
Abstract
Cystic fibrosis (CF) is the most common life-limiting autosomal recessive disease in the Republic of Ireland (ROI), with a previously quoted incidence of 1 in 1353 and carrier rate of 1 in 19. The National Newborn Screening (NBS) for CF was incorporated in July 2011 in the ROI. A cut-off point of the top 1% Immunoreactive Trypsinogen (IRT) was taken as an indication for 38 CFTR variant panel to maximise identification of affected CF cases and to minimise detection of carriers. All neonates from July 2011 to Dec 2017 with an elevated IRT on NBS were tested with 38 CFTR mutation panel and included. Clinical and laboratory database were analysed. In the first 6.5 years a total of 5,053 newborns (1.16% of total births) were screened with 38 CFTR panel. 170 CF affected cases, 320 unaffected carriers, 32 CF Screening Positive Inconclusive Diagnosis (CFSPID) were identified. There was one missed diagnosis. The most common disease-causing variant was c.1521_1523delCTT (p.(Phe508del)) followed by c.1652G>A (p.(Gly551Asp)). 95 out of 170 (55%) affected newborns were homozygous for c.1521_1523delCTT (p.(Phe08del)) and 25 (15%) carried at least one copy of c.1652G>A (p.(Gly551Asp)). Hence, 70% of affected newborns were eligible for CFTR modulator treatment. The NBS programme has identified almost triple the number of affected newborn with c.1652G>A (p.(Gly551Asp)) than previously quoted figures and identified less than 50% of carriers than predicted. The revised incidence and carrier frequency of CF in the ROI is 1 in 2570 and 1 in 25, respectively.
Similar content being viewed by others

Background
Cystic fibrosis (CF) is a life-limiting autosomal recessive multisystem disease caused by homozygous or compound heterozygous disease-causing variants in the CF transmembrane conductance regulator (CFTR) gene. CFTR encodes for a chloride ion channel that is expressed on epithelial cells of lungs, pancreas, liver, intestine, reproductive organ and in sweat glands. (MIM# 602421)
The Republic of Ireland (ROI) has previously reported the world’s highest CF incidence of 1 in 1353 and estimated carrier rate of 1 in 19 [1]. Newborn bloodspot screening for CF was incorporated in the National Newborn Bloodspot Screening (NBS) programme, commencing in July 2011. The benefit of early diagnosis is well documented [2, 3]. The purpose of newborn screening is to identify classic CF cases to allow early diagnosis and proactive multidisciplinary intervention which results in improved prognosis and better quality of life.
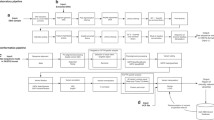
ROI CF NBS protocol is designed as followed and shown in Fig. 1. NBS card specimen is collected in the first 72–120 h of life and immunoreactive trypsinogen (IRT) is measured by the AutoDELFIA® Neonatal IRT assay kit in the NBS laboratory from dried bloodspot to screen for CF. Elevated trypsinogen, a pancreatic enzyme precursor, in the circulation reflects pancreatic duct obstruction and leakage [4]. In the ROI NBS programme, NBS cards with a raised IRT level and NBS cards alerted with clinical details indicative of meconium ileus or a suspicion of CF in the baby, regardless of IRT result, are referred to CFTR gene analysis. The IRT cut-off is based on the 99th centile (top 1%) and this cut-off has varied from 55 to 65 ng/ml since CF screening was implemented in July 2011. The cut-off is monitored quarterly as a key performance indicator. It is also reviewed with change of IRT kit lot post analysis of 10,000 samples. The cut-off is monitored every quarter to ensure that the percentage of patients being referred is within the target range of 1% [5]. If one or two CFTR variants are identified a sweat test is then carried out to confirm or out-rule CF in specialist CF centres.

Schematic diagram of ROI CF NBS protocol.
The NBS programme was designed to maximise identification of affected classic CF cases. Identifying 2 CFTR disease-causing variants and a positive sweat test confirms a diagnosis of CF. When only 1 CFTR variant is identified in the 38 CFTR variant panel in conjunction with a positive sweat test (sweat Cl− ≥ 60 mmol/L) a diagnosis of CF is made, and extended DNA sequencing is performed to confirm the 2nd variant. When only 1 CFTR variant is identified along with an intermediate sweat test result (sweat Cl− 30–59 mmol/L), a repeat sweat test is organised. When the repeat sweat test remains inconclusive, extended DNA sequencing is performed on peripheral blood, and the infant is assessed for clinical features of CF.
