies, such as ours, that seek to predict the functional impact of noncoding variants,38 which, following functional validation, will provide insights into their roles in disease causality. Although, the clinical utility of noncoding variation remains uncertain at present, the data created by GS may be reassessed once new tools for noncoding variant analysis and interpretation become available.
Presently, our results suggest that utilization of GS on a heterogeneous CHD cohort provides the greatest clinical benefit for patients with familial, surgically correctable CHD with ECA. However, GS, as utilized by our group, also provides an opportunity to deliver a genetic diagnosis for a significant proportion of familial cases with isolated CHD who do not currently receive advanced genetic testing. The comparably higher costs of GS and similar diagnostic yield39,40 may currently discourage its widespread use as a first-line tool for genetic diagnosis. However, the convenience of capturing all genomic variation by a singular mode of testing, coupled with decreasing sequencing costs and advances in bioinformatics techniques, should eventually see GS outperform all current diagnostic techniques to date. Ultimately, GS generates a data legacy that is an unparalleled clinical resource with utility for clinicians, patients, and researchers.
References
van der Linde D, Konings EE, Slager MA, et al. Birth prevalence of congenital heart disease worldwide: a systematic review and meta-analysis. J Am Coll Cardiol. 2011;58:2241–2247.
Marelli AJ, Ionescu-Ittu R, Mackie AS, Guo L, Dendukuri N, Kaouache M. Lifetime prevalence of congenital heart disease in the general population from 2000 to 2010. Circulation. 2014;130:749–756.
Blue GM, Kirk EP, Giannoulatou E, et al. Advances in the genetics of congenital heart disease: a clinician’s guide. J Am Coll Cardiol. 2017;69:859–870.
Lionel AC, Costain G, Monfared N, et al. Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genet Med. 2018;20:435–443.
Stavropoulos DJ, Merico D, Jobling R, et al. Whole genome sequencing expands diagnostic utility and improves clinical management in pediatric medicine. NPJ Genom Med. 2016;1:1–9.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424.
Szot JO, Cuny H, Blue GM, et al. A screening approach to identify clinically actionable variants causing congenital heart disease in exome data. Circ Genom Precis Med. 2018;11:1–11.
Blue GM, Kirk EP, Giannoulatou E, et al. Targeted next-generation sequencing identifies pathogenic variants in familial congenital heart disease. J Am Coll Cardiol. 2014;64:2498–2506.
Li H. Exploring single-sample SNP and INDEL calling with whole-genome de novo assembly. Bioinformatics. 2012;28:1838–1844.
Rimmer A, Phan H, Mathieson I, et al. Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat Genet. 2014;46:912–918.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164.
Aken BL, Achuthan P, Akanni W, et al. Ensembl 2017. Nucleic Acids Res. 2017;45:D635–D642.
Rausch T, Zichner T, Schlattl A, Stutz AM, Benes V, Korbel JO. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012;28:i333–i339.
Munro JE, Dunwoodie SL, Giannoulatou E. SVPV: a structural variant prediction viewer for paired-end sequencing datasets. Bioinformatics. 2017;33:2032–2033.
Kearney HM, Thorland EC, Brown KK, Quintero-Rivera F, South ST, Working Group of the American College of Medical Genetics Laboratory Quality Assurance C. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med. 2011;13:680–685.
Lin AE, Birch PH, Korf BR, et al. Cardiovascular malformations and other cardiovascular abnormalities in neurofibromatosis 1. Am J Med Genet A. 2000;95:108–117.
Stanczak P, Witecka J, Szydlo A, et al. Mutations in mammalian tolloid-like 1 gene detected in adult patients with ASD. Eur J Hum Genet. 2009;17:344–351.
Clark TG, Conway SJ, Scott IC, et al. The mammalian Tolloid-like 1 gene, Tll1, is necessary for normal septation and positioning of the heart. Development. 1999;126:2631–2642.
Sukalo M, Tilsen F, Kayserili H, et al. DOCK6 mutations are responsible for a distinct autosomal-recessive variant of Adams-Oliver syndrome associated with brain and eye anomalies. Hum Mutat. 2015;36:593–598.
Kosaki R, Gebbia M, Kosaki K, et al. Left-right axis malformations associated with mutations in ACVR2B, the gene for human activin receptor type IIB. Am J Med Genet. 1999;82:70–76.
Southgate L, Sukalo M, Karountzos ASV, et al. Haploinsufficiency of the NOTCH1 receptor as a cause of Adams-Oliver syndrome with variable cardiac anomalies. Circ Cardiovasc Genet. 2015;8:572–581.
Shi H, Enriquez A, Rapadas M, et al. NAD deficiency, congenital malformations, and niacin supplementation. N Engl J Med. 2017;377:544–552.
Roosing S, Hofree M, Kim S, et al. Functional genome-wide siRNA screen identifies KIAA0586 as mutated in Joubert syndrome. eLife. 2015;4:e06602.
