
Abstract
Purpose
SMARCB1 encodes a subunit of the SWI/SNF complex involved in chromatin remodeling. Pathogenic variants (PV) in this gene can give rise to three conditions. Heterozygous loss-of-function germline PV cause rhabdoid tumor predisposition syndrome and schwannomatosis. Missense PV and small in-frame deletions in exons 8 and 9 result in Coffin–Siris syndrome, which is characterized by intellectual disability (ID), coarse facial features, and fifth digit anomalies.
Methods
By a gene matching approach, individuals with a similar SMARCB1 PV were identified. Informed consent was obtained and patient data were collected to further establish genotype–phenotype relationship.
Results
A recurrent de novo missense PV (c.110G>A;p.Arg37His) in exon 2 of SMARCB1, encoding the DNA-binding domain, was identified in four individuals from different genetic centers. They shared a distinct phenotype consisting of profound ID and hydrocephalus due to choroid plexus hyperplasia. Other shared features include severe neonatal feeding difficulties; congenital heart, kidney, and eye anomalies; obstructive sleep apnea; and anemia.
Conclusion
The p.Arg37His PV in the DNA-binding domain of SMARCB1 causes a distinctive syndrome, likely through a gain-of-function or dominant-negative effect, which is characterized by severe ID and hydrocephalus resulting from choroid plexus hyperplasia. This report broadens the phenotypic spectrum associated with PV in SMARCB1.
Similar content being viewed by others


Introduction
The SMARCB1 (MIM 601607; GenBank: NM_003073.3) gene, located on chromosome 22q11.23 and consisting of nine exons, encodes a subunit of the switch/sucrose nonfermenting (SWI/SNF) complex involved in chromatin remodeling.1 The SWI/SNF complex, also known as the BAF complex, is an adenosine triphosphate (ATP)-dependent chromatin remodeling complex involved in the regulation of gene accessibility and expression.2 This complex is involved in mobilizing the histone–DNA bond using energy generated from ATP hydrolysis, causing nucleosome sliding, dissociation, or replacement.3 By altering histone configuration, SMARCB1 and the SWI/SNF complex are involved in both activation and repression of target genes.
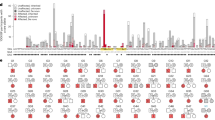
Three autosomal dominant conditions are caused by germline pathogenic variants (PV) in SMARCB1. Loss-of-function PV cause rhabdoid tumor predisposition syndrome type 1 (RTPS1) (MIM 609322),4,5,-6 which is characterized by the development of highly aggressive tumors in young children, mainly atypical teratoid/rhabdoid tumors (AT/RT) of the brain and malignant rhabdoid tumors of the kidney and soft tissue. Loss-of-function and missense alterations are also found in both families and sporadic patients with schwannomatosis (MIM 162091).7,8 Patients develop mainly benign cranial and peripheral nerve sheath tumors. Recent work in mice suggests that early SMARCB1 loss causes rhabdoid tumors, whereas a loss at later stages combined with NF2 gene inactivation causes schwannomas.9 Lastly, missense PV and small in-frame deletions in exons 8 and 9 of the gene are a cause of Coffin–Siris syndrome (CSS) (MIM 614608), a syndrome defined by intellectual disability (ID), coarse facial features, feeding difficulties and fifth digit anomalies. Because no truncating PV have been reported in CSS, these PV are thought to exert their effect through a gain-of-function or dominant-negative mechanism,10,11 but no functional studies have been performed. Approximately 7% of individuals with CSS carry a PV in SMARCB1.11,12,-13 CSS can also result from PV in other subunits of the SWI/SNF complex, but individuals with a PV in SMARCB1 are, in general, the most severely affected.10 Known pathogenic PV causing these three syndromes are shown in Fig. 1. Some patients and families carrying a SMARCB1 PV presented with more than one of the above described conditions, such as a family with both rhabdoid tumors and schwannomas14 and a patient with CSS who developed schwannomas.15

