
Abstract
Genome-wide association studies (GWAS) have identified over 100 loci containing single nucleotide variants (SNVs) that influence the risk of developing multiple sclerosis (MS). Most of these loci lie in non-coding regulatory regions of the genome that are active in immune cells and are therefore thought to modify risk by altering the expression of key immune genes. To explore this hypothesis we screened genes flanking MS-associated variants for evidence of allele specific expression (ASE) by quantifying the transcription of coding variants in linkage disequilibrium with MS-associated SNVs. In total, we were able to identify and successfully analyse 200 such coding variants (from 112 genes) in both CD4+ and CD8+ T cells from 106 MS patients and 105 controls. Fifty-six of these coding variants (from 43 genes) showed statistically significant evidence of ASE in one or both cell types. In the Lck interacting transmembrane adaptor 1 gene (LIME1), for example, we were able to show that in both cell types, the MS-associated variant rs2256814 increased the expression of some transcripts while simultaneously reducing the expression of other transcripts. In CD4+ cells from an additional independent set of 96 cases and 93 controls we were able to replicate the effect of this SNV on the balance of alternate LIME1 transcripts using qPCR (p = 5 × 10–24). Our data thus indicate that some of the MS-associated SNVs identified by GWAS likely exert their effects on risk by distorting the balance of alternate transcripts rather than by changing the overall level of gene expression.
Similar content being viewed by others


Introduction
Multiple Sclerosis (MS) is an autoimmune disease of the central nervous system with a complex aetiology. Through genome-wide association screening (GWAS) more than 100 susceptibility loci have been identified with the vast majority mapping to non-coding regulatory regions of the genome that are active in immune cells [1, 2]. The fact that very few of the identified associations seem to be driven by protein coding changes suggests that most of these variants likely exert their effects by altering the expression of key genes in critically important cell types [3]. The overlap between MS-associated loci and epigenetic markers of active regulation is particularly high in T cells suggesting that many genes likely exert their relevant effects within this cell type [4]. Collaborative efforts such as the International Human Epigenome Consortium (IHEC, http://ihec-epigenomes.org/) and the Genotype-Tissue Expression Consortium (GTEx) [5] have firmly established the pervasive effects of genetic variation on gene expression and have shown that while many single nucleotide variants (SNVs) influence tightly linked proximal genes (cis effects) in a range of cell types, others exert cell type specific and longer range (trans) effects [6]. It is also recognised that some of these genotype dependent regulatory effects are only apparent in particular contexts, such as when cells are stimulated [7] or come under the influence of particular environmental factors [8]. Very little of these epigenetic data are disease specific, leaving unanswered questions regarding the effects of associated variants in the context of the disease to which they predispose. To explore more specifically the effects of MS-associated variants on the transcriptional regulation of proximal genes, we quantified the allele specific expression (ASE) of coding variants mapping within such genes that are in high linkage disequilibrium (LD) with their respective MS SNVs using a targeted next generation sequencing approach, which not only increases the sensitivity of detecting cis-regulatory effects but also allows for the detection of low abundance mRNA that can be missed in RNAseq analysis [9]. In heterozygous individuals, the transcription of the risk allele is quantified relative to the transcription of the alternate allele, which provides an internal control correcting for external factors such as trans-acting factors and environmental influences that can confound cis-expression quantitative trait loci (eQTL) analysis [10], particularly in the presence of negative feedback mechanisms. The ASE approach has been successfully applied in other diseases [11, 12] and is expected to be more sensitive and specific in the context of disease [13, 14]. Based on this we elected to search for ASE in lymphocyte subtypes that are likely to be relevant in MS (CD4+ and CD8+ T cells) in both healthy individuals and patients with MS.
Materials and methods
Samples
We recruited 202 individuals with MS from our local specialist Clinic and 198 healthy control individuals from the NIHR Cambridge BioResource. Cases and controls were all of Northern European descent and were matched for gender and age. Sample characteristics are outlined in Table 1. In the screening stage, we included 106 cases and 105 controls. The remaining 189 individuals (96 cases and 93 controls) were utilised for replication analysis. The study was approved by the Ethics Committee of the University of Cambridge (REC-11/33/0007, controls) and the NRES Committee South Central—Berkshire (REC-06/MRE12/11, cases) and all subjects gave fully informed valid consent. All of these subjects had been previously genotyped enabling identification of heterozygotes at each SNV of interest.