The NBS programme is also designed to minimise detection of unaffected carriers and CFSPID (CF screening positive, inconclusive diagnosis). The term CFSPID is reserved for individuals who screen positive without clinical features consistent with a diagnosis of CF. These newborns can have a positive NBS plus a normal sweat test and two CFTR variants, at least one of which has unclear phenotypic consequences (variant of unknown significance), or a positive NBS plus an intermediate sweat test with <2 CF causing variant [6, 7]. If one CFTR variant is identified followed by a normal sweat test the newborn is discharged as a CF carrier and the parents are referred for genetic counselling, which may include cascade CF carrier testing. Cascade testing includes the c.350G>A (p.(Arg117His)) variant, which can be associated with a milder phenotype when found in trans with a disease-causing CFTR variant and is not included on the 38 variant panel when performed as part of the second tier of testing of the infant’s bloodspot. This can result in the identification of further cases of CFSPID.
Methods
All neonates from July 2011 to Dec 2017 who had an elevated IRT on NBS or all NBS cards noted with clinical details indicative of meconium ileus or query CF regardless of IRT result were tested with a 38 CFTR variant panel (Luminex xTAG® Cystic Fibrosis 39 kit v2) (variants listed in Supplementary data), with c.350G>A (p.(Arg117His)) masked at software level and therefore not included on initial NBS reports. However, it is included in cascade testing of parents and discussed later in this study. Clinical data were collected retrospectively from patient charts and clinical genetics database software, iGene. Genetic testing records were obtained from a prospectively maintained departmental database (Crumbase) and analysed. Data on IRT level and sweat test results were matched up with The National Newborn Bloodspot Screening Laboratory. This study was done as a part of clinical audit and usual practice [8] and designed to assess the outcome and efficacy of CF NBS. Standard used: ECFS best practice guidelines—2018 revision [2]. The International nomenclature recommended by the Human Genome Variation Society (http://www.hgvs.org/mutnomen/) was followed. The variants are described using CFTR sequence accession number NC_000007.14 (NM_000492.3)(CFTR). Data described here were submitted to Leiden Open Variation Database (LOVD v3.0) (www.lovd.nl) and the URL link to the submitted data is provided in Supplementary data.
Results
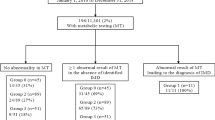
In the first 6 and a half years a total of 5053 newborns either had an elevated IRT or NBS cards noted with clinical details indicative of meconium ileus or query CF regardless of IRT result and all of them were screened with 38 CFTR disease-causing variants panel. This accounts for 1.16% of total 436,744 newborn who were screened with NBS (Total births of 436,940 and at least 196 NBS opted out during the study period). During this period, 170 CF affected cases, 320 unaffected carriers and 32 CFSPID were identified (Fig. 2).

Flow diagram of numbers of infants at each stage of CF NBS pathway.
To date, one additional CF case was missed by NBS. This newborn had co-existing malformations causing significant neonatal morbidity. Whole exome sequencing was performed to identify the cause of the malformations, revealed a rare homozygous CF variant that would not have been identified by our NBS 38 CFTR variant panel. As this case was identified outside of the NBS parameters it is not included in the final figures.
Of 170 newborns diagnosed as CF affected, 146 were picked up by the initial 38 CFTR variant panel and 24 patients required extended DNA sequencing (Fig 2). The most common CFTR variant identified in CF affected newborns was c.1521_1523delCTT (p.(Phe508del)). Of 170, 95 (56%) were homozygous and 59 (34.7%) were heterozygous for c.1521_1523delCTT (p.(Phe508del)) (Fig. 3).

Genotype identified in 170 CF affected newborns and the common 2nd CFTR variants in CF newborns with compound heterozygous with c.1521_1523delCTT (p.(Phe508del)).
The 2nd commonest disease-causing variant identified was c.1652G>A (p.(Gly551Asp)). Twenty five out of 170 (15%) affected newborns had c.1652G>A (p.(Gly551Asp)) on at least one allele; 3 were homozygous and 22 (13%) were compound heterozygous for c.1652G>A (p.(Gly551Asp)).
