
Abstract
Bryant-Li-Bhoj syndrome (BLBS), which became OMIM-classified in 2022 (OMIM: 619720, 619721), is caused by germline variants in the two genes that encode histone H3.3 (H3-3A/H3F3A and H3-3B/H3F3B) [1–4]. This syndrome is characterized by developmental delay/intellectual disability, craniofacial anomalies, hyper/hypotonia, and abnormal neuroimaging [1, 5]. BLBS was initially categorized as a progressive neurodegenerative syndrome caused by de novo heterozygous variants in either H3-3A or H3-3B [1–4]. Here, we analyze the data of the 58 previously published individuals along 38 unpublished, unrelated individuals. In this larger cohort of 96 people, we identify causative missense, synonymous, and stop-loss variants. We also expand upon the phenotypic characterization by elaborating on the neurodevelopmental component of BLBS. Notably, phenotypic heterogeneity was present even amongst individuals harboring the same variant. To explore the complex phenotypic variation in this expanded cohort, the relationships between syndromic phenotypes with three variables of interest were interrogated: sex, gene containing the causative variant, and variant location in the H3.3 protein. While specific genotype-phenotype correlations have not been conclusively delineated, the results presented here suggest that the location of the variants within the H3.3 protein and the affected gene (H3-3A or H3-3B) contribute more to the severity of distinct phenotypes than sex. Since these variables do not account for all BLBS phenotypic variability, these findings suggest that additional factors may play a role in modifying the phenotypes of affected individuals. Histones are poised at the interface of genetics and epigenetics, highlighting the potential role for gene-environment interactions and the importance of future research.
Similar content being viewed by others


Introduction
Bryant-Li-Bhoj syndrome (BLBS) (OMIM: 619720, 619721) is a multi-system disorder with profound neurodevelopmental and neurodegenerative phenotypes [1,2,3,4]. Germline variants in either H3-3A/H3F3A or H3-3B/H3F3B cause BLBS. Both H3-3A and H3-3B are highly intolerant to missense variants, with Genome Aggregation Database (gnomAD) v2.1.1 missense constraint metric z-scores of 3.16 and 2.88, respectively, where a z-score >2 indicates that a gene is highly intolerant to missense variants. gnomAD v2.1.1 is the most recent release with constraint metrics for these two genes [5]. Additionally, only one variant (M120K) observed in affected individuals is present in the non-neurologic phenotype gnomAD v2.1.1 release (Supplementary Fig. 1), which may be a technical mapping error, as it is only present on one strand and did not meet the previous Exome Aggregation Consortium (ExAC) reporting criteria [1]. This gnomAD analysis supports that reported BLBS variants are causative, rather than expected variation within the population.
In all prior reports, BLBS is reported to affect both male and females equally. Notably, H3-3A and H3-3B are located on autosomes 1 and 17, respectively. Even in cases in which a gene implicated in Mendelian neurodevelopmental disorders (NDDs) is on an autosome, there exists a 2–4:1 NDD diagnostic discrepancy rate between males and females in the United States [6, 7], supporting the exploration of sex as a contributor to phenotypic heterogeneity in BLBS.
From a fundamental biology perspective, it is important to consider which of the two H3.3 encoding genes (H3-3A or H3-3B) is perturbed, given unique properties that distinguish them from most other protein-coding genes. Systemic knockout of each gene individually in murine models leads to distinct phenotypes, suggesting that these genes are not functionally redundant [8,9,10,11]. Further, while H3-3A is constitutively expressed, H3-3B is expressed in response to cellular stress and stimuli. This differential expression impacts H3.3 incorporation into the nucleosome. Further, these genes contain distinct exonic and intronic sequences, yet encode an identical H3.3 protein (Fig. 1A). This absolute conservation at the protein level, in spite of the two independent genes and four alleles, is a rare protein phenomenon, but common amongst histones [12]. This underlying histone biology suggests that each gene has distinct functional significance and that phenotypic variation observed across individuals with BLBS might be due in part to whether H3-3A or H3-3B is affected.

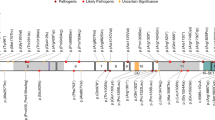
A 2D diagram of the genes that encode the histone H3.3 protein – H3-3A (top - ENST00000366815) and H3-3B (bottom – ENST00000254810). Green brackets and thicker boxes represent the coding sequence. Arrows represent the transcription start sites. B 2D diagram of histone H3.3 protein (green), including the location of the four alpha helices. Lollipops show the H3-3A derived (top) and the H3-3B derived (bottom) heterozygous germline variants. Length of lollipop corresponds to number of individuals who harbor a variant at that residue (e.g. H3-3A p.T45I represents four individuals with BLBS and H3-3A p.V46M represents one individual with BLBS). C 3D in silico structural model of the H3.3-containing nucleosome (PDB: 5X7X) with the two copies of H3.3 in green; other histones in gray; and DNA in black. The location of heterozygous germline variants in the crystallized histone core are highlighted in purple. D Circular boxplot visualizing BLBS phenotypes. Cyan = growth (height, weight and head circumference); blue = craniofacial anomalies; pink = abnormal neuroimaging findings and seizures; red = developmental milestones; yellow = tone anomalies and oculomotor features; green = review of systems.