Cell isolation and cDNA synthesis
We collected 50 ml of heparinised venous blood from each study subject and then isolated peripheral blood mononuclear cells using Ficoll density-gradient centrifugation. CD4+ and CD8+ T cells were isolated using magnetic activated cell sorting according to the manufacturers’ instructions (Miltenyi Biotec). Specifically, CD3+ cells were negatively selected using a Pan T-Cell Isolation Kit followed by positive selection of CD8+ cells with the remaining fraction representing CD4+ cells. The purity of the separated cells was checked by flow cytometry for a subset of the samples, with a mean purity of >90% for CD4+ T cells and >95% for CD8+ T cells. The isolated cells were immediately lysed in TRIzol® reagent (Thermo Fisher Scientific), total RNA extracted according to the standard TRIzol protocol and genomic DNA contamination removed using DNase I treatment (Thermo Fisher Scientific). The extracted RNA was cleaned using the RNeasy MinElute Cleanup Kit (Qiagen), assessed for integrity using an Agilent 2100 Bioanalyser and then quantified using a Nanodrop 1000. For each subject 2 µg of total RNA was reverse transcribed using SuperScript III (Thermo Fisher Scientific) according to the manufacturer’s protocol with a 1:1 mixture of random hexamers and oligo dT primers. In parallel genomic DNA was isolated from peripheral blood utilising the DNeasey Blood and Tissue kit (Qiagen) and quantified using a Nanodrop 1000.
SNV selection
Considering genes lying within 0.25 cM of the 110-associated SNVs identified in the MS Immunochip study [2], and using the 1000 genomes CEU/GBR Phase I population data together with the Variant Effect Predictor tool in Ensembl release 76, we identified 305 coding variants that were in high LD with an MS-associated SNV (r2 > 0.7). Primers were designed to amplify the sequence containing the coding variants of interest in both cDNA and genomic DNA (gDNA). For a subset of SNVs (n = 29) sequence restrictions meant that independent primers had to be designed to amplify cDNA and gDNA. All primers were designed using Primer3Plus [15] and checked using the UCSC in-silico PCR tool. Due to sequence constraints we were unable to design primers for 36 of the SNVs and initial testing failed for a further 9. The remaining 260 variants were taken forward for sequencing. A full list of the SNVs considered is shown in Supplementary Table S1.
PCR amplification and sequencing
For each of the 260 coding SNVs we utilised the existing genotyping data to identify heterozygous individuals and then amplified the sequence containing the SNV in CD4+ and CD8+ T cell derived cDNA and the gDNA. PCR amplification was performed using Amplitaq Gold 360 mastermix containing either 10 ng CD4 or CD8 derived cDNA or 20 ng gDNA, 1 μM forward and 1 μM reverse primer in a total volume of 5 μl. The reaction conditions were 95 °C for 10 mins followed by 40 cycles of: 95 °C for 30 s, 60 °C for 30 s, 72 °C for 30 s and a final extension stage of 72 °C for 7 min. For each individual the PCR products from all SNV containing sequence fragments were combined into cell type specific pools, such that three pools were generated from each individual; CD4+ cDNA, CD8+ cDNA, and gDNA. These pooled PCR products were cleaned using ExoSAP-IT (Affymetrix) according to the manufacturer’s instructions. A second PCR to add the indexed Illumina adaptors allowing 96-plex pooling was then completed. This indexing PCR was completed in a total volume of 10ul, containing 2 μl of the cleaned cell specific pooled PCR product and 200 nM each of forward and reverse Illumina indexed primers. The reaction conditions were 95 °C for 10 min followed by 20 cycles of: 95 °C for 30 s, 54 °C for 30 s, 72 °C for 1 min and a final extension stage of 72 °C for 5 min. The 96 indexed samples (comprising 32 individuals × 3 pools) were combined and cleaned using QiaQuick PCR purification kit (Qiagen). The samples were then sequenced using PE300 reads on the Illumina MiSeq. Due to low library diversity, a phiX spike in of 20% and between 5 and 10 pM of product was loaded onto the MiSeq.