Sanchez-Castro M, Pichon O, Briand A, et al. Disruption of the SEMA3D gene in a patient with congenital heart defects. Hum Mutat. 2015;36:30–33.
Degenhardt K, Singh MK, Aghajanian H, et al. Semaphorin 3d signaling defects are associated with anomalous pulmonary venous connections. Nat Med. 2013;19:760–765.
Wouters V, Limaye N, Uebelhoer M, et al. Hereditary cutaneomucosal venous malformations are caused by TIE2 mutations with widely variable hyper-phosphorylating effects. Eur J Hum Genet. 2010;18:414–420.
Dentici ML, Barresi S, Niceta M, et al. Clinical spectrum of Kabuki-like syndrome caused by HNRNPK haploinsufficiency. Clin Genet. 2018;93:401–407.
Koemans TS, Kleefstra T, Chubak MC, et al. Functional convergence of histone methyltransferases EHMT1 and KMT2C involved in intellectual disability and autism spectrum disorder. PLoS Genet. 2017;13:e1006864.
Jin SC, Homsy J, Zaidi S, et al. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat Genet. 2017;49:1593–1601.
Durst R, Sauls K, Peal DS, et al. Mutations in DCHS1 cause mitral valve prolapse. Nature. 2015;525:109–113.
van Bon BW, Balciuniene J, Fruhman G, et al. The phenotype of recurrent 10q22q23 deletions and duplications. Eur J Hum Genet. 2011;19:400–408.
Homsy J, Zaidi S, Shen Y, et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science. 2015;350:1262–1266.
Lewin MB, McBride KL, Pignatelli R, et al. Echocardiographic evaluation of asymptomatic parental and sibling cardiovascular anomalies associated with congenital left ventricular outflow tract lesions. Pediatrics. 2004;114:691–696.
Costain G, Jobling R, Walker S, et al. Periodic reanalysis of whole-genome sequencing data enhances the diagnostic advantage over standard clinical genetic testing. Eur J Hum Genet. 2018;26:740–744.
Nykamp K, Anderson M, Powers M, et al. Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria. Genet Med. 2017;19:1105–1117.
Tavtigian SV, Greenblatt MS, Harrison SM, et al. Modeling the ACMG/AMP variant classification guidelines as a Bayesian classification framework. Genet Med 2018.
Short PJ, McRae JF, Gallone G, et al. De novo mutations in regulatory elements in neurodevelopmental disorders. Nature. 2018;555:611–616.
Liu Y, Liang Y, Cicek AE, et al. A statistical framework for mapping risk genes from de novo mutations in whole-genome-sequencing studies. Am J Hum Genet. 2018;102:1031–1047.
Alfares A, Aloraini T, Subaie LA, et al. Whole-genome sequencing offers additional but limited clinical utility compared with reanalysis of whole-exome sequencing. Genet Med. 2018:1–6.
Christensen KD, Vassy JL, Phillips KA, et al. Short-term costs of integrating whole-genome sequencing into primary care and cardiology settings: a pilot randomized trial. Genet Med. 2018:1–10.
Acknowledgements
This work was supported by the National Health and Medical Research Council (NHMRC) (fellowships ID1135886, ID1042002 to S.L.D., ID573732 to R.P.H, and ID1105271 to J.W.K.H. and program grant ID1074386 to R.P.H., R.M.G., S.L.D.); Australian National Heart Foundation (fellowship ID100848 to J.W.K.H. and ID101204 to E.G.); Australian Postgraduate Award (UNSW) (J.O.S., E.I.); Office of Health and Medical Research NSW Government to S.L.D., R.P.H, R.M.G; Chain Reaction (The Ultimate Corporate Bike Challenge) to S.L.D.; Channel 7 Telethon to S.L.D., R.M.G.; and The Key Foundation to S.L.D. The authors would like to thank all families who took part in the study. The authors also thank Elizabeth Anderson for assistance with analysis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Alankarage, D., Ip, E., Szot, J.O. et al. Identification of clinically actionable variants from genome sequencing of families with congenital heart disease. Genet Med 21, 1111–1120 (2019). https://doi.org/10.1038/s41436-018-0296-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-018-0296-x
Keywords
This article is cited by
-
Expanding the phenotypic spectrum of NOTCH1 variants: clinical manifestations in families with congenital heart disease
European Journal of Human Genetics (2024)
-
ConanVarvar: a versatile tool for the detection of large syndromic copy number variation from whole-genome sequencing data
BMC Bioinformatics (2023)
-
The phenotypic spectrum of terminal 6q deletions based on a large cohort derived from social media and literature: a prominent role for DLL1
Orphanet Journal of Rare Diseases (2023)
-
Gendiagnostik bei kardiovaskulären Erkrankungen
Die Kardiologie (2023)
-
The clinical utility of exome and genome sequencing across clinical indications: a systematic review
Human Genetics (2021)
