
Abstract
Bicuspid aortic valve (BAV) is the most common congenital heart defect (CHD), affecting 1–2% of the population. BAV is associated with thoracic aortic aneurysms (TAAs). Deleterious copy number variations (CNVs) were found previously in up to 10% of CHD cases. This study aimed at unravelling the contribution of deleterious deletions or duplications in 95 unrelated BAV/TAA patients. Seven unique or rare CNVs were validated, harbouring protein-coding genes with a role in the cardiovascular system. Based on the presence of overlapping CNVs in patients with cardiovascular phenotypes in the DECIPHER database, the identification of similar CNVs in whole-exome sequencing data of 67 BAV/TAA patients and suggested topological domain involvement from Hi-C data, supportive evidence was obtained for two genes (DGCR6 and TBX20) of the seven initially validated CNVs. A rare variant burden analysis using next-generation sequencing data from 637 BAV/TAA patients was performed for these two candidate genes. This revealed a suggestive genetic role for TBX20 in BAV/TAA aetiology, further reinforced by segregation of a rare TBX20 variant with the phenotype within a BAV/TAA family. To conclude, our results do not confirm a significant contribution for deleterious CNVs in BAV/TAA as only one potentially pathogenic CNV (1.05%) was identified. We cannot exclude the possibility that BAV/TAA is occasionally attributed to causal CNVs though, or that certain CNVs act as genetic risk factors by creating a sensitised background for BAV/TAA. Finally, accumulative evidence for TBX20 involvement in BAV/TAA aetiology underlines the importance of this transcription factor in cardiovascular disease.
Similar content being viewed by others


Introduction
Bicuspid aortic valve (BAV) is the most common congenital heart malformation, affecting 1–2% of the general population. It is characterised by abnormal fusion of two aortic valve leaflets during embryonic development [1]. Although most BAV patients remain asymptomatic, 35% develop severe cardiovascular complications such as coarctation of the aorta (CoA), thoracic aortic aneurysms (TAAs) and dissections (TAADs) [2]. Owing to the current lack of curative therapies and accurate predictive biomarkers, the two latter manifestations are associated with high morbidity and mortality rates. Prophylactic aortic surgery can be life-saving, but requires early TAA identification and continuous cardiovascular surveillance [1]. To develop novel medical therapies, acquisition of more detailed insights in the aetiology of BAV/TAA is indispensable.
Historically, TAA development in BAV patients was considered to be the sole result of abnormal haemodynamic flows downstream of the BAV causing increased shear stress on the aortic wall. Meanwhile, many studies have pointed towards the involvement of genetic factors in the aetiology of BAV/TAA [1, 3, 4]. Despite the high heritability of human BAV (89%) [5] and numerous efforts to pinpoint the genetic causes for BAV/TAA, only a few disease genes have been published such as NOTCH1 [6] and SMAD6 [7]. Moreover, these genes explain only a small fraction of all BAV/TAA patients.
Although most genetic studies in hereditary diseases are focussing on single-nucleotide variation, copy number variations (CNVs) are a major source of human genetic variability [8], and have been shown to contribute to a multitude of diseases [9, 10]. An increased burden of (likely) pathogenic CNVs has been found in patients with congenital heart defects (7.8–8.8%) compared to controls (4.3–4.4%) [10, 11]. Family-based CNV studies for congenital heart diseases (CHDs) have also demonstrated that both inherited and de novo copy number events associate with disease [12, 13]. Overall, de novo and/or inherited CNVs are estimated to cause non-syndromic CHD in 5–10% of the probands [12,13,14]. Only a minority of the cases included in the above described studies presents with BAV, BAV/TAA or isolated TAA(D) (Supplementary Table 1). With regards to BAV/TAA, a recent study demonstrated an enrichment of the total number of CNVs per individual in BAV patients with early-onset TAAD compared to patients with a tricuspid aortic valve and early-onset TAAD [15]. Patients with Turner syndrome (TS; 45X0) have also been shown to present with larger (>200 kb) and more rare CNVs as compared to controls (p < 0.003 and p < 0.005, respectively) [16]. This is particularly relevant for BAV/TAA, since one-third of patients with TS presented with BAV, of which 25% develop TAA [17, 18].
We hypothesise that CNVs might explain, at least partially, the unknown genetic aetiology of BAV/TAA. We aimed at unravelling the contribution of unique and rare deleterious CNVs in patients with BAV/TAA using a genome-wide SNP array approach.
Methods and materials
Study population
The cohort consisted of 95 unrelated BAV/TAA patients who had been selected based on the following inclusion criteria: (1) BAV (~echocardiography) and (2) aortic diameter at the sinus of Valsalva or ascending aorta of at least 40 mm in adults, or a Z-score exceeding 3 in children (~echocardiography, computed tomography or magnetic resonance imaging). A positive family history was defined as having at least one first- or second-degree relative with BAV and/or TAA. Patients were recruited in four different centres (Antwerp, Montreal, Stockholm, Lübeck). Peripheral blood was collected for genomic DNA (gDNA) extraction from leukocytes. The local ethics committees approved the study protocol and all participants gave informed consent. The overall workflow is schematically shown in Fig. 1.