Data pre processing and ASE analysis
Adaptor sequences were removed using CutAdapt v1.16 [16] and the paired reads merged using FLASH v1.2.9 [17], low quality bases were trimmed using Trimmomatic v0.36 [18] and aligned to the human reference genome (Homo_sapiens.GRCh38.dna.primary_assembly) using BWA-MEM v0.7.17 [19]. Variant calling and allele read counts were generated using SAM tools v1.8 and BCF tools v1.8 [20]. At each variant site, samples with read counts <30 were excluded from analysis. The ratio of the risk allele to total read count was established for each SNV in the CD4, CD8, and gDNA sample. Given that the expected ratio is 0.5 in the gDNA sample, the observed ratio in this sample provided a means to compensate for any allele specific PCR bias in sequencing and/or reference allele alignment bias. Individual samples were excluded if the gDNA assay failed or if the observed gDNA allelic ratio deviated by more than 12.5% from the median across all other samples for that SNV; resulting in an average of three samples being excluded per tested coding SNV. The CD4+ and CD8+ ratios were then normalised according to the mean risk allele ratio seen in the gDNA for each SNV and the ASE effect size defined as the ‘normalised risk allele ratio—0.5’. Statistical support for ASE was tested using a paired two-tailed t test between the normalised gDNA and cDNA risk allele ratios. Statistical support for case-control and cell type specific differences in ASE was assessed using an un-paired two-tailed t test. A conservative Bonferroni correction factor of 1000 (200 SNPs × 5 tests) was applied giving a nominal statistical significance level of p < 5 × 10–5. These data have been deposited in the European Genome-phenome Archive (EGA) which is hosted by the EBI and the CRG, under accession number EGAS00001004139.
Replication analysis
To validate and replicate the ASE we observed in the LIME1 gene we repeated the analysis using qPCR in an independent second cohort of 96 cases and 93 controls. Specifically, we designed primers to detect rs914559 carrying transcripts (forward primer—GGCCCGAGGACGCTGTA, reverse primer—AGGTGGGTCCGCCTCAGTA, probe—6FAM-CGACCAGCCTTCCT) and rs2236510 carrying transcripts (forward primer—GGCAGAGCAGCCCTAGTTCA, reverse primer—TCTCCCGTTCCAGCTCTGACT, probe—6FAM-CCCCGCCAGAAGG) using Taqman methodology on a Quantstudio 7K Flex and data analysed using the Quantstudio Real-Time PCR Software. The PCR reaction was set up according to the manufacturer’s instructions with 20 ng of cDNA and normalised against the combined expression of the β-actin (ACTB) gene (Hs99999903_m1, Thermo Fisher Scientific) and 18S ribosomal RNA (Hs99999901_s1, Thermo Fisher Scientific) [21]. All samples were processed blind to genotype. A regression analysis of the resulting data including age and gender as covariates was completed in PLINK [22].
Results
To investigate the cis-regulatory effects of the MS-associated SNVs, we searched for evidence of ASE in coding SNVs that were strong proxies for MS-associated variants in CD4+ and CD8+ T cells collected from 106 MS patients and 105 controls. Targeted re-sequencing was attempted for 260 proxy coding variants. Following quality control (as described in the methods) we identified and excluded a total of 60 SNVs with inadequate data quality; 41 SNVs where there were <5 individuals with paired CD4 or CD8 and gDNA data, two SNVs with very extreme sequencing bias in the gDNA sample (risk allele ratio was <0.3 or >0.7) and 17 SNVs where it was impossible to accurately align sequence (most frequently because of pseudogenes). We successfully analysed the remaining 200 coding SNVs (from 112 genes) that were in high LD (r2 > 0.7) with 60 of the 110 MS-associated variants identified in the MS Immunochip study [2]. At a Bonferroni corrected conservative significance threshold of p < 5 × 10–5, we found statistically significant evidence for ASE at 38 of these SNVs in both cell types (see Table 2), 7 in CD4+ alone and 11 in CD8+ alone (see Supplementary Table S2). These 56 significant ASE variants were located in 29 of the 60 MS regions investigated and implicated 43 genes. The full list of all SNVs investigated and the final number of heterozygous samples analysed for each SNV is provided in Supplementary Table S2 and Supplementary Figures.
The strongest ASE we identified in CD4+ T cells was for the known splice site variant within the TNF Receptor Superfamily Member 1A gene (TNFRSF1A) rs1800693 (NC_000012.12:g.6330843T>C). Carriage of the risk allele (rs1800693_C) has previously been shown to increase the expression of a transcript lacking exon 6, which codes for a soluble version of the receptor [23, 24]. Using our approach, we could not directly interrogate this Δ6 transcript but were able to quantify the expression of the non-coding transcript ENST00000535038.1 as this includes the retained intron in which rs1800693 is located and therefore transcribed. We found that the MS risk allele results in increased expression of this non-coding transcript, with consistent ASE identified across all the heterozygous samples in both CD4+ cells (n = 97, ASEeffect = 0.17, p = 8.6 × 10–53) and CD8+ cells (n = 98, ASEeffect = 0.18, p = 1.1 × 10–53). Within the same region we also investigated a second proxy coding SNV within TNFRSF1A (rs767455; NC_000012.12:g.6341779T>C) which lies within exon 1 and captures all 11 known transcripts of TNFRSF1A and is in tight LD with rs1800693 (r2 = 0.79). However, for this SNV the evidence for ASE was not consistent across all heterozygous individuals and did not reach significance (see Supplementary Table S2). These data, illustrate the sensitivity of ASE to identify disease relevant transcripts in MS.