The H3.3 protein encoded by these two genes is comprised of a disordered tail, four α helices, and two loop domains. As in prior reports, the disordered tail is defined as amino acids (AA) 1–43 and the core (four α helices and two loop domains) as AA 44–135 (Fig. 1B), with the initiating methionine excluded from numbering [1, 4, 13,14,15,16,17,18]. Phenotypic variability could also be affected by which protein domain, specifically the tail or core, is affected by the variant. Certain “hotspot” somatic variants in H3.3, which are restricted to residues that include H3.3 p.K27M and p.G34R/V, cause pediatric brain tumors [17,18,19,20,21,22]. While these somatic variants are restricted to the H3.3 tail, the causative germline variants in BLBS are distributed throughout H3.3 (Fig. 1B). Currently, there is no evidence that germline H3.3 variants are oncogenic, but this is an area of ongoing investigation [4, 23].
It also important to distinguish between the histone core and tail because of the histone code. The histone code, written by post-translational modifications (PTMs) of amino acid residues that play a fundamental role in transcriptional regulation, is most commonly associated with the modification of residues on the N-terminal tail of H3.3 [24]. It is unsurprising that germline perturbation of residues in the tail are associated with disease pathogenesis [1]. However, affected individuals demonstrate that germline variants in the core also cause BLBS. Interestingly, in Tessadori-Bicknell-van Haaften (TBvH) NDD, caused by germline variants in histone H4, all known variants are restricted to the histone core [25, 26]. The mechanisms by which germline variants in the H3.3 and H4 cores cause BLBS and TBvH NDD remain poorly understood.
Here, 38 previously unpublished, unrelated individuals with heterozygous germline variants in H3-3A/H3F3A or H3-3B/H3F3B nearly double the number of known individuals affected by BLBS. All 58 previously published individuals are included in this analysis, totaling 96 individuals, which enables the interrogation of the effect of 1) the sex of the individual; 2) the gene harboring the germline variant; or 3) the location of the variant in the histone tail versus the histone core on BLBS phenotypes [1,2,3,4]. Then, potential genotype-phenotype relationships are interrogated by sub-stratifying the cohort into 1) individuals with the same missense variant in different genes; 2) individuals with different missense variants affecting the same residue in different genes; and 3) individuals with either germline or somatic variants in different genes affecting the same residue. As with many ultra-rare diseases, there are analytical limits that arise from the small number of affected individuals. Since a single individual can drastically affect statistical significance, it is cautioned that utilizing p-values to denote significance may not reflect the trends within the overall population [27, 28], statistical significance may not always reflect biological significance. For these reasons, trends, rather than statistical significance, are reported here.
Subjects and methods
The Institutional Review Board of the Children’s Hospital of Philadelphia approved this study. Informed consent was obtained from all individuals included in the study. Individuals were referred by clinicians to EJKB through GeneMatcher [29]. Individual phenotypes and genetic sequencing information were provided by the referring clinicians. Analyses and graphs were made in Microsoft Excel, GraphPad Prism v8, and R v4.3.2 using circular barplot code from R-graph-gallery (https://github.com/holtzy/R-graph-gallery). Graphics were generated with BioRender.
Phenotyping criteria
The denominators presented in Table 1 for each sub-analysis are not always equal to the total number of individuals with BLBS (96). A denominator less than 96 reflects the subset of individuals for whom phenotypic information for a particular sub-analysis was available or consideration of developmental stage. For instance, when analyzing attainment of independent sitting, walking or first words, individuals who have not yet surpassed the expected window of achievement for these milestones are excluded (e.g. an individual who is 18 months old would be included in the independent sitting metric, but not the independent walking or speech metrics).
In alignment with field standards, overgrowth (height/weight) and macrocephaly were defined as measurements that were equal to or above 95th percentile, or greater than 2 standard deviations (SD) above the mean, compared to age- and sex-matched controls [30]. Undergrowth (height/weight) and microcephaly were similarly defined as measurements that were equal to or below the 5th percentile, or greater than 2 SD below the mean, compared to age- and sex-matched controls.
Delayed attainment of developmental milestones was defined based on established developmental trajectories within pediatrics [31]. An individual was classified as demonstrating delayed independent sitting if they had not yet achieved that milestone at 12 months of age. Delayed independent walking was identified if an individual had not yet achieved that milestone at 20 months of age. Delayed speech was indicated if an individual had not yet achieved their first word at 20 months of age.
PyMOL in silico 3D structural protein modeling
Utilizing PyMOL Molecular Graphics System Version 2.5.5, the crystallized structure of the nucleosome containing H3.3 at 2.18 Å resolution was imported from the Research Collaboratory for Structural Bioinformatics Protein Data Bank by referencing ID 5X7X [32]. The H3.3 protein sequence identity was verified by cross-referencing UniProt Knowledgebase sequences (Human H3.3 - P84243). H3.3 is color-coded “smudge green” and denoted as green; all other histones are color-coded “gray70” and denoted as gray; the DNA double helix is color-coded “gray10” and denoted as black; and BLBS-causing variants are color-coded “purpleblue” and denoted as purple.