a Detailed view of the SMARCB1 gene (GenBank: NM_003073.3) and the reported deletions and duplications in rhabdoid tumor predisposition syndrome type 1 (RTPS1). ¥ = one of the duplications of exon 6 represents a family with an overlapping phenotype of both atypical teratoid/rhabdoid tumors and schwannomas. b Overview of SMARCB1 including the DNA-binding and SNF5 domain and the location of known pathogenic variants on a protein level in patients with RTPS1 (in yellow, based on report of Eaton et al.,30) schwannomatosis (in green, based on reports of Smith et al.,31,32 Sestini et al.,28 Hadfield et al.,7 Boyd et al.33 and Rousseau et al.,34) Coffin–Siris syndrome (in red, based on reports of Tsurusaki et al.,11,35 Santen et al.,12 and Wieczorek et al.13), and an overlapping phenotype of multiple of these phenotypes (in purple). The patients with overlapping phenotypes are two families with both atypical teratoid/rhabdoid tumors (AT/RTs) and schwannomas (p.Gln123* and p.Arg158*) and a male with both Coffin–Siris syndrome and schwannomas (p.Arg374Gln)
Previously, we identified a de novo p.Arg37His PV in SMARCB1 in an individual with a clinical presentation overlapping with Kleefstra syndrome (KS) (MIM 610253),16 We have now identified three additional patients with the same PV, who show a strikingly similar phenotype consisting of severe intellectual disability (ID), hydrocephalus due to a choroid plexus hyperplasia, various congenital anomalies, and feeding difficulties. In this report, we describe the four individuals, which broadens the spectrum of disorders associated with PV in SMARCB1.
Subjects and methods
The first patient was identified by exome sequencing (ES) in a cohort of EHMT1-negative patients within the Kleefstra syndrome phenotypic spectrum (KSS).16 Approval for this study was obtained from the institutional ethical board at Radboud University Medical Center, Commissie Mensgebonden Onderzoek Regio Arnhem-Nijmegen (NL36191.091.11). Approval was also obtained to publish patient photos. Three additional individuals were identified in different genetic centers by trio ES in a diagnostic setting as previously described.17 Details are available upon request. In all four patients, ES was indicated because of severe intellectual disability in combination with congenital anomalies. ES was performed when patients visited the clinical genetics department at ages 9 years, 5 years, 12 years, and 17 months, respectively. Informed consent was obtained from parents and clinical information and photographs were collected (Fig. 2). Magnetic resonance images (MRIs) were collected and re-evaluated.

Frontal pictures of the four patients with a de novo p.Arg37His pathogenic variant in SMARCB1. Patients share a hypotonic facies, upslanted palpebral fissures, round nasal tip, and large mouth. In patients 1 and 3, diastema of the upper incisors is seen. In patients 1 and 4, midface hypoplasia and tongue protrusion is seen
Results
By ES, four females were identified with a recurrent de novo missense PV in SMARCB1 (c.110G>A; p.Arg37His). No other possible explanatory variants were found with ES. The four patients share a distinctive phenotype, which is discussed in more detail in the following case reports and summarized in Table 1.
Patient 1 was previously described by Kleefstra et al.16 She was born to healthy non consanguineous parents at 36 weeks of gestation with a birth weight of 2400 grams after an uncomplicated pregnancy. The neonatal period was complicated by feeding difficulties. She developed hydrocephalus as a result of an abnormally high production of cerebrospinal fluid (CSF) due to choroid plexus hyperplasia, for which a ventriculoperitoneal shunt was placed at the age of 2.5 years. At that age, she had severe ID and was not able to sit or speak any words. A brain MRI showed moderately enlarged ventricles, decreased volume of the pons and brainstem and an enlarged choroid plexus (Fig. 3). Multiple shunt revisions were performed, and a partial choroid plexectomy was needed at age 8 years to reduce the CSF production. Histopathological examination of intraventricular tissue removed at surgery was consistent with a diagnosis of choroid plexus papilloma (Fig. 4). Immunohistochemical staining of the papilloma with INI1/SMARCB1 antibodies (purified mouse anti-BAF47, BD Transduction Laboratories, product number 612110) was performed, to determine if there was loss of INI1/SMARCB1 expression in the tumor as seen in AT/RTs. We observed weak staining of INI1/SMARCB1, with no difference between the tumor cells and the endothelial cells (data not shown). The child has visual impairment, strabismus, and myopia (approximately -7 diopters bilaterally). She has signs of sleep apnea with pauses in breathing at night, but does not require oxygen. She has a history of unexplained intermittent microcytic anemia (lowest Hb 4.3 mmol/L at age 2 and 4.9 mmol/L at age 14, mean corpuscular volume (MCV) 68).