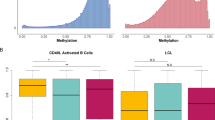
On Chromosome 20 we identified a novel region of ASE, where the genomic architecture of local genes conveniently intersected with the position of usable proxy variants thereby allowing us to quantify individual transcripts and subsets of transcripts. Within this region the MS-associated SNV rs2256814 (NC_000020.11:g.63742630G>A) is tagged by six proxy coding SNVs from three genes: two in the zinc finger CCCH-type and G-patch domain containing gene (ZGPAT); two in the Lck interacting transmembrane adaptor 1 gene (LIME1); and one in the SLC2A4 regulator gene (SLC2A4RG), this last variant being the MS-associated SNV rs2256814 itself. We found no statistically significant evidence for ASE in either ZGPAT or SLC2A4RG, whereas both the proxy coding variants in LIME1, rs914559 (NC_000020.11:g.63737947C>G) and rs2236510 (NC_000020.11:g.63737451C>T), showed statistically significant evidence for ASE (see Fig. 1). Notably these proxy variants lie in different transcripts and show opposite effects with respect to the MS risk allele, this allele increasing expression of transcripts containing rs914559 (ENST00000487026.5, ENST00000465591.1) and reducing expression of transcripts containing rs2236510 (ENST00000493265.2 and ENST00000621325.1). To confirm these initial findings, we designed transcript specific primers and completed an expression analysis using Taqman methodology. In an independent replication cohort of 96 cases and 93 controls we confirmed the effects of the MS risk variant on LIME1 transcript balance in CD4+ cells (see Fig. 2). Due to the exon structure surrounding the SNVs of interest, we could not establish whether the ASE was restricted to the protein coding or non-coding transcripts.

Box-whisker plots represent median, quartiles and 1.5× interquartile range. Statistically significant ASE is shown in grey, with the transcripts captured by the rs914559 SNV showing increased expression of the risk allele and those transcripts captured by rs2236510 showing decreased expression of risk allele.

Box-whisker plots represent median, quartiles and 1.5x interquartile range. a Quantitative PCR relative gene expression capturing ENST00000487026.5 and ENST00000465591.1 LIME1 transcripts with increased expression in rs914559_C risk allele carriers. b Quantitative PCR relative gene expression capturing ENST00000493265.2 and ENST00000621325.1 LIME1 transcripts with decreased expression in rs2236510_T risk allele carriers. c Ratio of the ENST00000487026.5/ENST00000465591.1: ENST00000493265.2/ENST00000621325.1 correlated with rs914559 genotype.
For 29 ASE SNVs common primers could not be designed for both the cDNA and gDNA (see Supplementary Table 1), therefore potential amplification bias or copy number variation could potentially bias the results. Of these 29 SNVs, 21 were successfully analysed and five showed significant evidence of ASE in either CD4 or CD8 cells: rs28445040 (NC_000002.12:g.230245867C>T) in SP140; rs1054283 (NC_000008.11:g.78698475A>G) in ZC2HC1A; rs6897932 (NC_000005.10:g.35874473C>T) in IL7R; rs1883832 (NC_000020.11:g.46118343T>C) in CD40 and rs8033595 (NC_000015.10:g.90540121G>A) in CRTC3. All of these SNVs have been identified as eQTLs or splice-QTLs in GTEx V8 (see Supplementary Table 2) with the direction of effect consistent to that we observed for all expect CRTC3.
Amongst our top ASE findings 17 relate to long non-coding RNA (lncRNA) transcripts. Such molecules are often only expressed at low levels [25] and are suggested to be involved in transcriptional regulation, particularly of the protein coding genes that they overlap in a cell type specific manner [26]. One of the most significant of such findings related to the SNV rs4078410 (NC_000002.12:g.112055265T>C) that we used as a proxy for the nearby MS-associated SNV (rs17174870; NC_000002.12:g.111907624C>T) and in the latest Gencode V32 release lies in the lncRNA AC093675.1. This lncRNA overlaps in an antisense direction in the 5’UTR of the transmembrane protein 87B gene (TMEM87B). Little is known about TMEM87B beyond its potential interaction with Human Papilloma Virus Type 18 [27] and its possible relevance in modulating endosome to trans-Golgi network transport [28]. At the same locus, and centromeric to TMEM87B, lies the MER proto-oncogene, tyrosine kinase gene (MERTK), which is perhaps a more obvious candidate for MS given its established role in the clearance of apoptotic cells by phagocytosis [29]. Future studies on the role of these lncRNAs in disease is required to establish their functionality and the gene(s) they may regulate.