Results
Thirty-eight previously unpublished, unrelated individuals with BLBS, along with the 58 previously reported individuals, represent a global cohort of 96 individuals who harbor 70 unique causative variants (Fig. 1B and C, Table 1, Supplementary Table 1) [1,2,3,4]. Individuals range in age from 10 weeks to 39 years at the time of their most recent evaluation, and include 47 males and 49 females (Table 1, Supplementary Table 1). In total, 65 individuals harbor variants in H3-3A and 31 individuals harbor variants in H3-3B. At the time of this report, all H3-3A variants are heterozygous de novo missense variants when parents are available, though inheritance was undetermined for three individuals (Supplementary Table 1). Conversely, while most variants in H3-3B are heterozygous de novo missense variants, more variability in variant type and modes of inheritance was observed. One individual harbors a variant that is synonymous in the canonical H3-3B transcript and leads to a stop-gain in a non-canonical transcript (H3-3B p.V117V/S147*) [1]. Another individual harbors a two-nucleotide deletion that ablates the stop codon (H3-3B p.C136*ext9) (Fig. 1B, Supplementary Table 1) [3]. Two individuals with variants in H3-3B have unknown inheritance and one individual has a maternally inherited H3-3B heterozygous missense variant (H3-3B p.N108S) (Supplementary Table 1). The referring geneticist has confirmed the maternal genotype and is working to fully phenotype and genotype this individual’s siblings, mother, and maternal grandparents.
BLBS phenotypic variability
BLBS is predominantly characterized by developmental delay/intellectual disability (DD/ID), growth anomalies, craniofacial anomalies, abnormal neuroimaging, and hypo/hypertonia (Fig. 1D, Table 1, Supplementary Table 1). Phenotypic heterogeneity is noted across the BLBS population. For example, individuals may present with microcephaly, macrocephaly, or a head circumference within the normal range (Table 1, Supplementary Table 1). Potential sources underlying this variation were interrogated by stratifying by 1) the sex of the individual; 2) which of the two genes harbors the variant; and 3) the location of the variant in the histone tail or core.
BLBS and growth
Over half of individuals with BLBS demonstrate typical age- and sex-based growth (Table 1, Fig. 1D, Supplementary Table 1). Forty-one percent of individuals have height trajectories outside of the normal range. Most of these individuals exhibit undergrowth (35%) (Table 1, Supplementary Table 1). This trend holds when the population is stratified by sex or gene (Fig. 2, Supplementary Table 1). When variants are stratified by location in H3.3, this trend is even more pronounced, with 44% of individuals harboring variants in the tail presenting with undergrowth while only 6% presenting with overgrowth (Fig. 2, Supplementary Table 1). Even though individuals with variants in the core demonstrate a similar distribution between overgrowth and undergrowth, 63% of individuals with variants in the core are reported to be of average height, compared to 50% of those with variants in the tail (Fig. 2, Supplementary Table 1). Similarly, individuals with variants in H3-3A (46%) are more likely to present with undergrowth or overgrowth than those with variants in H3-3B (31%).

Phenotypic categories (rows) analyzed across all 96 individuals with BLBS include growth; craniofacial features; neuroimaging findings and seizures; attainment of developmental milestones; tone anomalies; and general review of systems. Phenotypic analyses were performed by stratifying the cohort of individuals (columns) based on their sex (reported as male or female) (column 1); on the localization of their causative variant to either H3-3A or H3-3B (column 2); or on the location of their causative variant to the histone tail or histone core (column 3). Each graph is representative of the percentages of individuals with BLBS for which this category was reported. The colored cells highlight stratifications emphasized in the text. The color-coding is based on the phenotypic overview in Fig. 1D, where cyan = growth (height, weight and head circumference); blue = dysmorphic craniofacial features; pink = neuroimaging findings and seizures; red = developmental milestones; yellow = tone anomalies and oculomotor features; green = review of systems.
Of the 38% of individuals with weight metrics reported outside of the normal range, 14% of reported weights were ≤5th and 14% of reported weights were ≥95th percentile (Table 1, Supplementary Table 1). As with height, individuals with variants in the histone tail show a similar phenotypic distribution in relation to their weight, with more individuals being underweight (32%) than overweight (7%) (Fig. 2, Supplementary Table 1). Interestingly, the pattern is inverted for individuals with variants in the core: 25% of individuals are reported as being overweight while 12% are reported as being underweight (Fig. 2, Supplementary Table 1).
BLBS and craniofacial development
In addition to growth trajectories outside of the standard range for age and sex, 92% of individuals with BLBS present with craniofacial anomalies (Table 1, Fig. 1D, Supplementary Table 1). Commonly reported features are broad/prominent forehead, broad nasal bridge, thin upper lip, hypertelorism, epicanthal folds, and ear and tooth anomalies.Dysmorphic features are slightly more common in individuals with variants in H3-3A (95%) than H3-3B (86%) (Fig. 2, Supplementary Table 1), though it is important to note that these individuals have all been phenotyped by different geneticists and the assessment of facial feature analysis is strongly influenced by the ancestral and ethnic background of both the evaluator and the affected individual [33].
Notably, 46% of individuals with craniofacial anomalies present with concurrent microcephaly or macrocephaly (Table 1, Fig. 1D, Supplementary Table 1). Among these individuals, microcephaly is more common than macrocephaly (32 and 15%, respectively), which is consistent across all variables interrogated (Table 1). The occurrence of micro- or macrocephaly is most different between variants in the tail or core (60 and 38%, respectively) (Fig. 2). Microcephaly is more common in individuals with variants in the tail (43%) than the core (25%). Discrepancies in the prevalence of micro- and macrocephaly also depend upon the affected gene (43% H3-3A and 53% H3-3B, respectively) (Fig. 2).