a, e Patient 1, MRIs at age 8 years. a T1 sagittal section shows the thin corpus callosum, the drain and enlarged cisterna magna. e T2 axial section shows enlarged ventricles, choroid plexus hyperplasia and reduced peripheral cerebral spinal fluid spaces. b, c, f, g Patient 3, MRIs at (b, f) age 2 months and (c, g) age 12 years. b, c T1 sagittal midline sections show a thin corpus callosum, thin pons and mild cerebellar vermis atrophy. f, g T2 axial sections show a progressive enlargement of the lateral ventricles and hyperplasia of the choroid plexus in time. d, h Patient 4, MRIs at age 7 months. d Sagittal cube fluid-attenuated inversion recovery (FLAIR) section shows a thin and stretched corpus callosum; thin pons, mesencephalon, and medulla oblongata; and small cerebellar vermis with enlarged fourth ventricle and cisterna magna. h T1 axial section shows dilated lateral ventricles and choroid plexus hyperplasia

Microphotographs are shown at a magnification of 400× of the choroid plexus after it was partially resected in patient 1 at the age of 8 years. Thin sections from paraffin embedded tumor were used for immunohistochemistry with the following antibodies: Ki-67 (monoclonal mouse antihuman Ki-67 antigen, dilution 1:100, Dako-Denmark Agilent, product number M7240, Glostrup, Denmark), transthyretin (polyclonal rabbit antihuman prealbumin, dilution 1:3000, Dako-Denmark Agilent, product number A0002, Glostrup, Denmark) and Ini-1 (purified mouse anti-BAF47, concentration 250 µg/mL, dilution 1:100, BD Transduction Laboratories, product number 612110). a Hematoxylin-eosin staining showing that the tumor has the morphology of a choroid plexus papilloma. b Ki-67 shows a proliferative activity that was lower than normal in choroid plexus papillomas. c The tumor was positive for transthyretin, as expected for a choroid plexus papilloma
At the age of 9.5 years, her height was 144 cm (0 SD), weight 31 kg (−1 SD) and head circumference 52.4 cm (0 SD). She has an extra skin fold adjacent to one of her labia minora. A cardiac ultrasound showed no abnormalities. She has truncal hypotonia, and does not walk, but is independently mobile in a wheelchair. She does not speak, but has well-developed nonverbal communication skills. She is a socially aware and friendly teenager.
Patient 2 was born to healthy non consanguineous parents after a pregnancy complicated by gestational diabetes. A prenatal ultrasound showed mild ventriculomegaly. She was born at 37 weeks gestation with a birth weight of 2990 grams. She had severe feeding difficulties and has required total parental nutrition and gastrojejunal tube feeding since the age of 15 months. She developed hydrocephalus for which she was treated with a ventriculoperitoneal shunt at the age of 8 months. She has undergone multiple shunt revisions. A brain MRI showed hydrocephalus with choroid plexus hyperplasia, in addition to a thin corpus callosum and hippocampal atrophy. When examined at the age of 6 years, she had severe ID, was nonambulatory, and spoke no words. She had cortical visual impairment, no light perception, vertical intermittent nystagmus and multiple additional eye problems such as a cataract (diagnosed at age 3) and detached retina of the right eye (both surgically corrected) and a hyphema of the left eye of unknown origin. She had generalized hypotonia with joint hypermobility and a persistent knee dislocation. Cardiac ultrasound showed a bicuspid aortic valve, mild aortic dilation, and an atrial septal defect. With abdominal ultrasound, severe bilateral vesicoureteral reflux was diagnosed. At night, she required bilevel positive airway pressure (BiPAP) for central sleep apnea. She had positional deformities of the feet, a history of kidney stones, and precocious puberty with acne and pubic hair at the age of 4 years. She had a history of mild macrocytic anemia (lowest Hb 6.3 mmol/L at age 4, MCV 90.8), for which she was placed on iron suppletion. Physical examination at the age of 6 years showed a height of 122 cm (96th centile), weight of 20.8 kg (67th centile), and head circumference of 49 cm (7th centile). She had limited social awareness and was nonverbal, but she could vocalize. She passed away at the age of 6 years after having progressive gastrointestinal dysmotility with inability to tolerated enteral nutrition.