Flowchart of our strategy and tools used to identify causative CNVs for BAV/TAA. Genome-wide raw microarray data of 95 unrelated BAV/TAA patients were analysed with GenomeStudio software and an in-house developed online tool called the CNV-Webstore. Rare and unique CNVs with protein-coding genes with a potential role in the cardiovascular system and with a low frequency in healthy individuals (MAF < 1%) were prioritised using data of control samples, NCBI gene, OMIM, PubMed, Human Protein Atlas, Zebrafish Information Network and Mouse Genome Informatics. Only validated CNVs were further investigated. Additional genetic evidence was searched by looking for overlapping CNVs in similar cases in DECIPHER and in-house WES (whole-exome sequencing) data of 67 BAV/TAA patients on which CNV-calling was performed (unpublished data). For CNVs not affecting our gene of interest, public available Hi-C data were consulted for the presence of a potential topological associated domain (TAD), minor allele frequency (MAF) boundary (promoter.bx.psu.edu/hi-c/). In case of supportive evidence, more genetic data for the involvement of the gene of interest in the BAV/TAA pathology by means of a variant burden analysis on rare deleterious next-generation sequencing (NGS) variants detected in 637 BAV/TAA patients and the gnomAD database. BAV bicuspid aortic valve, TAA thoracic aortic aneurysm, SNP single-nucleotide polymorphism, CNV copy number variation, MLPA multiplex ligation-dependent probe amplification, MAQ multiplex amplicon quantification, qPCR quantitative polymerase chain reaction, NCBI National Center for Biotechnology Information, OMIM Online Mendelian Inheritance in Man, TAD topological associated domain, MAF minor allele frequency
CNV analysis and data filtering
CNV analysis was carried out with the HumanCytoSNP-12 BeadChip (Illumina, San Diego, USA), which allows genotyping of 298 563 SNP markers (mean probe spacing, 7.5kb). The BeadChip was scanned by the iScan Reader (Illumina, San Diego, USA) and the raw data were processed using the Illumina GenomeStudio software package. CNVs were subsequently identified with CNV-Webstore [19] (http://cnv-webstore.ua.ac.be/cnv-webstore), an in-house developed tool for CNV detection, visualisation, interpretation and storage. This web-based tool enables the identification of rare small scale CNVs of at least three consecutive SNPs by combining existing methods (QuantiSNPv1.1, PennCNV rev081119 and VanillaICEv1.4.0) in a majority vote-based calling as described elsewhere [19]. The tool reports minimal affected regions, corresponding to the first and last affected probes, and maximal affected regions, corresponding to the last and first unaffected probes, respectively. Unique and rare CNVs (i.e. minor allele frequency below 1% in the HapMap project database and the Toronto database of Genomic Variants [20]) affecting protein-coding genes with a role in the cardiovascular system were selected for further genetic follow-up. Selection of protein-coding genes with a role in the cardiovascular system was done through manual inspection of the literature (https://www.ncbi.nlm.nih.gov/pubmed) as well as databases on established genotype–phenotype correlations (https://www.ncbi.nlm.nih.gov/gene, http://omim.org/), gene expression profiles (http://www.proteinatlas.org/) and animal models (http://www.informatics.jax.org/, https://zfin.org/).
CNV validation, fine mapping and genetic follow-up
Validation and breakpoint refinement of selected CNVs involved both confirmation of the called CNV, and verification of actual involvement of the gene of interest in the CNV, by experimentally refining the called CNV boundaries. Three independent methods were available: Multiplex Amplicon Quantification (MAQ) (Flagged You-MAQ control Assay, Agilent, Niel, Belgium), Multiplex Ligation-dependent Probe Amplification (MLPA, SALSA MLPA EK reagent kit, MRC-Holland, Amsterdam, Nederland) or quantitative PCR (qPCR, qPCR Mastermix Plus for SYBR Green I, Eurogentec S.A., Seraing, Belgium). Primers and probes were developed with online available tools (https://tools.multiplicom.com/maq-design/index.html; http://osa-bioinform.uhmc.sunysb.edu/mlpa2/cgi-bin/mlpa.cgi; http://primer3.ut.ee/) and specialised software packages were used for data analysis (MAQ-S Software, Agilent, Niel, Belgium; GeneMarker, SoftGenetics, LLC, State College, USA; qBasePLUS, http://www.qbaseplus.com).
Additional genetic evidence for pathogenicity of the selected CNVs was acquired by looking into the presence of overlapping CNVs in patients with a similar phenotype using (1) DECIPHER [21] and (2) in-house available CNV-called whole-exome sequencing (WES) data of 67 unrelated BAV/TAA patients using eXome-Hidden Markov Model (XHMM) with default parameters on the joined coverage estimates and minimum quality of 70 (unpublished data) [22,23,24,25]. Moreover, if validated CNVs were not affecting the gene of interest, online available Hi-C data of Human Umbilical Vein Endothelial Cell (HUVEC) (promoter.bx.psu.edu/hi-c/) were checked for CNV-afflicted disturbances of topological associated domains (TADs) and boundaries. The candidate CNVs listed in Table 3 were submitted to the ClinVar repository (SCV000854402—SCV000854408).