BLBS and neuroradiological imaging
Seventy-nine percent of individuals with BLBS underwent diagnostic magnetic resonance imaging (MRI). Fifty-eight percent of individuals with a reported MRI were diagnosed with at least one abnormal finding (Table 1). While not all referring clinicians elaborated on the MRI results, common findings included delayed myelination or hypomyelination; dysgenesis of the corpus collosum; dilated ventricles; and hemispheric asymmetry across multiple structures (Table 1, Supplementary Table 1). These findings were expanded upon by Alves et al., where 18 MRIs from previously reported individuals with BLBS were carefully analyzed [1, 34]. Within this subgroup, 72% of individuals presented with small posterior fossa, 28% presented with dysgenesis of the corpus collosum, and 44% presented with cortical developmental abnormalities [34].
When analyzing MRI findings beyond these 18 individuals, abnormal findings were more commonly reported for individuals with variants in the tail (63%) than for individuals with variants in the core (54%). Abnormal findings were seen in more males (63%) than females (53%). Forty nine percent of individuals also present with seizures (Table 1). Of these individuals, 20% experienced febrile seizures. Seizures were more frequently reported in individuals harboring variants in H3-3B (59%) than those harboring variants in H3-3A (45%) (Fig. 2).
BLBS and developmental milestones
Ninety-nine percent of individuals with BLBS have DD/ID (Table 1, Fig. 1D, Supplementary Table 1). The only individual not reported to have DD/ID harbors the H3-3B p.V117V/S147* variant [1]. While he is the only individual reported with a synonymous variant, he exhibited delayed attainment of speech (first word at 24 months); presented with an “expressive language disorder with neurologic progression” when evaluated at 15 years; and had neuroimaging anomalies consistent with the other individuals in this cohort.
In addition to DD/ID, many individuals have co-existing neurodevelopmental diagnoses, including 8% with autism spectrum disorder, 3% with attention deficit disorder, and 3% with anxiety diagnoses. Others have neurobehavioral diagnoses, including behavioral issues (4%) and stereotyped repetitive movements (7%) (Supplementary Table 1). None of these phenotypes are correlated to sex, gene, or location (Fig. 2, Supplementary Table 1).
A subset of individuals displayed delays in developmental milestones including independent sitting, independent walking, and/or speech development (Table 1, Fig. 1D, Supplementary Table 1). Individuals with variants in the tail (59%) more frequently had delayed or yet to be achieved independent sitting compared to individuals with variants in the core (47%) (Fig. 2). Individuals with variants in H3-3A (56%) more frequently had delayed or yet to be achieved independent sitting compared to individuals with variants in H3-3B (43%) (Fig. 2). More individuals harboring variants in H3-3A (85%) had delayed or yet to be achieved independent walking when compared to individuals harboring variants in H3-3B (65%). Differences were not seen between variants in the tail versus core (Fig. 2). Sex captured some phenotypic variability related to independent walking (86% of males had delayed or yet to be achieved independent walking compared to 71% females) and speech development (33% of females had not yet achieved one word compared to 49% of males) (Fig. 2).
BLBS and hyper/hypotonia
Eighty-four percent of individuals present with hypotonia, hypertonia, or a combination of axial hypotonia with peripheral hypertonia (Table 1, Fig. 1D, Supplementary Table 1). Sixty-two percent of individuals presented with hypotonia whereas 12% presented with hypertonia (Table 1, Fig. 2). Five of the reported 55 individuals presented with resolved hypotonia at their most recent evaluation. Interestingly, 10% of individuals have concordant axial hypotonia and peripheral hypertonia, or dystonia, which is exclusively present in individuals with variants in the core (Fig. 2, light gray). Hypotonia is also more commonly reported for individuals with variants in the tail (73%) compared to those with variants in the core (56%) (Fig. 2). While not directly queried here, two individuals were reported to demonstrate an ataxic gait, while Okur et al. reported that individuals in their cohort universally presented with gait anomalies [1, 3].
In conjunction with global tone abnormalities, 54% of individuals report oculomotor dysfunction, 82% of whom demonstrate eye rolling and strabismus, which may be attributed to abnormal muscle tone (Table 1, Fig. 1D, Supplementary Table 1). More females (64%) present with oculomotor dysfunction than males (44%) (Fig. 2).
BLBS and review of systems
Beyond the neurological features, individuals also variably present with phenotypes resulting in abnormalities within the musculoskeletal, dermatologic, cardiac, and genital systems (Fig. 1D). Over half of individuals (58%) present with musculoskeletal anomalies such as club foot, scoliosis, hip dysplasia, subluxation of various joints/hypermobility, kyphosis, and femoral anteversion (Figs. 1D and 2, Table 1, Supplementary Table 1).
Fifty-three percent of individuals present with dermal phenotypes such as eczema, hypoplastic nails, fetal finger pads, nipple abnormalities and 2/3 toe syndactyly (Fig. 1D, Table 1, Supplementary Table 1). Dermal features are more likely found in individuals with variants in the tail (64%) than the core (45%) (Fig. 2).
Fourteen percent of individuals present with cardiac anomalies, including atrial septal defects (Fig. 1D). Cardiac anomalies are twice as likely to be reported in individuals with core variants (17%) than tail variants (7%) (Table 1, Fig. 2, Supplementary Table 1).