Patient 3 was born to healthy non consanguineous parents at 41 weeks gestation with a birth weight of 3145 grams and a head circumference of 34 cm, after a pregnancy complicated by intrauterine growth retardation. She was admitted to the hospital neonatally because of respiratory and feeding difficulties and hypotonia. A brain MRI showed ventriculomegaly with choroid plexus hyperplasia, a thin corpus callosum, thin pons, and mild cerebellar vermis atrophy (Fig. 3). She has high myopia (-14/-15 diopters), strabismus, limited abduction of the left eye, and nystagmus. In the first months of her life she had mild sleep apnea, which resolved subsequently. She had severe vesicoureteral reflux requiring nephrectomy at the age of 3 years. In addition, she had a history of transient microcytic iron deficiency anemia (lowest Hb 5.5 mmol/L at age 3, MCV 71). At the age of 6 years, eye fundus examination showed papillary edema that later resolved, compatible with mild increased intracranial pressure for which no surgical intervention was needed. Repeat MRIs at the age of 6 years and 12 years showed global atrophy, enlarged CSF spaces, and choroid plexus hyperplasia. Physical examination at the age of 12 years showed a height of 150 cm (−1 SD) and head circumference of 52 cm (−1 SD). There is no speech development, but she can reproduce musical sounds of songs. She is able to stand with support.
Patient 4 was born to healthy non consanguineous parents after a pregnancy complicated by polyhydramnios. A prenatal ultrasound showed borderline enlarged cerebral ventricles. After an uncomplicated delivery at 39 weeks of gestation, she was admitted to the neonatal intensive care unit, where she was diagnosed with laryngotracheomalacia and partial vocal cord paralysis, which was surgically corrected. A cardiac ultrasound showed an atrial septal defect and a patent ductus arteriosus, which were also corrected surgically. Brain MRI showed a foreshortened corpus callosum, and choroid plexus hyperplasia with hydrocephalus. At age 7 months, her hydrocephalus was treated with a shunt, later revised at two occasions. Severe central obstructive sleep apnea was diagnosed, which required tracheostomy in addition to BiPAP and oxygen supply at night. She had feeding difficulties that required G-tube placement. She underwent exploratory surgery for concerns for a malrotated gut, during which an ectopic appendix was identified and removed. She has poor visual tracking and myopia, in addition to moderate sensorineural hearing loss. Furthermore, she has hypotonia, joint hypermobility, congenital hip dysplasia that required splinting, and mild hydronephrosis. She was admitted to the hospital because thrombocytopenia was identified by a complete blood count performed because of lethargy. Immune thrombocytopenic purpura (ITP) was diagnosed, and she received intravenous immunoglobulins, after which the platelet count normalized. Physical examination at the age of 2 years showed a height of 80.5 cm (52nd centile), weight of 11.4 kg (82nd centile), and head circumference of 50 cm (99th centile).
In all four girls, a recurrent de novo missense PV was identified (c.110G>A; p.Arg37His) by ES. This PV affects a highly conserved residue in exon 2 of the SMARCB1 gene. It is predicted to result in the replacement of an arginine residue by histidine, leading to a loss of charge.18 In silico analysis was performed with SIFT,19 Mutation Taster,20 and PolyPhen-221 prediction programs, which all predicted the PV to be deleterious. The PV was not reported before in our in-house database of 15,576 exomes, nor in the databases ExAC, gnomAD, GoNL, or the Exome Variant Server (EVS).