Notably we saw no evidence for any corrected statistically significant difference in ASE between cases and controls, in either CD4+ or CD8+ T cells. However, one of the most highly ranked SNVs showing differential expression in CD4+ T cells between cases and controls was rs914559 in LIME1 (p = 0.005) where the ASE effect size was greater in MS patients (ASEeffect = 0.17) than in healthy controls (ASEeffect = 0.12). Likewise, only one corrected statistically significant cell type specific ASE was observed, which related to the coding proxy rs2272697 (NC_000004.12:g.102634835A>G) in the Mannosidase Beta gene (MANBA) where a significantly greater ASE bias was found in CD4+ cells compared with CD8+ cells (p = 5.3 × 10–6)). Amongst all the significant ASE identified the ASEeffect was always in the same direction in both CD4+ and CD8+ T cells. For the 18 SNVs where the evidence for ASE was significant in only one of the cell types (CD4 or CD8), the direction of ASE effect was consistent in the other cell type.
Discussion
In this targeted re-sequencing effort focused on coding proxies for MS-associated variants we have been able to identify statistically significant evidence for MS risk dependent ASE in 43 genes; thereby suggesting a potential role for these genes in the aetiology of the disease. The most striking novel ASE discovery was on chromosome 20q13 where the fortuitous existence of two coding proxies, each tagging different LIME1 mRNA transcripts, provides us with novel insights in to the effects of the MS-associated variant rs2256814 on LIME1 expression. LIME1 is a lipid raft-associated transmembrane adaptor protein which is highly expressed on T cells, interacts with Lck in mediating TCR signalling [30,31,32] and has also been implicated in BCR signalling [33]. Very little is known about the function of LIME1 however several mechanisms for its involvement in TCR signalling have been proposed. In one model LIME1 recruits signalling molecules for T-cell activation by functioning as a docking protein at the immunological synapse, with phosphorylation of tyrosine residues in the cytoplasmic tail of LIME1 following TCR stimulation leading to the recruitment of signalling molecules including PI3K, SHP2, Grb2 and Gads [32]. In a second model LIME1 interacts with the guanine nucleotide exchange factor VAV following stimulation which in turn converts Rac1 and Cdc42 to a GTP-bound active form required for cytoskeletal rearrangement and immunological synapse formation [31]. A role for LIME1 in controlling T cell responses by limiting the over-activation of antigen experienced T cells has also been suggested [30]. Within LIME1 we were able to show that the coding proxy risk variants rs914559 and rs2236510 alter the balance of transcripts from the LIME1 gene. The balance of RNA transcripts in a cell is a known and important regulatory mechanism for controlling (amongst other pathways) the immune response, for example within the interferon regulatory transcription factor 3 (IRF3) gene, multiple alternatively spliced transcripts provide a range of regulatory mechanisms to downregulate IRF3 responses [34,35,36]. The transcript structure of the LIME1 gene contained in Gencode V32 remains incomplete, so establishing the potential functional consequences of each transcript is not possible at present. Tissue specific differences in the expression of the multiple LIME1 transcripts is found in GTEx, with the highest expression of the non-coding LIME1 transcript captured by rs2236510 occurring in the cerebellum, and the highest expression of the protein coding transcript captured by rs914559 occurring in whole blood. The results from the splice QTL analysis in GTEx V8 correlate with our data, suggesting that the SNVs we have identified either directly influence alternative splicing of LIME1 transcripts or tag other SNVs that do this. In the 15 cell types contained in the Database of Immune Cell Expression, eQTLs, and Epigenomics project (DICE), the expression of LIME1 was highest in CD4 cells, in particular in T follicular helper cells, with little to no expression seen in monocytes and B cells [37]. Given the higher ASE bias in CD4+ T cells from MS cases, and the crucial role of this gene in regulating TCR signalling and T-cell responses, further exploring the differential expression of LIME1 transcripts, particularly in the context of stimulation is required.