Twenty percent of individuals present with genital anomalies (Fig. 1D, Table 1, Supplementary Table 1). Genital anomalies are more often reported in males (37%) than females (2%), though this may be related to the necessity of intervention associated with a particular anomaly, such as cryptorchidism. More individuals with variants in H3-3B (28%) present with genital anomalies compared to individuals with variants in H3-3A (17%).
While urinary anomalies were not specifically queried here, some clinicians reported phenotypes such as small right kidney, horseshoe kidney, solitary kidney, nephrocalcinosis, and chronic urinary tract infections (Supplementary Table 1). This suggests that genitourinary surveillance may be important for individuals with BLBS going forward.
Exploration of potential genotype-phenotype correlations in BLBS
Potential genotype-phenotype correlation was explored given our hypothesis that individuals harboring similar variants would present with similar phenotypes. A similar analysis was previously performed for the four individuals harboring H3-3A p.T45I variants, which showed phenotypic variation (Fig. 3A) [1]. With this cohort expansion, additional analyses were performed to explore the possibility of a genotype-phenotype correlation in other subgroups who harbored 1) the same variant in the same residue of different genes (H3-3A vs H3-3B) (Fig. 3B); 2) different variants in the same residue of different genes (Fig. 3C); and 3) variants arising in germline versus somatic cells (Fig. 3D).

A–C Representation of the phenotypic variation across individuals who (A) share the same missense variant in the same residue of the same gene, (B) the same missense variant in the same residue of different genes, or (C) different missense variants affecting the same residue of different genes. The color-coding in the key corresponds to the phenotypic overview in Fig. 1D and Fig. 2. A Representation of the phenotypic variation across the four individuals who share the H3-3A p.T45I variant. B Representation of phenotypic variation across the eight individuals who share the H3.3 p.Q125R variant. Five individuals harbor a nucleotide substitution in H3-3A while three individuals harbor a nucleotide substitution in H3-3B. C Representation of phenotypic variation across the eight individuals who harbor variants affecting the H3.3 p.P121 residue. Four individuals harbor a nucleotide substitution in H3-3A (leading to either p.P121L or p.P121R missense variants) while four individuals harbor a nucleotide substitution in H3-3B (leading to either p.P121L or p.P121R missense variants). D Phenotypic variation across individuals with BLBS-causing germline variants throughout the disordered histone tail region and histone core (top) compared to hotspot high grade glioma-causing somatic mutations (bottom). Black = amino acids with associated germline variants; magenta = amino acids with associated germline and somatic variants; blue = amino acids with associated somatic variants.
Same missense variant, same residue, different genes
Eight individuals harbor the H3.3 p.Q125R variant (Fig. 3B). This protein alteration results from a nucleotide change in H3-3A for 5 individuals and a change in H3-3B for 3 individuals. In this subcohort, individuals span the ages of 10 months to 15 years. All individuals present with DD/ID. Only two individuals exhibit overgrowth, a phenotype that seems to segregate with variants in H3-3A in this sub-cohort (Fig. 3B). Conversely, two individuals present with macrocephaly, a phenotype only associated with H3-3B in this sub-cohort (Fig. 3B). Finally, all five individuals with the H3-3A variant present with seizures, while seizures are only reported in one individual with an H3-3B variant (Fig. 3B).
Different missense variant, same residue, different genes
Eight unrelated individuals between the ages of 2.5 months to 18 years harbor variants that affect the p.P121 residue (Fig. 3C). Four individuals harbor a c.365C>T change, resulting in p.P121R variants, while the other four individuals harbor a c.365C>G change, resulting in p.P121L variants. All individuals have DD/ID and either hypotonia or a combination of axial hypotonia and peripheral hypertonia (Fig. 3C). Additionally, seven of the eight individuals present with seizures (Fig. 3C, Supplementary Table 1).
Different cells of origin
BLBS arises from germline variants in H3.3 while high grade gliomas arise from somatic variants in H3.3. These somatic variants have a specific genotype-phenotype relationship, exhibiting precise spatiotemporal relationships (Fig. 3D) [4, 22, 23]. H3.3 p.K27M somatic variants are associated with universally fatal diffuse midline gliomas in children under 12, while H3.3 p.G34R/V somatic variants are associated with diffuse hemispheric brain tumors, with age of diagnosis between 12–35 [4, 22, 23]. Two individuals with BLBS harbor germline variants affecting the p.G34 residue (p.G34R and p.G34V) [4]. Interestingly, these individuals harbor germline substitutions in H3-3B, while individuals with diffuse hemispheric brain tumors harbor somatic variants in H3-3A (Fig. 3D) [4]. Currently, there are no reported cases of individuals with BLBS with any oncologic diagnoses, including high grade gliomas.
Discussion
With this expanded cohort of individuals, the genetic causes and resultant phenotypes of the BLBS population can be more thoroughly examined. These updated analyses highlight that the four most common features of BLBS are DD/ID, craniofacial anomalies, abnormal neuroimaging findings, and tonal anomalies (Fig. 1D, Table 1, Supplementary Table 1). However, the presentation of these features is highly variable across individuals, and the addition of each individual continues to deepen our understanding of the phenotypic spectrum of BLBS. The variability suggests that molecular testing will continue to play a role in diagnosing affected individuals, though a characteristic phenotype for individuals with BLBS may yet emerge.