Discussion
By ES and a subsequent gene matching approach, we have identified four patients with a recurrent de novo p.Arg37His PV in SMARCB1. They share a specific phenotype consisting of severe ID and hydrocephalus due to choroid plexus hyperplasia, in addition to anemia, sleep apnea, and ophthalmologic problems. PV in SMARCB1 have previously been reported as a cause of Coffin–Siris syndrome.11,12,13 PV causing this syndrome are localized in the SNF5 domain on the C-terminal end of the gene, between amino acids 363 and 377, and are presumably activating or dominant-negative PV (Fig. 1).10,11 Because missense PV in the same region are also found somatically in schwannomas, and because a CSS patient with schwannomas has been described, a dominant-negative effect might be more likely. However, no functional studies have been performed to confirm this. The PV reported here, however, is located at the N-terminal end of the gene, within the DNA-binding domain of the protein, suggesting that this PV might have a different effect and cause a different phenotype.
Some similarities exist between individuals with CSS and the patients described in this report, such as severe ID, congenital heart defects, kidney anomalies, and feeding difficulties. On the other hand, features that are frequently noted in SMARCB1-based CSS are impaired growth, microcephaly, fifth digit anomalies, scoliosis, and epilepsy, none of which are present in the individuals we describe. Furthermore, the most distinctive phenotypic feature associated with the p.Arg37His variant is enlargement of the central CSF spaces, often leading to high-pressure hydrocephalus. This is associated with choroid plexus hyperplasia and overproduction of cerebrospinal fluid, which has not been described in patients diagnosed with CSS. This implies that our patients represent a different entity within the spectrum of syndromes associated with PV in SMARCB1.
In patients heterozygous for a p.Arg37His PV in SMARCB1, clinicians should be aware of signs of increased and progressive intracranial pressure. The most striking shared feature in the cohort we describe is high-pressure hydrocephalus, which required shunting and multiple shunt revisions in three individuals, as well as partial ablative surgery in one. Neuroimaging reports were available for three patients, showing choroid plexus hyperplasia in addition to hydrocephalus, indicating that the hydrocephalus is a consequence of increased production of cerebrospinal fluid (Fig. 3). The choroid plexus was unilaterally resected in patient 1, and histopathological analysis showed an underlying choroid plexus papilloma. In the past, families with PV in SMARCB1 and choroid plexus carcinomas have been described,5,22 but the current opinion is that these tumors were actually atypical teratoid/rhabdoid tumors (AT/RTs), with loss of SMARCB1 as the distinguishing feature between these two types of tumors.23 Furthermore, no evidence has been found for PV in SMARCB1 in choroid plexus papilloma.24
Another rare feature present in all patients is sleep apnea, which is most likely of central origin. Two patients require BiPAP and oxygen supply at night, one of whom underwent a tracheostomy in addition. This might be caused by increased intracranial pressure due to hydrocephalus, however in two patients the sleep apnea persisted after shunting of the hydrocephalus. Furthermore, we noted a reduced volume of the brainstem and pons, i.e., the structure containing centers controlling the respiration (Fig. 3), which is an explanation for sleep apnea. As sleep apnea can cause serious health problems, performing a polysomnogram should be considered in these patients.
Several additional congenital anomalies and medical problems were noted. Two girls had severe vesicoureteral reflux disease, resulting in hydronephrosis and requiring a nephrectomy in one, implying that performing an abdominal ultrasound could be of added value. All four patients were admitted to the hospital in infancy due to neonatal feeding difficulties, and three continued to require gastrojejunal (GJ) tube feeds afterward. In one individual, the inability to tolerate enteral nutrition was the cause of death, emphasizing the potential severity of feeding difficulties. Diverse eye problems were reported in all children, including severe myopia, absent vision, and eye movement disorders, in addition to acute conditions like a detached retina, hyphema, and papillary edema. Finally, three girls had a period of transient anemia (lowest Hb values ranging from 4.3 to 6.3 mmol/L) and the fourth had severe ITP requiring treatment with intravenous immunoglobulins, which indicates that a routine complete blood count can be considered.