Of those 59 affected newborns with compound heterozygous with c.1521_1523delCTT (p.(Phe508del)), the most common variant in the other allele identified was c.1652G>A (p.(Gly551Asp)) (17, 10%) followed by c.489+1G>A (4, 2.36%), c.1679G>C (p.(Arg560Thr)) (4, 2.36%) and c.3718-2477C>T (3, 1.76%). (Fig. 3) Combining 25 newborns with at least one allele of c.1652G>A (p.(Gly551Asp)) and 95 newborns who were homozygous for c.1521_1523delCTT (p.(Phe508del)), a total of 120 out of 170 (70%) of CF affected newborns identified by NBS are potentially eligible for currently available CFTR modulators, such as either Ivacaftor or Lumacaftor/Ivacaftor.
The variants seen in CF carrier newborns were similar to that of CF affected newborns and the commonest variant was c.1521_1523delCTT (p.(Phe508del)) (78%) followed by c.1652G>A (p.(Gly551Asp)) (11%), c.489+1G>T (2%), c.1679G>C (p.(Arg560Thr)) (2%), c.1585-1G>A (1%) and further details are described (Fig. 4).

Breakdown of variants identified in CF carrier newborns.
There are 18 out of 170 (10.6%) affected infants born to at least one parent of self-reported non-Irish ethnicities identified in this period and the identified variants in this cohort is shown in Table 1. In this group, initial 38 NBS panel identified 2 CFTR variants in 14 affected newborns and 4 newborns required extended DNA sequencing to identify the 2nd CFTR variant and those variants are highlighted in Table 1 in bold.
Of 32 newborns identified as CFSPID; 6 newborns with 2 CFTR variants had at least one variant of unknown significant in addition to the disease-causing variant and all had a final sweat test which was normal; 17 had an intermediate sweat test in conjunction with one CF causing variant, triggering extended gene testing in which no 2nd variants were found. In addition, a further 9 newborns who were initially identified as CF carriers (one CFTR variant with a normal sweat test) were subsequently found to also carry the c.350G>A (p.(Arg117His)) variant in trans following cascade testing in their parents. All of them had 7T/9T variant. These have been grouped under CFSPID. The second commonest variant seen in CFSPID newborns was c.350G>A (p.(Arg117His)). (Supplementary Fig. 2)
Discussion
In the period of July 2011 to December 2017, the national newborn bloodspot screening programme identified 170 CF affected newborns, 320 unaffected carriers, 32 CFSPID and missed one affected CF case.
A cut-off point of the top 1% IRT level followed by a second tier of testing on a 38 CFTR gene panel molecular screening was recommended. The IRT cut-off level is a compromise between maximising detection of newborns with CF and minimising detection of unaffected carriers and CFSPID as well as scheme cost. The steering committee decided to prioritise sensitivity to detect CF cases over the avoidance of carriers and CFSPID detection. An IRT cut-off of top 1% allows detection of almost all newborns who will have classical CF, while minimising carrier detection. It was predicted in the modelling of the programme by the steering committee, that 53 infants with CF would be identified per annum, based on a livebirth rate of 75,000 per annum. This equates to a predicted estimate of 344 CF affected case expected in this study period. Clearly, the number of affected cases is lower than predicted likely due to several factors including incremental reduction in the birth rate (2017 livebirth rate was 62,000) and increasing number of births to non-Irish parents from countries with a low incidence of CF [9].
It was predicted that the programme would miss 0.8 affected CF cases per annum. This would equate to 5 cases over the study period, whereas we are only aware of one missed case. It was also predicted that 94 carriers would be identified per annum, but we only identified 320 over the study period which equates to 49 per annum. Although the overall numbers detected are considerably less than predicted in the original model, the proportion of 1 infant diagnosed with CF, for every 1.8 infants identified as carriers, is close to that predicted by the model of 1 case for every 1.6 carriers identified.