The current stratification by sex, affected gene, or affected protein domain does not account for all phenotypic variation observed in individuals with BLBS. This suggests that the remainder of phenotypic heterogeneity may be attributed to other molecular mechanisms, such as the incorporation of H3.3 with the causative variant into nucleosomes, or the altered deposition of PTMs on H3.3, leading to a disrupted histone code and aberrant gene regulation [24, 35]. Additional functional work will be crucial both for diagnosis and the development of therapeutic interventions [36].
In ultra-rare Mendelian NDDs, every affected individual impacts the way translational research and medical communities understand a syndrome. Collaboration and data-sharing between groups around the world is imperative to ensure that the generous gift to medicine and science that each affected individual’s family makes shapes the trajectory of the field. In a five-year span from 2019 to 2024, we have moved from the first single-individual BLBS case report to now analyzing a cohort of almost 100 individuals, which has enabled the deep interrogation of trends. This patient-guided approach, coupled with ongoing functional work, will hopefully enable more conclusive guidance in the near-future.
Follow-up phenotyping
This analysis of BLBS phenotypes highlights the need for repeat phenotyping of individuals throughout their lives. Longitudinal follow-up is currently only accessible for a few of the individuals presented in this cohort (H3-3A p.L61R, H3-3B p.P121R and p.Q125R) [1, 2, 3, 37]. In infancy, individuals who harbor the H3-3B p.P121R and p.Q125R variants were diagnosed with an unspecified overgrowth disorder but, at follow up years later, they presented with normal height or undergrowth [1, 37]. Longitudinal follow-up will also allow for direct comparison between evaluation timepoints, elucidating more detail about the temporal phenotype, including the previously established neurodegenerative component of this syndrome [1, 4]. This long-term follow-up could facilitate a deeper understanding of the tonal anomalies associated with BLBS. For some individuals, there is a progressive transition between generalized hypotonia to axial hypotonia with peripheral hypertonia, while for others, there is a trend of resolved hypotonia. This suggests that there may be an underlying neurological progression. Additionally, 25% of individuals are diagnosed with concurrent neurodevelopmental and neurobehavioral diagnoses. This concurrence can only be diagnosed once children reach a certain age or developmental stage. Identifying individuals with syndromic features in addition to neurobehavioral diagnoses would not only facilitate access to resources like behavioral intervention programs, but could also aid in the referral of these individuals for genetic testing [38]. This would be most beneficial in cases of individuals with less severe phenotypes who would not otherwise be referred for whole exome or genome sequencing.
Further, when individuals have access, and based on considerations such as the need for sedation, repeat neuroimaging could facilitate the management of progressive disease, as suggested in the 2023 Gene Reviews entry by Bryant and Bhoj [38]. Many individuals present with dysgenesis/hypoplastic/thinning corpus collosum as well as hypomyelination [34]. These phenotypes exhibit overlap with leukodystrophy disorders, which are characterized by structural brain and muscle tone anomalies [34, 39, 40]. Repeat brain MRIs could help determine whether this is a progressive neurodegenerative disorder, as is suggested by the tonal and the abnormal gait/progressive gait ataxia phenotype [3]. Further delineating the neurodegenerative characteristics of BLBS would help clinicians provide prognostic information to families.
Finally, somatic variants in H3.3 are driver mutations in cancers like high-grade gliomas [23]. Current phenotypic evaluations do not suggest that individuals with BLBS have a cancer predisposition. It is possible that some individuals have since received a cancer diagnosis, as malignancy was not a specific query of these surveys. Longitudinal phenotyping of individuals with BLBS could enable more definitive answers related to co-morbidities for individuals and their families.
BLBS, not just de novo missense variants
In the initial characterization of BLBS, all individuals harbored heterozygous de novo missense variants. The individual with the reported synonymous H3-3B p.V117V variant was excluded from the phenotyping analysis at that time. It has since been confirmed that this individual harbors a synonymous variant in the canonical H3-3B transcript, which maps to a truncating variant in a non-canonical transcript [1]. Investigations into the implications of this variant on understanding the mechanism of pathogenesis in BLBS are ongoing. Additionally, an individual with a stop-loss variant in H3-3B was reported [3]. These variants highlight that the genotypic spectrum of the disorder is not isolated to missense variants.
This cohort also includes the first known individual with a BLBS-causing inherited variant. The individual harbors a maternally inherited p.N108S variant in H3-3B demonstrating that BLBS does not always arise from de novo variants. A full phenotypic evaluation of the individuals’ mother, maternal grandparents, and siblings is ongoing. Given the shared genetic background, families harboring inherited variants will play a crucial role in elucidating the additional genetic and environmental modifiers of BLBS.
In sum, this expanded cohort provides new detail about BLBS and extends its characterization to a neurodevelopmental and neurodegenerative disorder with variable multi-systemic effects. Ongoing functional work is needed to clearly determine how the factors reviewed here – sex, gene, and variant location – affect phenotypic variability. Additionally, future functional work is needed to elucidate how other factors impact the severity of this disorder. The information presented here, coupled with ongoing functional work, will aid in shortening the diagnostic odysseys for future individuals with BLBS and their families.
Data availability
All data analyzed in this study are available in the manuscript and represented in Supplementary Information.