Although our approach cannot distinguish cis expression QTLs from cis splice QTLs a number of the most extreme ASE we identified are with proxy SNVs that have previously been shown to alter splicing of MS relevant genes. Alongside rs1800693_C in the TNFRSF1A gene [23, 24] these include the synonymous coding variant rs28445040 within the SP140 nuclear body protein (SP140) gene [38], rs6897932 in the interleukin-7 receptor (IL7R) [39] and rs1883832 (which is in complete LD with the previously described rs4810485;NC_000020.11:g.46119308T>G) in the CD40 gene [40]. All of the above SNPs (aside from rs1883832 in the CD40 gene) have also been implicated as splice-QTLs in whole blood in GTEx (V8). Validation of these previously known findings provide a positive control for the sensitivity and specificity of the ASE assay we have used.
In several regions where more than one gene was investigated we could prioritise the potentially MS relevant gene. In one such region on Chromosome 12 where we were able to interrogate 11 suitable coding SNVs from five genes, we only found statistically significant evidence for ASE of the EEF1A lysine methyltransferase 3 (EEF1AKMT3, previously referred to as METTL21B) gene: with the MS risk haplotype reducing the expression of the gene, in concordance with previous findings [41]. Each of these five genes might be genetically considered to be equally implicated by the extensive LD flanking the local MS-associated variant rs10431552, but the ASE analysis suggest that it is only the expression of EEF1AKMT3 that is altered by this risk haplotype (at least in these cell types). In other regions, pinpointing the gene influenced by the associated variants has been more difficult. This is most evident in the IKAROS family zinc finger 3 (IKZF3)—ORMDL sphingolipid biosynthesis regulator 3 (ORMDL3) region on chromosome 17q12-q21 in the region implicated by the MS-associated variant rs12946510 (NC_000017.11:g.39756124C>T). Two previous studies in MS have explored this region and reported correlation of the disease associated variant with either IKZF3 [42] or gasdermin B (GSDMB) [43] but neither study explored the other genes in the region. In our study we identified ASE for all three tested genes in the region, IKZF3, GSDMB and the zona pellucida binding protein gene 2 (ZPBP2), with the greatest ASE bias observed in GSDMB. Detailed exploration of this region in asthma established that the common genetic variant associated with asthma (rs7216389; NC_000017.11:g.39913696C>T r2 = 0.7 with the MS SNV rs12946510) is involved in regulating the expression of multiple genes within this region through nucleosome repositioning and CTCF binding [44]. In such regions where extensive LD and multiple regulatory variants exist, correlating the disease associated variants with gene expression is particularly challenging and caution needs to be taken in the interpretation of the results [45, 46].
As we only considered CD4 and CD8 T-cell types we were not able to directly assess regulatory effects in other cell types such as monocytes and B cells. It has been shown that the difference in ASE between individuals is greater than it is between cell types within an individual, thus suggesting a common regulatory factor across cell types within an individual [47]. Interestingly several of our top findings relate to genes that are most highly expressed in B cells (for example FCRL3 and SP140). Further work is therefore required to investigate the role of these SNVs in different cell types.
Our findings confirm that many of the disease associated SNVs identified by GWAS influence gene expression, in many instances by altering the balance of the alternate transcripts produced. Although our approach only allows the assessment of ASE in genes that contain common coding variants in LD with associated SNVs it has proven to be informative with regard to allelic imbalance that could otherwise have been missed in global transcriptomic studies and provides a list of prioritised candidate genes. The identification of divergent ASE in specific mRNA transcripts of the LIME1 gene highlights the value and importance of assessing the expression of individual transcripts and adds to the growing evidence suggesting the importance of altered transcriptional balance as a mechanism by which associated variants exert their effects on proximal gene expression. Increasing our understanding of these mechanisms will help drive the translation of GWAS discoveries in MS.
References
IMSGC and WTCCC2 Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011;476:214–9.
Beecham AH, Patsopoulos NA, Xifara DK, Davis MF, Kemppinen A, Cotsapas C, et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet. 2013;45:1353–60.
Sawcer S, Franklin RJ, Ban M. Multiple sclerosis genetics. Lancet Neurol. 2014;13:700–9.
Raj T, Rothamel K, Mostafavi S, Ye C, Lee MN, Replogle JM, et al. Polarization of the effects of autoimmune and neurodegenerative risk alleles in leukocytes. Science. 2014;344:519–23.
GTEx Consortium. Genetic effects on gene expression across human tissues. Nature. 2017;550:204–13.
Grundberg E, Small KS, Hedman AK, Nica AC, Buil A, Keildson S, et al. Mapping cis- and trans-regulatory effects across multiple tissues in twins. Nat Genet. 2012;44:1084–9.