Replication of candidate BAV/TAA genes
For the most interesting candidate genes, i.e. those implicated in more than one CNV and those for which an adjacent TAD boundary was disturbed by the CNV, additional genetic evidence for involvement in the BAV/TAA pathology was pursued via a single nucleotide variant burden analysis. In an in-house available WES (n = 196) and targeted gene panel (n = 441) dataset of 637 BAV/TAA patients (unpublished data; identical patient inclusion criteria), were filtered and analysed as previously described [7]. The gene panel consisted of validated CNV-selected protein-coding genes (Table 3, genes in bold). The genomes from the gnomAD database (mean coverage > 40× per gene) were filtered in an identical manner and served as the control cohort. Statistics involved the two-tailed χ2 test with Yates correction. Prediction programmes (CADD, MutationTaster2, Polyphen2, SIFT) were used to evaluate the pathogenic effect of the identified variants. Finally, segregation analysis of identified variants was performed if DNA of affected or unaffected relatives was available. Variants identified in the candidate BAV/TAA gene(s) were deposited in the ClinVar database (SCV000854409—SCV000854412).
Results
CNV analysis: filtering and validation
A genome-wide microarray-based CNV analysis was performed in 95 unrelated BAV/TAA patients in order to reveal the contribution of rare CNVs to the genetic aetiology of BAV/TAA (Table 1). The average age at inclusion of cases was 49.5 ± 13.1 years and 79% of patients were male, complying with the known BAV 3:1 male preponderance. For the patients with known family history, positive family history was reported for 24% of cases (n = 11/46). Left–right fusion was most commonly observed (40%, n = 38), followed by the left–right/right-non-coronary subtype (21%, n = 20). Besides the aortic aneurysm, 86% of the patients presented with at least one additional cardiovascular finding, i.e. aortic valve stenosis (n = 48), aortic valve insufficiency (n = 60), coarctation of the aorta (n = 3), hypertrophic cardiomyopathy (n = 2) and left ventricular hypertrophy (n = 1).
Upon inspection of the general characteristics of the high-quality yet otherwise unfiltered CNVs, we observed on average 15 CNVs in each BAV/TAA patient (Table 2). Duplications were more common (56% versus 44%) and larger than deletions ((134–948 kb) versus (48–120 kb)). Per patient, the copy number of on average 17.2 genes was altered, of which on average 12.2 genes were duplicated/deleted in more than one patient. The latter genes were located in known hypervariable regions coding for olfactory receptors, components of the immune system and microRNAs. Subsequent filtering for unique or rare CNVs (MAF <1%) of which the encoded protein exerts a function in the cardiovascular system (Supplementary Table 2) resulted in a selection of 16 prioritised CNVs. Further validation with an independent technique (MLPA, MAQ or qPCR) confirmed seven of these CNVs (6 duplications and 1 deletion), and fine mapping revealed that four CNVs truly affect the coding region of the gene of interest (KLF12, FBN3, ZNF626, DGCR6) whereas three other CNVs did not (RHOU with WNT3A, TBX20 and NOMO3) (Table 3). All protein-coding genes within rare CNVs not surviving our gene-based filter strategy are summarised in Supplementary Table 3.
From candidate CNVs to candidate genes
Candidate CNVs affecting the coding region of the gene of interest
The three genes affected by a duplication were KLF12, FBN3 and ZNF626, while the deletion contained the DGCR6 gene (Table 3). The first duplication was affecting the 3′ UTR, last exon and partially last intron of KLF12, a gene coding for a transcriptional regulator predominantly expressed in arterial endothelial cells [26, 27]. KLF12 was considered an interesting candidate gene as it was reported to be downregulated in aneurysmal tissue of early ruptured intracranial aneurysms (IA) as compared to unruptured IA [28]. The second region was a duplication partially encompassing the 5′ UTR and exon 1 of FBN3, which encodes an extracellular matrix protein expressed in the endocardial cushions and blood vessels [29]. The last duplicated region contained exon 1 and the 5′ UTR of ZNF626, of which little is known besides that it codes for a transcription factor with expression in the cardiovascular system. In DECIPHER, no supportive evidence for a role in CHD or aortopathy was found for the CNVs affecting KLF12 and FBN3, as overlapping CNVs (deletions or duplications) were only observed in highly dissimilar diseases without cardiovascular involvement. Conversely, two similar ZNF626 duplications as the one we found, were either identified in a patient with a dilatation of the ascending aorta and abnormality of the mitral valve and in another patient presenting with an unspecified abnormality of the cardiovascular system morphology in addition to multi-systemic features like butterfly vertebrae, failure to thrive and horseshoe kidney. In DECIPHER, 14 other CNVs overlapping ZNF626 were reported as likely benign or benign in patients with non-cardiovascular disease (Supplementary Figure 1C). In-house CNV-calling on WES data of 67 BAV/TAA patients identified no extra CNVs for KLF12, FBN3 and ZNF626. In addition, Hi-C data of HUVECs (promoter.bx.psu.edu/hi-c/) did not indicate the presence of a TAD for any of the three aforementioned regions, suggesting no regulatory effect via the disruption of chromatin structure for any of these duplications (Supplementary Figure 2A, B, C).