Change history
31 July 2024
An author was not named.
26 July 2024
A Correction to this paper has been published: https://doi.org/10.1038/s41431-024-01659-y
References
Bryant L, Li D, Cox SG, Marchione D, Joiner EF, Wilson K, et al. Histone H3.3 beyond cancer: Germline mutations in Histone 3 Family 3A and 3B cause a previously unidentified neurodegenerative disorder in 46 patients. Sci Adv. 2020;6:eabc9207.
Maver A, Čuturilo G, Ruml SJ, Peterlin B. Clinical Next Generation Sequencing Reveals an H3F3A Gene as a New Potential Gene Candidate for Microcephaly Associated with Severe Developmental Delay, Intellectual Disability and Growth Retardation. Balk J Med Genet. 2019;22:65–8.
Okur V, Chen Z, Vossaert L, Peacock S, Rosenfeld J, Zhao L, et al. De novo variants in H3-3A and H3-3B are associated with neurodevelopmental delay, dysmorphic features, and structural brain abnormalities. npj Genom Med. 2021;6:104.
Khazaei S, Chen CCL, Andrade AF, Kabir N, Azarafshar P, Morcos SM, et al. Single substitution in H3.3G34 alters DNMT3A recruitment to cause progressive neurodegeneration. Cell. 2023;186:1162–78.e20.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–43.
May T, Adesina I, McGillivray J, Rinehart NJ. Sex differences in neurodevelopmental disorders. Curr Opin Neurol. 2019;32:622–6.
Bölte S, Neufeld J, Marschik PB, Williams ZJ, Gallagher L, Lai M-C. Sex and gender in neurodevelopmental conditions. Nat Rev Neurol. 2023;19:136–59.
Witt O, Albig W, Doenecke D. Transcriptional regulation of the human replacement histone gene H3.3B. FEBS Lett. 1997;408:255–60.
Frank D, Doenecke D, Albig W. Differential expression of human replacement and cell cycle dependent H3 histone genes. Gene. 2003;312:135–43.
Delaney K, Mailler J, Wenda JM, Gabus C, Steiner FA. Differential Expression of Histone H3.3 Genes and Their Role in Modulating Temperature Stress Response in Caenorhabditis elegans. Genetics. 2018;209:551–65.
Klein RH, Knoepfler PS. Knockout tales: the versatile roles of histone H3.3 in development and disease. Epigenetics Chromatin. 2023;16:38.
Muhire BM, Booker MA, Tolstorukov MY. Non-neutral evolution of H3.3-encoding genes occurs without alterations in protein sequence. Sci Rep. 2019;9:8472.
Bryant L, Sangree A, Clark K, Bhoj E. Histone 3.3-related chromatinopathy: missense variants throughout H3-3A and H3-3B cause a range of functional consequences across species. Hum Genet. 2023. https://doi.org/10.1007/s00439-023-02536-2.
Ahmad K, Henikoff S. The Histone Variant H3.3 Marks Active Chromatin by Replication-Independent Nucleosome Assembly. Mol Cell. 2002;9:1191–1200.
Funk OH, Qalieh Y, Doyle DZ, Lam MM, Kwan KY. Postmitotic accumulation of histone variant H3.3 in new cortical neurons establishes neuronal chromatin, transcriptome, and identity. Proc Natl Acad Sci USA. 2022;119:e2116956119.
Goldberg AD, Banaszynski LA, Noh K-M, Lewis PW, Elsaesser SJ, Stadler S, et al. Distinct Factors Control Histone Variant H3.3 Localization at Specific Genomic Regions. Cell. 2010;140:678–91.
Jang C-W, Shibata Y, Starmer J, Yee D, Magnuson T. Histone H3.3 maintains genome integrity during mammalian development. Genes Dev. 2015;29:1377–92.
Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 1997;389:251–60.
Behjati S, Tarpey PS, Presneau N, Scheipl S, Pillay N, Van Loo P, et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat Genet. 2013;45:1479–82.
Boileau M, Shirinian M, Gayden T, Harutyunyan AS, Chen CCL, Mikael LG, et al. Mutant H3 histones drive human pre-leukemic hematopoietic stem cell expansion and promote leukemic aggressiveness. Nat Commun. 2019;10:2891.
Schwartzentruber J, Korshunov A, Liu X-Y, Jones DTW, Pfaff E, Jacob K, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–31.
Voon HPJ, Hii L, Garvie A, Udugama M, Krug B, Russo C, et al. Pediatric glioma histone H3.3 K27M/G34R mutations drive abnormalities in PML nuclear bodies. Genome Biol. 2023;24:284.
Andrade AF, Chen CCL, Jabado N. Oncohistones in brain tumors: the soil and seed. Trends Cancer. 2023;9:444–55.
Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45.
Tessadori F, Duran K, Knapp K, Fellner M, Deciphering Developmental Disorders Study, Smithson S, et al. Recurrent de novo missense variants across multiple histone H4 genes underlie a neurodevelopmental syndrome. Am J Hum Genet. 2022;109:750–8.
Knapp K, Naik N, Ray S, van Haaften G, Bicknell LS. Histones: coming of age in Mendelian genetic disorders. J Med Genet. 2023;60:213–22.
Mitani AA, Haneuse S. Small Data Challenges of Studying Rare Diseases. JAMA Netw Open. 2020;3:e201965.