Fairfax BP, Humburg P, Makino S, Naranbhai V, Wong D, Lau E, et al. Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science. 2014;343:1246949.
Moyerbrailean GA, Richards AL, Kurtz D, Kalita CA, Davis GO, Harvey CT, et al. High-throughput allele-specific expression across 250 environmental conditions. Genome Res. 2016;26:1627–38.
Zhang R, Li X, Ramaswami G, Smith KS, Turecki G, Montgomery SB, et al. Quantifying RNA allelic ratios by microfluidic multiplex PCR and sequencing. Nat Methods. 2014;11:51–54.
Pastinen T. Genome-wide allele-specific analysis: insights into regulatory variation. Nat Rev Genet. 2010;11:533–8.
Locke JM, Hysenaj G, Wood AR, Weedon MN, Harries LW. Targeted allelic expression profiling in human islets identifies cis-regulatory effects for multiple variants identified by type 2 diabetes genome-wide association studies. Diabetes. 2015;64:1484–91.
Xu X, Wang H, Zhu M, Sun Y, Tao Y, He Q, et al. Next-generation DNA sequencing-based assay for measuring allelic expression imbalance (AEI) of candidate neuropsychiatric disorder genes in human brain. BMC Genomics. 2011;12:518.
Tan AC, Fan JB, Karikari C, Bibikova M, Garcia EW, Zhou L, et al. Allele-specific expression in the germline of patients with familial pancreatic cancer: an unbiased approach to cancer gene discovery. Cancer Biol Ther. 2008;7:135–44.
Baranzini SE, Mudge J, van Velkinburgh JC, Khankhanian P, Khrebtukova I, Miller NA, et al. Genome, epigenome and RNA sequences of monozygotic twins discordant for multiple sclerosis. Nature. 2010;464:1351–6.
Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R, Leunissen JA. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007;35:W71–74.
Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal. 2011;17:10–12.
Magoc T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–63.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9.
Riedel G, Rudrich U, Fekete-Drimusz N, Manns MP, Vondran FW, Bock M. An extended DeltaCT-method facilitating normalisation with multiple reference genes suited for quantitative RT-PCR analyses of human hepatocyte-like cells. PLoS ONE. 2014;9:e93031.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Gregory AP, Dendrou CA, Attfield KE, Haghikia A, Xifara DK, Butter F, et al. TNF receptor 1 genetic risk mirrors outcome of anti-TNF therapy in multiple sclerosis. Nature. 2012;488:508–11.
Ottoboni L, Frohlich IY, Lee M, Healy BC, Keenan BT, Xia Z, et al. Clinical relevance and functional consequences of the TNFRSF1A multiple sclerosis locus. Neurology. 2013;81:1891–9.
Palazzo AF, Lee ES. Non-coding RNA: what is functional and what is junk? Front Genet. 2015;6:2.
Hon CC, Ramilowski JA, Harshbarger J, Bertin N, Rackham OJ, Gough J, et al. An atlas of human long non-coding RNAs with accurate 5' ends. Nature. 2017;543:199–204.
Li S, Liu P, Xi L, Jiang X, Wu M, Deng D, et al. Expression of TMEM87B interacting with the human papillomavirus type 18 E6 oncogene in the Hela cDNA library by a yeast two-hybrid system. Oncol Rep. 2008;20:421–7.
Hirata T, Fujita M, Nakamura S, Gotoh K, Motooka D, Murakami Y, et al. Post-Golgi anterograde transport requires GARP-dependent endosome-to-TGN retrograde transport. Mol Biol Cell. 2015;26:3071–84.
Scott RS, McMahon EJ, Pop SM, Reap EA, Caricchio R, Cohen PL, et al. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature. 2001;411:207–11.
Brdickova N, Brdicka T, Angelisova P, Horvath O, Spicka J, Hilgert I, et al. LIME: a new membrane Raft-associated adaptor protein involved in CD4 and CD8 coreceptor signaling. J Exp Med. 2003;198:1453–62.
Son M, Park I, Lee OH, Rhee I, Park C, Yun Y. LIME mediates immunological synapse formation through activation of VAV. Mol Cells. 2012;33:407–14.
Hur EM, Son M, Lee OH, Choi YB, Park C, Lee H, et al. LIME, a novel transmembrane adaptor protein, associates with p56lck and mediates T cell activation. J Exp Med. 2003;198:1463–73.
Ahn E, Lee H, Yun Y. LIME acts as a transmembrane adapter mediating BCR-dependent B-cell activation. Blood. 2006;107:1521–7.