Clustering of PV or recurrence of a specific PV can be indicative of a gain-of-function or dominant-negative effect on the protein.25,26 In the N-terminal DNA-binding domain in which the p.Arg37His PV is located, several conserved residues including Arg37 are suspected to be functionally important.27 A histidine substitution can potentially influence the DNA-binding capacity. As shown in Fig. 1, the Arg37His PV affects an area of the protein where both missense and truncating PV in schwannomatosis and RTPS1 patients have been described frequently. The tumors of these patients are characterized by loss of the second allele of SMARCB1, and in schwannomas in addition biallelic somatic NF2 PV are observed.7,28,29 The two- and four-hit loss-of-function mechanisms in these syndromes plead against a dominant-negative effect of the p.Arg37His PV, because a tumor phenotype like in the schwannomatosis and RTPS1 patients would be expected if this was the case. Therefore, we hypothesize that the de novo p.Arg37His PV in SMARCB1 results in a gain of function.
Conclusion
We have identified four patients with a de novo p.Arg37His PV in SMARCB1, with a distinctive phenotype different from Coffin–Siris syndrome, consisting of severe ID, hydrocephalus due to choroid plexus hyperplasia, and various additional congenital defects. Patients identified with this specific de novo PV need careful clinical monitoring.
References
Kalimuthu SN, Chetty R. Gene of the month: SMARCB1. J Clin Pathol. 2016;69:484–9.
Sokpor G, Xie Y, Rosenbusch J, et al. Chromatin remodeling BAF (SWI/SNF) complexes in neural development and disorders. Front Mol Neurosci . 2017;10:243.
Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer . 2011;11:481–92.
Biegel JA, Zhou JY, Rorke LB, et al. Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res. 1999;59:74–79.
Sevenet N, Sheridan E, Amram D, et al. Constitutional mutations of the hSNF5/INI1 gene predispose to a variety of cancers. Am J Hum Genet . 1999;65:1342–8.
Versteege I, Sevenet N, Lange J, et al. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature. 1998;394:203–6.
Hadfield KD, Newman WG, Bowers NL, et al. Molecular characterisation of SMARCB1 and NF2 in familial and sporadic schwannomatosis. J Med Genet. 2008;45:332–9.
Hulsebos TJ, Plomp AS, Wolterman RA, et al. Germline mutation of INI1/SMARCB1 in familial schwannomatosis. Am J Hum Genet . 2007;80:805–10.
Vitte J, Gao F, Coppola G, et al. Timing of Smarcb1 and Nf2 inactivation determines schwannoma versus rhabdoid tumor development. Nat Commun. 2017;8:300.
Kosho T, Okamoto N. Genotype-phenotype correlation of Coffin-Siris syndrome caused by mutations in SMARCB1, SMARCA4, SMARCE1, and ARID1A. Am J Med Genet C Semin Med Genet. 2014;166c:262–75.
Tsurusaki Y, Okamoto N, Ohashi H, et al. Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat Genet. 2012;44:376–8.
Santen GW, Aten E, Vulto-van Silfhout AT, et al. Coffin-Siris syndrome and the BAF complex: genotype-phenotype study in 63 patients. Hum Mutat . 2013;34:1519–28.
Wieczorek D, Bogershausen N, Beleggia F, et al. A comprehensive molecular study on Coffin-Siris and Nicolaides-Baraitser syndromes identifies a broad molecular and clinical spectrum converging on altered chromatin remodeling. Hum Mol Genet. 2013;22:5121–35.
Swensen JJ, Keyser J, Coffin CM, et al. Familial occurrence of schwannomas and malignant rhabdoid tumour associated with a duplication in SMARCB1. J Med Genet . 2009;46:68–72.
Gossai N, Biegel JA, Messiaen L, et al. Report of a patient with a constitutional missense mutation in SMARCB1, Coffin-Siris phenotype, and schwannomatosis. Am J Med Genet A. 2015;167a:3186–91.
Kleefstra T, Kramer JM, Neveling K, et al. Disruption of an EHMT1-associated chromatin-modification module causes intellectual disability. Am J Hum Genet. 2012;91:73–82.
de Ligt J, Willemsen MH, van Bon BW, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012;367:1921–9.
Venselaar H, Te Beek TA, Kuipers RK, et al. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinformatics. 2010;11:548.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–81.