The fourth candidate CNV was a deletion encompassing several genes, of which DGCR6 was the most interesting one (Table 3). Since two functional copies of DGCR6 exist due to a duplication of an ancestral locus [30], this deletion was expected to result in 25% protein loss. Numerous investigations have implied a potential role for DGCR6 in neural crest cell migration [31], of which some studies hypothesised that this gene can act as a modifier for the cardiovascular phenotype observed in patients with DiGeorge syndrome [32, 33]. This syndrome is caused by a 1.5 to 3.0 Mb hemizygous deletion of chromosome 22q11.2, and haplo-insufficiency of TBX1 is thought to be the major disease cause [34, 35]. In the literature, one paper described a patient with DiGeorge syndrome and BAV [36]. In our BAV/TAA patient, TBX1 was not structurally affected by the CNV. The DECIPHER database showed that 230 out of 469 (49%) of the overlapping CNVs occur in highly dissimilar diseases without cardiovascular abnormalities. In-house CNV-calling on WES data of 67 BAV/TAA patients identified three MAQ-validated additional DGCR6-containing duplications leading to a total CNV incidence affecting this gene of 2.5% (4 out of 162) in our BAV/TAA series. Finally, Hi-C data of HUVECs predicted a TAD boundary at the locations of the DGCR6 deletion and duplications (Fig. 2a). DNA of only one affected relative of the four families was available for the segregation analysis. No supportive evidence for full pathogenicity was shown as this BAV/TAA individual did not carry the structural variant containing DGCR6.

Extra genetic evidence for CNV involvement: DGCR6 and TBX20. a Location of DGCR6 gene on chromosome 22. CNV-calling on WES data of 67 BAV/TAA patient identified three validated DGCR6-containing duplications, as marked with a blue bar. The red bar represents the initial deletion identified in a BAV/TAA patient using microarray. Hi-C data of HUVECs predicted a TAD boundary at the locations of the DGCR6 deletion and duplication. b Location of TBX20 gene on chromosome 7. In DECIPHER, two overlapping deletions, marked by red bars, were identified in patients with a cardiovascular feature, i.e. atrial septal defect and ventricular septal defect. Hi-C data in HUVECs were suggestive for a TAD boundary near TBX20 that is affected by the CNV
Candidate CNVs not affecting the coding region of the gene of interest
For three out of the seven candidate CNVs, we were not able to demonstrate that these affect the coding region of the genes of interest, i.e. RHOU, WNT3A, NOMO3 and TBX20 (Table 3). The genes adjacent to the first duplication were RHOU and WNT3A, for which both cardiovascular features in zebrafish models were described including heart looping abnormalities [37, 38]. The second duplicated region was closely located to NOMO3, a gene known to encode for an antagonist of the Nodal signalling pathway. As a member of the transforming growth factor-β super family, Nodal signalling tightly regulates embryonic heart morphogenesis [39]. The third and last region was a duplication 89 kb upstream of the T-box family transcription factor TBX20. In DECIPHER, no additional CNVs near RHOU, WNT3A or NOMO3 were found in patients with cardiovascular disease, but we did identify nine overlapping CNVs (deletions/duplications) for the region adjacent to TBX20. Two out of nine reported patients (n = 2/9, 22%), carrying a bigger deletion (5.36 and 21.92 Mb, respectively) than our initial finding, presented with syndromic CHD including an atrial septal defect or a ventricular septal defect. In-house CNV-calling on WES data of 67 BAV/TAA patients identified no extra CNVs for the aforementioned genes. Furthermore, Hi-C data in HUVECs were suggestive for a TAD boundary near RHOU, NOMO3 and TBX20, that might be affected by the CNV (Supplementary Figure 1A, B). Owing to the segmental duplications, exact fine mapping of duplication breakpoints was not possible for RHOU and NOMO3. Only for the 5′ end of the TBX20 duplication, we were able to confirm it does affect the predicted TAD boundary (Fig. 2b). Unfortunately, no DNA of relatives of the TBX20 proband was available to investigate segregation of the CNV with the phenotype.