There is a steady increase in CF NBS across Europe and many countries incorporate CF NBS as part of a national screening programme [10]. It is reported that most of the programmes measure IRT with floating cut-off level ranging from 99.0th to 99.5th while some use fixed cut-off value between 60 and 200 ng/ml of IRT measured on dried bloodspot or a combination of IRT and pancreatitis associated protein measurements as the starting point. Significant variation in 2nd tier testing has been observed as some programmes assess 2nd IRT level on day 21 while others use sweat test or DNA analysis approach using a variant panel [10]. When compared the performances it is reported that the programme includes DNA variant panel resulted in improved positive predictive value while it will recognise more newborns with CFSPID [9]. In the ROI CF NBS positive predictive value derived from this period is 0.841 (84.1%) which is calculated as (total newborns affected with CF = 170)/(total newborns affected with CF (170) + total number of newborns with positive NBS i.e. elevated IRT and had positive sweat test and/or positive gene analysis [32] = 202). In addition, there is significant variation in the approach to the “safety net” with different countries employing a different cut-off to define the safety net threshold if tier 2 is negative (e.g., IRT > 99.9th centile, or an absolute IRT value greater than 60–200 ng/ml), and then employing different strategies to test the infants (repeat IRT on day 21, sweat test, extended gene analysis), with no clear consensus on the best approach, or whether it truly improves detection rates. In fact, there is some clear evidence that the “safety net” approach has a low yield [10, 11].
Our NBS has identified a total of 32 CFSPID in this given period and this represents a ratio of 19:1 (CF: CFSPID) or 0.63% of a total newborns screened. The sensitivity (true positive rate) calculated for this period according to the ECFS guideline is 99.42% (170/171) [2]. NBS is designed to identify classic CF cases who would benefit most from early intervention, those infants identified with CFSPID, or those infants who has c.350G>A (p.(Arg117His)) and other CF causing variant are considered false positives for the purpose of this study. The values for both positive predictive value and the sensitivity are well above the minimum recommended value by ECFS best practice guidelines [2].
More than 2000 CFTR disease-causing variants are reported and these are grouped in different classes due to their impact on the CFTR protein. In recent years genotype-directed disease-modifying small molecule therapies, CFTR modulators, have been approved in the European Union in 2012 and are available to treat CF patients [12]. Ivacaftor monotherapy are licenced for use in patients with at least one copy of a Class III CFTR affecting variant such as c.1652G>A (p.(Gly551Asp)). Lumacaftor combined with Ivacaftor is approved to use in CF patients homozygous for c.1521_1523delCTT (p.(Phe508del)) [13]. In our study, 95 out of 170 newborns were homozygous for c.1521_1523delCTT (p.(Phe508del)) and 25 affected newborns had at least one copy of c.1652G>A (p.(Gly551Asp)) variant. Hence, 120 of 170 (70%) of newborns identified in this period through NBS are potentially eligible for such CFTR modulator treatments.
The c.1652G>A (p.(Gly551Asp)) variant, which resulting in Class III or gating CFTR receptor defect in homozygous or compound heterozygous form, is reported to be in the top 5 common variants seen in CF patients in European countries and in the United States of America (USA) and its prevalence ranges from 1.4% to 5% [14, 15]. In contrast, the allele frequency of c.1652G>A (p.(Gly551Asp)) in CF affected newborns during this 6.5 year period in the ROI identified through NBS is 8.2% (28 alleles out of 340). There were a total of 25 out of 170 (15%) affected newborns with at least one copy of c.1652G>A (p.(Gly551Asp)) identified in this period; 3 newborns were homozygous for c.1652G>A (p.(Gly551Asp)), 17 newborns were compound heterozygous for c.1652G>A (p.(Gly551Asp)) and c.1521_1523delCTT (p.(Phe508del)), and 5 newborns were heterozygous for c.1652G>A (p.(Gly551Asp)) and another variants. Hence, we identified almost triple the number of affected newborns with c.1652G>A (p.(Gly551Asp)) variant than previously quoted figures from elsewhere in Europe and in the USA [14, 15]. As these children are potentially eligible for CFTR modulator treatments our data will help inform health ministry negotiating drug reimbursement nationally.
The observed incidence of CF in the NBS programme is 1 in 2570 (170 cases in 436,940 births) yielding an estimated carrier frequency of 1 in 25.3. The revised incidence is less than previous report of 1 in 1353. This is likely explained by recent net immigration resulting in 18 to 22% of births to parents of non-Irish origin over this study period [9]. However, we recommend that in case of counselling a couple of Irish origin carrier rate of 1 in 19 should still be used for more accurate counselling purposes.
Change history
05 July 2020
An amendment to this article has been published and can be accessed via a link at the top of the article.
References
Farrell P, Joffe S, Foley L, Canny GJ, Mayne P, Rosenberg M. Diagnosis of cystic fibrosis in the Republic of Ireland: epidemiology and costs. Ir Med J. 2007;100:557–60.