Schwarz JM, Cooper DN, Schuelke M, et al. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11:361–2.
Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9.
Taylor MD, Gokgoz N, Andrulis IL, et al. Familial posterior fossa brain tumors of infancy secondary to germline mutation of the hSNF5 gene. Am J Hum Genet . 2000;66:1403–6.
Judkins AR, Burger PC, Hamilton RL, et al. INI1 protein expression distinguishes atypical teratoid/rhabdoid tumor from choroid plexus carcinoma. J Neuropathol Exp Neurol . 2005;64:391–7.
Mueller W, Eum JH, Lass U, et al. No evidence of hSNF5/INI1 point mutations in choroid plexus papilloma. Neuropathol Appl Neurobiol. 2004;30:304–7.
Hoischen A, van Bon BW, Gilissen C, et al. De novo mutations of SETBP1 cause Schinzel-Giedion syndrome. Nat Genet . 2010;42:483–5.
Schuurs-Hoeijmakers JH, Landsverk ML, Foulds N, et al. Clinical delineation of the PACS1-related syndrome--Report on 19 patients. Am J Med Genet A. 2016;170:670–5.
Allen MD, Freund SM, Zinzalla G, et al. The SWI/SNF subunit INI1 contains an N-terminal winged helix DNA binding domain that is a target for mutations in schwannomatosis. Structure. 2015;23:1344–9.
Sestini R, Bacci C, Provenzano A, et al. Evidence of a four-hit mechanism involving SMARCB1 and NF2 in schwannomatosis-associated schwannomas. Hum Mutat. 2008;29:227–31.
Kehrer-Sawatzki H, Farschtschi S, Mautner VF, et al. The molecular pathogenesis of schwannomatosis, a paradigm for the co-involvement of multiple tumour suppressor genes in tumorigenesis. Hum Genet . 2017;136:129–48.
Eaton KW, Tooke LS, Wainwright LM, et al. Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatr Blood Cancer. 2011;56:7–15.
Smith MJ, Wallace AJ, Bowers NL, et al. SMARCB1 mutations in schwannomatosis and genotype correlations with rhabdoid tumors. Cancer Genet. 2014;207:373–8.
Smith MJ, Wallace AJ, Bowers NL, et al. Frequency of SMARCB1 mutations in familial and sporadic schwannomatosis. Neurogenetics. 2012;13:141–5.
Boyd C, Smith MJ, Kluwe L, et al. Alterations in the SMARCB1 (INI1) tumor suppressor gene in familial schwannomatosis. Clin Genet . 2008;74:358–66.
Rousseau G, Noguchi T, Bourdon V, et al. SMARCB1/INI1 germline mutations contribute to 10% of sporadic schwannomatosis. BMC Neurol . 2011;11:9.
Tsurusaki Y, Okamoto N, Ohashi H, et al. Coffin-Siris syndrome is a SWI/SNF complex disorder. Clin Genet. 2014;85:548–54.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Rights and permissions
About this article

Cite this article
Diets, I.J., Prescott, T., Champaigne, N.L. et al. A recurrent de novo missense pathogenic variant in SMARCB1 causes severe intellectual disability and choroid plexus hyperplasia with resultant hydrocephalus. Genet Med 21, 572–579 (2019). https://doi.org/10.1038/s41436-018-0079-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-018-0079-4
Keywords
This article is cited by
-
The genetic basis of hydrocephalus: genes, pathways, mechanisms, and global impact
Fluids and Barriers of the CNS (2024)
-
GEMC1 and MCIDAS interactions with SWI/SNF complexes regulate the multiciliated cell-specific transcriptional program
Cell Death & Disease (2023)
-
A Carboxy-terminal Smarcb1 Point Mutation Induces Hydrocephalus Formation and Affects AP-1 and Neuronal Signalling Pathways in Mice
Cellular and Molecular Neurobiology (2023)
-
This was the year that was: brain barriers and brain fluid research in 2019
Fluids and Barriers of the CNS (2020)
-
Mutations in SMARCB1 and in other Coffin–Siris syndrome genes lead to various brain midline defects
Nature Communications (2019)