Replication of candidate BAV/TAA genes
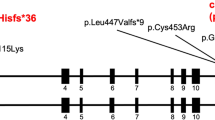
For further replication of the candidate BAV/TAA genes, we selected the two genes (DGCR6 and TBX20) for which we identified additional CNVs (either in DECIPHER or own data) as well as supportive evidence for TAD-boundary involvement. To consolidate proof for their involvement in BAV/TAA pathogenesis, a single-nucleotide variant burden analysis was performed in 637 unrelated BAV/TAA patients for these two candidate genes. Results of this analysis on rare deleterious heterozygous single-nucleotide variants show a significant burden for TBX20 variants (p = 0.033) (Table 4). Only rare deleterious next-generation sequencing variants (n = 7) were identified in our targeted gene panel dataset of BAV/TAA patients (c.117C>G (p.(Ile39Met)), c.374C>A (p.(Ser125*)), c.527A>C (p.(Asp176Ala) (4×)) and c.533C>T (p.(Pro178Leu))) (NM_001077653.2). All variants are absent from the gnomAD database, except for one (p.(Pro178Leu), 7/270 496 or 0.0026%). Three out of the four different variants (c.374C>A (p.(Ser125*)), c.527A>C (p.(Asp176Ala)) and c.533C>T (p.(Pro178Leu))) were located within the functionally important T-box domain of the TBX20 protein (Fig. 3b). In-silico programmes predicted a pathogenic effect of the variants (Fig. 3c). For one TBX20 mutation carrier (c.117C>G), gDNA of his sibling and children was available for segregation analysis. Sanger sequencing chromatograms confirmed the presence of this TBX20 variant in the proband, and its absence in the unaffected (n = 3) family members, i.e. no BAV and/or TAA (Fig. 3a).

TBX20 variants identified in BAV/TAA cases: segregation analysis and next-generation sequencing variants. a Segregation analysis of p.(Ile39Met) in BAV/TAA family. The arrow indicates the family’s proband; squares are males; circles are females; full-filled symbols indicate BAV/TAA (bicuspid aortic valve-related thoracic aortic aneurysm); plus symbol indicates presence of the variant; minus symbol represents absence of the variant. b Overview of all TBX20 variants identified within our BAV/TAA cohort using next-generation sequencing plotted on the protein structure. c Overview of frequencies and in-silico predictions of TBX20 variants using gnomAD database, CADD, MutationTaster2, Polyphen2 and SIFT. NA not applicable
Discussion
Prior studies have suggested a potential role for causative CNVs in the genetic aetiology of BAV/TAA. Up to now most CNV studies have included patients with a variety of congenital heart malformations like tetralogy of Fallot, atrial septal defects and ventricular septal defects [10,11,12,13,14]. So far, only a minority of the studied patients presented isolated BAV/TAA. Generally, the CNV-uptake in non-syndromic CHD probands caused by de novo and/or recurrent CNVs was estimated between 5 and 10% [12,13,14]. Due to lack of BAV/TAA-specific clinical information within the reported CNV studies for CHD and cardiovascular disease, the current CNV-uptake number in BAV/TAA patients could not be determined [12, 15, 40, 41]. Within these CHD cohorts, harbouring rare CNVs encompassed genes playing an important role in vascular biology and/or angiogenesis with a strong focus on genes regulating ECM (HAS2) [15] and its interaction with vascular smooth muscle cells in addition to known disease-causative genes for aortic valve defects (HOXA3) [15] and arterial/aortic aneurysms (MYH11) [40, 41]. In our study population, we did not identify CNVs that affect previously identified BAV/TAA genes or loci (Supplementary Table 4). We did not observe a major contribution of deleterious CNVs to the BAV/TAA phenotype, as only one (1/162; 0.6%) potential pathogenic CNV (affecting TBX20) was identified within our cohort. Based on the identification of overlapping CNVs in patients with similar cardiovascular phenotypes and the potential effect of the CNVs on TADs, we sought further supporting genetic evidence for pathogenicity for two candidate CNVs (DGCR6 and TBX20). A single-nucleotide variant burden analysis strengthened the genetic evidence for the involvement of TBX20 (p = 0.03) in the BAV/TAA pathology. For DGCR6, burden analysis did not show a significant number of variants in the BAV/TAA cohort and segregation analysis demonstrated that the CNV affecting DGCR6 was absent in an affected relative in one of the four BAV/TAA families with DGCR6 CNVs.
The role of TBX20 has been extensively studied in CHD pathogenesis [42,43,44,45]. In 2007, one nonsense and two missense mutations in TBX20 were first described in patients with diverse cardiac pathologies including defects of septation and valvulogenesis (most prominently mitral valve disease) and cardiomyopathy [43]. Also, one proband presented with mild coarctation of the aorta in addition to an atrial septal defect. Subsequent TBX20 mutational screening studies expanded the clinical spectrum of (likely) pathogenic TBX20 mutation carriers with tetralogy of Fallot, total anomalous pulmonary venous connection, double outlet right ventricle and persistent truncus arteriosus [42, 44, 45]. So far no CNVs (deletions and/or duplications) of TBX20 have been described in literature, apart from the ones reported in the DECIPHER database as described in the results section. The mutation spectrum includes mostly missense mutations (n = 11/14) affecting the T-box domain of TBX20 (n = 8/11). In our BAV/TAA cohort, four unrelated patients carried the missense variant (c.527A>C (p.(Asp176Ala))) at the exact same position (in a CpG island) as a previously reported mutation (c.527A>C (p.(Asp176Ala))) in a patient with atrial septal defect, dilated right atrium and ventricle [44]. The human transcription factor TBX20 is expressed in the aorta (Genotype-Tissue Expression database version 7, https://www.gtexportal.org/home/), pulmonary valve (Functional Annotation of Mammalian Genomes 5, http://fantom.gsc.riken.jp/5/) and heart muscle (Human Protein Atlas dataset, https://www.proteinatlas.org/). More in-depth data from mice showed expression of Tbx20 in cardiac progenitor cells, developing myocardium, endocardial cushions of the outflow tract, atrioventricular canal, and leading edge of the interatrial septum primum [46]. Previously, a link between Smad6a and tbx20 expression has been shown during cardiogenic differentiation in vivo [47]. This is highly interesting as SMAD6 has recently been identified as the most important gene in the aetiology of BAV/TAA [7].