Castellani C, Duff AJA, Bell SC, Heijerman HGM, Munck A, Ratjen F, et al. ECFS best practice guidelines: the 2018 revision. J Cyst Fibros. 2018;17:153–78. https://doi.org/10.1016/j.jcf.2018.02.006.
Dankert-Roelse JE, Vernooij-van Langen A. Newborn screening for cystic fibrosis: pros and cons. Breathe. 2011;8:24–30. https://doi.org/10.1183/20734735.004111.
Castellani C, Massie J, Sontag M, Southern KW. Newborn screening for cystic fibrosis. Lancet Respiratory Med. 2016;4:653–61. https://doi.org/10.1016/S2213-2600(16)00053-9.
A Practical Guide to Newborn Bloodspot Screening In Ireland. https://www.hse.ie/eng/health/child/newbornscreening/newbornbloodspotscreening/information-for-professionals/a-practical-guide-to-newborn-bloodspot-screening-in-ireland.pdf.
Ren CL, Borowitz DS, Gonska T, Howenstine MS, Levy H, Massie J, et al. Cystic fibrosis transmembrane conductance regulator-related metabolic syndrome and cystic fibrosis screen positive, inconclusive diagnosis. J Pediatr. 2017;181S:S45–51.e1. https://doi.org/10.1016/j.jpeds.2016.09.066.
Munck A, Mayell SJ, Winters V, Shawcross A, Derichs N, Parad R, et al. Cystic Fibrosis Screen Positive, Inconclusive Diagnosis (CFSPID): a new designation and management recommendations for infants with an inconclusive diagnosis following newborn screening. J Cyst Fibros. 2015;14:706–13. https://doi.org/10.1016/j.jcf.2015.01.001.
Health Research Board, Ireland. www.hrb.ie.
Central Statistic Office, Vital Statistic Annual Report. 2016 https://www.cso.ie/en/releasesandpublications/ep/p-vsar/vitalstatisticsannualreport2016/births2016/.
Barben J, Castellani C, Dankert-Roelse J, Gartner S, Kashirskaya N, Linnane B, et al. The expansion and performance of national newborn screening programmes for cystic fibrosis in Europe. J Cyst Fibro. 2017;16:207–13. https://doi.org/10.1016/j.jcf.2016.12.012.
Massie J, Curnow L, Tzanakos N, Francis I, Robertson CF. Markedly elevated neonatal immunoreactive trypsinogen levels in the absence of cystic fibrosis gene mutations is not an indication for further testing. Arch Dis Child. 2006;91:222–5. https://doi.org/10.1136/adc.2005.081349.
Ong T, Ramsey BW. New therapeutic approaches to modulate and correct cystic fibrosis transmembrane conductance regulator. Pediatr Clin N Am. 2016;63:751–64. https://doi.org/10.1016/j.pcl.2016.04.006.
Ramsey BW, Davies J, McElvaney N, Tullis E, Bell SC, Drevinek P, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365:1663–72. https://doi.org/10.1056/NEJMoa1105185.
ECFS patient registry annual data report. 2017. https://www.ecfs.eu/sites/default/files/general-content-images/working-groups/ecfs-patient-registry/ECFSPR_Report2017_v1.3.pdf.
US Cystic Fibrosis Foundation National Registry annual report. 2017 https://www.cff.org/Research/Researcher-Resources/Patient-Registry/2017-Patient-Registry-Annual-Data-Report.pdf.
Acknowledgements
We would like to express our deep gratitude to Prof. Philip Mayne, the former Head of the National Newborn Bloodspot Screening Laboratory, Department of Paediatric Laboratory Medicine at Children’s Health Ireland at Temple Street Hospital.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Sasaki, E., Kostocenko, M., Lang, N. et al. National Newborn Screening for cystic fibrosis in the Republic of Ireland: genetic data from the first 6.5 years. Eur J Hum Genet 28, 1669–1674 (2020). https://doi.org/10.1038/s41431-020-0661-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-020-0661-5
This article is cited by
-
Evaluation of specificity and sensitivity of IRT/IRT protocol in the cystic fibrosis newborn screening program: 6-year experience of three tertiary centers
European Journal of Pediatrics (2022)
-
Setting a new standard in cystic fibrosis newborn screening illustrates controversial issues as new data emerge
European Journal of Human Genetics (2020)