Karpova AY, Ronco LV, Howley PM. Functional characterization of interferon regulatory factor 3a (IRF-3a), an alternative splice isoform of IRF-3. Mol Cell Biol. 2001;21:4169–76.
Li C, Ma L, Chen X. Interferon regulatory factor 3-CL, an isoform of IRF3, antagonizes activity of IRF3. Cell Mol Immunol. 2011;8:67–74.
Li Y, Hu X, Song Y, Lu Z, Ning T, Cai H, et al. Identification of novel alternative splicing variants of interferon regulatory factor 3. Biochim Biophys Acta. 2011;1809:166–75.
Schmiedel BJ, Singh D, Madrigal A, Valdovino-Gonzalez AG, White BM, Zapardiel-Gonzalo J, et al. Impact of genetic polymorphisms on human immune cell gene expression. Cell. 2018;175:1701–15 e1716.
Matesanz F, Potenciano V, Fedetz M, Ramos-Mozo P, Abad-Grau Mdel M, Karaky M, et al. A functional variant that affects exon-skipping and protein expression of SP140 as genetic mechanism predisposing to multiple sclerosis. Hum Mol Genet. 2015;24:5619–27.
Gregory SG, Schmidt S, Seth P, Oksenberg JR, Hart J, Prokop A, et al. Interleukin 7 receptor alpha chain (IL7R) shows allelic and functional association with multiple sclerosis. Nat Genet. 2007;39:1083–91.
Field J, Shahijanian F, Schibeci S, Australia, MSGC NewZealand, Johnson L, et al. The MS risk allele of CD40 is associated with reduced cell-membrane bound expression in antigen presenting cells: implications for gene function. PLoS ONE. 2015;10:e0127080.
Shahijanian F, Parnell GP, McKay FC, Gatt PN, Shojoei M, O'Connor KS, et al. The CYP27B1 variant associated with an increased risk of autoimmune disease is underexpressed in tolerizing dendritic cells. Hum Mol Genet. 2014;23:1425–34.
Keshari PK, Harbo HF, Myhr KM, Aarseth JH, Bos SD, Berge T. Allelic imbalance of multiple sclerosis susceptibility genes IKZF3 and IQGAP1 in human peripheral blood. BMC Genet. 2016;17:59.
Cardamone G, Paraboschi EM, Rimoldi V, Duga S, Solda G, Asselta R. The characterization of GSDMB splicing and backsplicing profiles identifies novel isoforms and a circular RNA that are dysregulated in multiple sclerosis. Int J Mol Sci. 2017;18:576.
Verlaan DJ, Berlivet S, Hunninghake GM, Madore AM, Lariviere M, Moussette S, et al. Allele-specific chromatin remodeling in the ZPBP2/GSDMB/ORMDL3 locus associated with the risk of asthma and autoimmune disease. Am J Hum Genet. 2009;85:377–93.
Locke JM, Wei FY, Tomizawa K, Weedon MN, Harries LW. A cautionary tale: the non-causal association between type 2 diabetes risk SNP, rs7756992, and levels of non-coding RNA, CDKAL1-v1. Diabetologia. 2015;58:745–8.
Farh KK, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2015;518:337–43.
Gutierrez-Arcelus M, Ongen H, Lappalainen T, Montgomery SB, Buil A, Yurovsky A, et al. Tissue-specific effects of genetic and epigenetic variation on gene regulation and splicing. PLoS Genet. 2015;11:e1004958.
Acknowledgements
We gratefully acknowledge the participation of all NIHR Cambridge BioResource volunteers and thank the NIHR Cambridge BioResource centre and staff for their contribution. We also thank the National Institute for Health Research and NHS Blood and Transplant.
Funding
This work was supported by the UK Medical Research Council (grant reference: G1100125) and the Cambridge NIHR Biomedical Research Centre.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Compliance with ethical standards
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Ban, M., Liao, W., Baker, A. et al. Transcript specific regulation of expression influences susceptibility to multiple sclerosis. Eur J Hum Genet 28, 826–834 (2020). https://doi.org/10.1038/s41431-019-0569-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-019-0569-0
This article is cited by
-
SLAMF7 modulates B cells and adaptive immunity to regulate susceptibility to CNS autoimmunity
Journal of Neuroinflammation (2022)
-
The genetics of autoimmune Addison disease: past, present and future
Nature Reviews Endocrinology (2022)
-
Transcript-specific regulation in T-cells in multiple sclerosis susceptibility
European Journal of Human Genetics (2020)