Overall, 1% of the BAV/TAA patients are currently explained by a TBX20 CNV (n = 1/162) or a TBX20 SNV (n = 7/637). Yet, further work is essential to elucidate the potential involvement of this gene in the BAV/TAA pathology. Interestingly, our TBX20 proband also presented with the CNV affecting the DGCR6 gene, and, as such, an extra genetic burden for this patient might contribute to the BAV/TAA phenotype.
Despite many efforts in gene discovery, the aetiology of BAV/TAA remains mainly elusive [48]. A very heterogeneous genetic architecture for the BAV/TAA pathology has been revealed by these studies [48], and to some extent by the CNV work presented here. In the future, whole genome sequencing may provide crucial missing information in the elucidation of the genetic causes. In addition, epigenetic controlling mechanisms may be involved [49]. Ultimately, the clinical presentation of a BAV/TAA patient may likely be explained best by the interplay of genetic variants and epigenetic regulatory modifications with valve-mediated hemodynamic blood flow disturbances.
This study should be interpreted in light of a few limitations. First, the sample size of our initial CNV-cohort was rather small, in particular for a genetically complex phenotype such as BAV/TAA. Second, CNVs might map to essential non-coding sequences such as cardiac enhancers as numerous studies have been recognising their importance in embryonic cardiogenesis and disease progression [50]. However, the current version of the CNV-Webstore does not annotate these enhancers. And finally, duplications might not be in tandem and cause disease due to disruption of genes at a different genomic location. Depending on the size of the CNV, fluorescence in-situ hybridisation technique could detect such events. Yet, the number of duplications identified within our cohort is too large to investigate in a cost-efficient manner. Moreover, we did not have access to patient-specific cells.
In conclusion, our BAV/TAA cohort is not strongly enriched for deleterious CNVs that are directly causing the BAV/TAA phenotype. This finding is in contrast to prior studies of other left-sided CHDs, in which CNVs were identified as a significantly contributing factor. However, we cannot exclude the possibility that certain copy numbers contribute as modifiers, creating a sensitised background for BAV/TAA.
References
Braverman AC, Guven H, Beardslee MA, Makan M, Kates AM, Moon MR. The bicuspid aortic valve. Curr Probl Cardiol. 2005;30:470–522.
Michelena HI, Khanna AD, Mahoney D, Margaryan E, Topilsky Y, Suri RM, et al. Incidence of aortic complications in patients with bicuspid aortic valves. J Am Med Assoc. 2011;306:1104–12.
Hinton RB. Bicuspid aortic valve and thoracic aortic aneurysm: three patient populations, two disease phenotypes, and one shared genotype. Cardiol Res Pract. 2012;2012:926975.
Loscalzo ML, Goh DL, Loeys B, Kent KC, Spevak PJ, Dietz HC. Familial thoracic aortic dilation and bicommissural aortic valve: a prospective analysis of natural history and inheritance. Am J Med Genet A. 2007;143A:1960–7.
Cripe L, Andelfinger G, Martin LJ, Shooner K, Benson DW. Bicuspid aortic valve is heritable. J Am Coll Cardiol. 2004;44:138–43.
Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, et al. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437:270–4.
Gillis E, Kumar AA, Luyckx I, Preuss C, Cannaerts E, van de Beek G. Candidate gene resequencing in a large bicuspid aortic valve-associated thoracic aortic aneurysm cohort: SMAD6 as an important contributor. Front Physiol. 2017;8:400.
Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–54.
Marshall CR, Noor A, Vincent JB, Loinel AC, Feuk L, Skaug J, et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008;82:477–88.
Soemedi R, Wilson JJ, Bentham J, Darlay R, Töpf A, Zelenika D, et al. Contribution of global rare copy-number variants to the risk of sporadic congenital heart disease. Am J Hum Genet. 2012;91:489–501.
Silversides CK, Lionel AC, Costain G, Merico D, Migita O, Liu B, et al. Rare copy number variations in adults with tetralogy of Fallot implicate novel risk gene pathways. PLoS Genet. 2012;8:e1002843.
Hitz MP, Lemieux-Perreault LP, Marshall C, Feroz-Zada Y, Davies R, Yang SW, et al. Rare copy number variants contribute to congenital left-sided heart disease. PLoS Genet. 2012;8:e1002903.
Warburton D, Ronemus M, Kline J, Jobanputra V, Williams I, Anyane-Yeboa K, et al. The contribution of de novo and rare inherited copy number changes to congenital heart disease in an unselected sample of children with conotruncal defects or hypoplastic left heart disease. Hum Genet. 2014;133:11–27.