Wasserstein RL, Schirm AL, Lazar NA. Moving to a World Beyond “ p < 0.05”. Am Statistician. 2019;73:1–19.
Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: A Matching Tool for Connecting Investigators with an Interest in the Same Gene. Hum Mutat. 2015;36:928–30.
Burkardt DD, Tatton-Brown K, Dobyns W, Graham JM. Approach to overgrowth syndromes in the genome era. Am J Med Genet C Semin Med Genet. 2019;181:483–90.
Scharf RJ, Scharf GJ, Stroustrup A. Developmental Milestones. Pediatr Rev. 2016;37:25–37. quiz 38, 47.
Taguchi H, Xie Y, Horikoshi N, Maehara K, Harada A, Nogami J, et al. Crystal Structure and Characterization of Novel Human Histone H3 Variants, H3.6, H3.7, and H3.8. Biochemistry. 2017;56:2184–96.
Lumaka A, Cosemans N, Lulebo Mampasi A, Mubungu G, Mvuama N, Lubala T, et al. Facial dysmorphism is influenced by ethnic background of the patient and of the evaluator. Clin Genet. 2017;92:166–71.
Alves CAPF, Sherbini O, D’Arco F, Steel D, Kurian MA, Radio FC, et al. Brain Abnormalities in Patients with Germline Variants in H3F3 : Novel Imaging Findings and Neurologic Symptoms Beyond Somatic Variants and Brain Tumors. AJNR. Am J Neuroradiol. 2022;43:1048–53.
Maze I, Noh K-M, Soshnev AA, Allis CD. Every amino acid matters: essential contributions of histone variants to mammalian development and disease. Nat Rev Genet. 2014;15:259–71.
Fahrner JA, Bjornsson HT. Mendelian disorders of the epigenetic machinery: postnatal malleability and therapeutic prospects. Hum Mol Genet. 2019;28:R254–R64.
Shah PS, Weksberg R, Chitayat D. Overgrowth with severe developmental delay following IVF/ICSI: A newly recognized syndrome? Am J Med Genet A. 2006;140:1312–5.
Bryant L, Bhoj E. Bryant-Li-Bhoj Neurodevelopmental Syndrome. In: Adam MP, Feldman J, Mirzaa GM et al. editors. GeneReviews®. University of Washington, Seattle: Seattle (WA); 1993. http://www.ncbi.nlm.nih.gov/books/NBK595206/. Accessed 20 Dec 2023.
Modesti NB, Evans SH, Jaffe N, Vanderver A, Gavazzi F. Early recognition of patients with leukodystrophies. Curr Probl Pediatr Adolesc Health Care. 2022;52:101311.
Vanderver A, Prust M, Tonduti D, Mochel F, Hussey HM, Helman G, et al. Case definition and classification of leukodystrophies and leukoencephalopathies. Mol Genet Metab. 2015;114:494–500.
Acknowledgements
We would like to thank the individuals and families that have shared their clinical data with us. It is our sincere hope that the sharing of this information will lead to informed and expedited diagnosis for future families that are awaiting diagnostic answers. We would also like to thank the Germline Histone Mutations in Human Disease International Consortium, created by EEL, for their discussion and input on the phenotypic similarities across neurodevelopmental disorders caused by germline histone variants.
Funding
This study was supported by NHGRI T32 5T32HG009495 and the Eagles Autism Foundation (DELC), NICHD F30 1F30HD112125 (EEL), NIGMS T32GM008638 (LMB, ELD), NICHD P50HD109879 (WKC), NU22-07-00165 (MH, SB, DP, AB). Additionally, this was funded by Burroughs Wellcome Fund, Hartwell Foundation, and the Chan Zuckerberg Initiative (EJKB).
Author information
Authors and Affiliations
Contributions
DLC, LMB, EW, and EJKB collected and compiled clinical data for all participants. DLC, EEL and AKS analyzed and interpreted the data. DLC, EEL and AKS drafted the manuscript. KJC, ELD, EMG, SS, RA, and XMW contributed to review of the clinical data and critical review of the manuscript. RML, LD, ND, DV, IV, MCS, CZ, JJH, KC, DS, MK, KB, DC, AID, MJMN, MH, SB, ZS, AB, CBN, JD, WKC, NL, KP, CK, LA, DW, IK, CZ, AR, TB, MS, HYK, AW, GV, NMM, DV, CS, KK, JTS, KAV, MLV, PG, ND, EC, CB, BA, MD, MW, LG, FM, NS, SW, ASS, RFK, MM, BTC, KT, CEP, MTM, PJ, ERR, RL, NAB, WT, KK, RS, MS, EN, SB, GLEG, JK, CM, and DAC provided clinical data, wrote clinical case descriptions, diagnosed patients, clinically reviewed and edited manuscript. EJKB conceived of the study and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
The Institutional Review Board of the Children’s Hospital of Philadelphia approved this study. Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Layo-Carris, D.E., Lubin, E.E., Sangree, A.K. et al. Expanded phenotypic spectrum of neurodevelopmental and neurodegenerative disorder Bryant-Li-Bhoj syndrome with 38 additional individuals. Eur J Hum Genet 32, 928–937 (2024). https://doi.org/10.1038/s41431-024-01610-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-024-01610-1
This article is cited by
-
Summer reading in EJHG
European Journal of Human Genetics (2024)