Greenway SC, Pereira AC, Lin JC, DePalma SR, Israel SJ, Mesquita SM, et al. De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nat Genet. 2009;41:931–5.
Prakash S, Kuang SQ, Gen TACRI, Regalado E, Guo D, Milewicz D. Recurrent rare genomic copy number variants and bicuspid aortic valve are enriched in early onset thoracic aortic aneurysms and dissections. PLoS ONE. 2016;11:e0153543
Prakash SK, Bondy CA, Maslen CL, Silberbach M, Lin AE, Perrone L, et al. Autosomal and X chromosome structural variants are associated with congenital heart defects in Turner syndrome: The NHLBI GenTAC registry. Am J Med Genet A. 2016;170:3157–64.
Mortensen KH, Andersen NH, Gravholt CH. Cardiovascular phenotype in Turner syndrome—integrating cardiology, genetics, and endocrinology. Endocr Rev. 2012;33:677–714.
Olivieri LJ, Baba RY, Arai AE, Bandettini WP, Rosing DR, Bakalov V, et al. Spectrum of aortic valve abnormalities associated with aortic dilation across age groups in Turner syndrome. Circ Cardiovasc Imaging. 2013;6:1018–23.
Vandeweyer G, Reyniers E, Wuyts W, Rooms L, Kooy RF. CNV-WebStore: online CNV analysis, storage and interpretation. BMC Bioinformatics. 2011;12:4.
MacDonald JR, Ziman R, Yuen RK, Feuk L, Scherer SW. The database of genomic variants: a curated collection of structural variation in the human genome. Nucleic Acids Res. 2014;42:D986–992.
Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, et al. DECIPHER: database of chromosomal imbalance and phenotype in humans using Ensembl resources. Am J Hum Genet. 2009;84:524–33.
Fromer M, Moran JL, Chambert K, Banks E, Bergen SE, Ruderfer DM, et al. Discovery and statistical genotyping of copy-number variation from whole-exome sequencing depth. Am J Hum Genet. 2012;91:597–607.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303.
Poultney CS, Goldberg AP, Drapeau E, Kou Y, Harony-Nicolas H, Kajiwara Y, et al. Identification of small exonic CNV from whole-exome sequence data and application to autism spectrum disorder. Am J Hum Genet. 2013;93:607–19.
Roth C, Schuierer M, Gunther K, Buettner R. Genomic structure and DNA binding properties of the human zinc finger transcriptional repressor AP-2rep (KLF12). Genomics. 2000;63:384–90.
Shinoki N, Kawasaki T, Minamino N, Okahara K, Ogawa A, Ariyoshi H, et al. Shear stress down-regulates gene transcription and production of adrenomedullin in human aortic endothelial cells. J Cell Biochem. 1998;71:109–15.
Nakaoka H, Tajima A, Yoneyama T, Hosomichi K, Kasuya H, Mizutani T, et al. Gene expression profiling reveals distinct molecular signatures associated with the rupture of intracranial aneurysm. Stroke. 2014;45:2239–45.
Sabatier L, Miosge N, Hubmacher D, Lin G, Davis EC, Reinhardt DP. Fibrillin-3 expression in human development. Matrix Biol. 2011;30:43–52.
Edelmann L, Stankiewicz P, Spiteri E, Pandita RK, Shaffer L, Lupski JR, et al. Two functional copies of the DGCR6 gene are present on human chromosome 22q11 due to a duplication of an ancestral locus. Genome Res. 2001;11:208–17.
Van Mierop LH, Kutsche LM. Cardiovascular anomalies in DiGeorge syndrome and importance of neural crest as a possible pathogenetic factor. Am J Cardiol. 1986;58:133–7.
Gao W, Higaki T, Eguchi-Ishimae M, Iwabuki H, Wu Z, Yamamoto E, et al. DGCR6 at the proximal part of the DiGeorge critical region is involved in conotruncal heart defects. Hum Genome Var. 2015;2:15004.
Hierck BP, Molin DG, Boot MJ. Poelmann RE, Gittenberger-de Groot AC. A chicken model for DGCR6 as a modifier gene in the DiGeorge critical region. Pediatr Res. 2004;56:440–8.
Kobrynski LJ, Sullivan KE. Velocardiofacial syndrome, DiGeorge syndrome: the chromosome 22q11.2 deletion syndromes. Lancet. 2007;370:1443–52.
Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa S, Minoshima S, et al. Role of TBX1 in human del22q11.2 syndrome. Lancet. 2003;362:1366–73.
Sundaram UT, McDonald-McGinn DM, Huff D, Emanuel BS, Zackai EH, Driscoll DA, et al. Primary amenorrhea and absent uterus in the 22q11.2 deletion syndrome. Am J Med Genet A. 2007;143A:2016–8.
Dickover M, Hegarty JM, Ly K, Lopez D, Yang H, Zhang R, et al. The atypical Rho GTPase, RhoU, regulates cell-adhesion molecules during cardiac morphogenesis. Dev Biol. 2014;389:182–91.
Sun X, Zhang R, Lin X, Xu X. Wnt3a regulates the development of cardiac neural crest cells by modulating expression of cysteine-rich intestinal protein 2 in rhombomere 6. Circ Res. 2008;102:831–9.
David NB, Rosa FM. Cell autonomous commitment to an endodermal fate and behaviour by activation of Nodal signalling. Development. 2001;128:3937–47.
Kuang SQ, Guo DC, Prakash SK, McDonald ML, Johnson RJ, Wang M, et al. Recurrent chromosome 16p13.1 duplications are a risk factor for aortic dissections. PLoS Genet. 2011;7:e1002118.
Prakash SK, LeMaire SA, Guo DC, Russell L, Regalado ES, Golabbakhsh H, et al. Rare copy number variants disrupt genes regulating vascular smooth muscle cell adhesion and contractility in sporadic thoracic aortic aneurysms and dissections. Am J Hum Genet. 2010;87:743–56.
Huang RT, Wang J, Xue S, Qiu XB, Shi HY, Li RG, et al. TBX20 loss-of-function mutation responsible for familial tetralogy of Fallot or sporadic persistent truncus arteriosus. Int J Med Sci. 2017;14:323–32.
Kirk EP, Sunde M, Costa MW, Rankin SA, Wolstein O, Castro ML, et al. Mutations in cardiac T-box factor gene TBX20 are associated with diverse cardiac pathologies, including defects of septation and valvulogenesis and cardiomyopathy. Am J Hum Genet. 2007;81:280–91.
Liu C, Shen A, Li X, Jiao W, Zhang X, Li Z. T-box transcription factor TBX20 mutations in Chinese patients with congenital heart disease. Eur J Med Genet. 2008;51:580–7.
Pan Y, Geng R, Zhou N, Zheng GF, Zhao H, Wang J, et al. TBX20 loss-of-function mutation contributes to double outlet right ventricle. Int J Mol Med. 2015;35:1058–66.
Stennard FA, Costa MW, Elliott DA, Rankin S, Haast SJ, Lai D, et al. Cardiac T-box factor Tbx20 directly interacts with Nkx2-5, GATA4, and GATA5 in regulation of gene expression in the developing heart. Dev Biol. 2003;262:206–24.
de Pater E, Ciampricotti M, Priller F, Veerkamp J, Strate I, Smith K, et al. Bmp signaling exerts opposite effects on cardiac differentiation. Circ Res. 2012;110:578–87.
Giusti B, Sticchi E, De Cario R, Magi A, Nistri S, Pepe G. Genetic bases of bicuspid aortic valve: the contribution of traditional and high-throughput sequencing approaches on research and diagnosis. Front Physiol. 2017;8:612.
Serra-Juhe C, Cusco I, Homs A, Flores R, Toran N, Perez-Jurado LA. DNA methylation abnormalities in congenital heart disease. Epigenetics. 2015;10:167–77.
Postma AV, Bezzina CR, Christoffels VM. Genetics of congenital heart disease: the contribution of the noncoding regulatory genome. J Hum Genet. 2016;61:13–19.
Acknowledgements
We would like to thank the patients, families and our collaborating partners for their participation and contribution to this work. This study makes use of data generated by the DECIPHER community. A full list of centres who contributed to the generation of the data is available from http://decipher.sanger.ac.uk and via email from decipher@sanger.ac.uk. Funding for the project was provided by Wellcome.
Members of MIBAVA Leducq consortium
Rustam Zhurayev12, Dmytro Zerbino12, Seema Mital13, Luc Mertens13, Anders Franco-Cereceda14, Judith M.A. Verhagen15, Ingrid M.B.H. van de Laar15, Marja W. Wessels15, Michaela Nemcikova16 and Alice Krebsova17
Funding
This work is supported by the Fund for Scientific Research Flanders [G.0221.12]; the Foundation Leducq [12 CVD 03] and the European Research Council [ERC-StG-2012–30972]. BL.L. is a senior clinical investigator of the Fund for Scientific Research Flanders. G.A. is a Senior Research Scholar of the Fonds de Recherche Québec—Santé. A.V. and G.V. are postdoc researchers and supported by the Fund for Scientific Research Flanders. I.L. is supported by a PhD grant from the Agency for Innovation by Science and Technology (IWT).
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Luyckx, I., Kumar, A.A., Reyniers, E. et al. Copy number variation analysis in bicuspid aortic valve-related aortopathy identifies TBX20 as a contributing gene. Eur J Hum Genet 27, 1033–1043 (2019). https://doi.org/10.1038/s41431-019-0364-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-019-0364-y
This article is cited by
-
Variants in structural cardiac genes in patients with cancer therapy-related cardiac dysfunction after anthracycline chemotherapy: a case control study
Cardio-Oncology (2024)
-
Heritable thoracic aortic disease: a literature review on genetic aortopathies and current surgical management
General Thoracic and Cardiovascular Surgery (2024)
-
Site-specific genetic and functional signatures of aortic endothelial cells at aneurysm predilection sites in healthy and AngII ApoE−/− mice
Angiogenesis (2024)
-
Genetics and mechanisms of thoracic aortic disease
Nature Reviews Cardiology (2023)
