
Abstract
Melanin-concentrating hormone (MCH) is a cyclic neuropeptide that regulates food intake, energy balance, and other physiological functions by stimulating MCHR1 and MCHR2 receptors, both of which are class A G protein-coupled receptors. MCHR1 predominately couples to inhibitory G protein, Gi/o, and MCHR2 can only couple to Gq/11. Here we present cryo-electron microscopy structures of MCH-activated MCHR1 with Gi and MCH-activated MCHR2 with Gq at the global resolutions of 3.01 Å and 2.40 Å, respectively. These structures reveal that MCH adopts a consistent cysteine-mediated hairpin loop configuration when bound to both receptors. A central arginine from the LGRVY core motif between the two cysteines of MCH penetrates deeply into the transmembrane pocket, triggering receptor activation. Integrated with mutational and functional insights, our findings elucidate the molecular underpinnings of ligand recognition and MCH receptor activation and offer a structural foundation for targeted drug design.
Similar content being viewed by others

Introduction
Melanin-concentrating hormone (MCH) is a cyclic neuropeptide of 19 amino acids, predominantly synthesized by neurons in the hypothalamus and the zona incerta of the brain1,2,3,4,5. Originally identified in fish due to its role in melanin aggregation within skin melanophores, MCH in mammals orchestrates a myriad of physiological functions1. These range from energy homeostasis, appetite regulation, and sleep–wake cycles to mood modulation, stress responses, and reproductive functions6,7,8,9,10,11,12,13,14. Disruptions in MCH signaling pathways have been associated with obesity, psychiatric conditions, and sleep disorders15,16,17,18,19. Mice lacking the MCH system display a lean phenotype, diminished appetite, increased mobile activity, and metabolic shift20,21,22,23. Consequently, the MCH system is increasingly recognized as a promising therapeutic target for conditions such as obesity, depression, and sleep disorders.
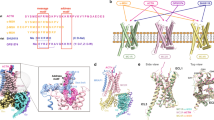
MCH exerts its effects through two specific G protein-coupled receptors: MCHR1 and MCHR2, both predominantly found in the central nervous system24. The binding of MCH to these receptors triggers conformational changes, initiating intracellular signaling cascades via heterotrimeric G proteins. Specifically, MCHR1 predominantly couples with inhibitory G proteins, Gi/o24,25,26,27, whereas MCHR2 is exclusively coupled to Gq/1124,28 (Fig. 1a). While MCHR1 antagonists are primarily explored for obesity treatment, they also show promise in treating anxiety and depression19,29,30,31,32,33. However, a challenge arises as some antagonists interact with the human ether-a-go-go-related gene (hERG) channel, leading to cardiovascular complications31. To design drugs with enhanced specificity and minimal off-target effects, a comprehensive understanding of the structural mechanisms of MCH receptor activation is paramount. In this paper, we present cryo-EM structures of MCH-activated MCHR1‒Gi and MCHR2‒Gq complexes, with resolutions of 3.01 Å and 2.40 Å, respectively. These structures elucidate the molecular details of ligand binding, G protein coupling, and receptor activation, facilitating structure-based drug design targeting MCH receptors.

a Depiction of the MCH sequence and a schematic representation illustrating the G-protein coupling of MCHR1 and MCHR2. b, c Cryo-EM density map (b) and the corresponding molecular model (c) of the MCH‒MCHR1‒Gi complex. d, e Cryo-EM density map (d) and the corresponding molecular model (e) of the MCH‒MCHR2‒Gq complex.
Results
Structure determination and overall structures
In our efforts to stabilize the MCHR1 and MCHR2 complexes for structure determination, we employed multiple strategies to overcome challenges. To enhance cell-surface expression, we implemented an N-terminal truncation of 69 amino acids from MCHR15, following previously reported methods. Additionally, we fused a b562RIL (BRIL) to the N-terminus of both MCHR1 and MCHR234, which has shown to be effective in stabilizing GPCRs. Furthermore, to stabilize the complex, we employed the NanoBiT tethering strategy, attaching the large BiT (LgBiT) to the C-terminus of MCHR1 (residues 70–405) and MCHR2 (residues 1‒331). This arrangement allowed for the formation of a complex with a small BiT (HiBiT) attached to the C-terminus of Gβ35,36,37, as detailed in our methods. Additionally, we appended a dual maltose-binding protein (MBP) tag after the LgBiT to augment receptor expression and facilitate protein purification38.
For the assembly of the MCH‒MCHR1‒Gi complex, MCHR1, Gαi, Gβ, Gγ subunits, and scFv16 were co-expressed in Hi5 insect cells and subsequently incubated with MCH. Accordingly, the MCH‒MCHR2‒Gq complex was formed by co-expressing MCHR2, Gαq, Gβ, and Gγ subunits in Hi5 insect cells, followed by MCH incubation. The Gαi in the MCHR1 complex harbors dominant-negative mutations (S47N, G203A, A326S, E245A)39, which diminish nucleotide-binding affinity and stabilize the Gαβγ heterotrimer complex. The Gαq in the MCHR2 complex is a mini-Gαq, which permits the addition of nanobody (Nb35) to bolster the stability of the receptor‒G protein complex. The Gβ subunit in both MCHR1 and MCHR2 complexes contains a C-terminal HiBiT, which interacts with the LgBiT at the C-terminus of receptors, effectively anchoring the G protein complex to the receptor.
The structures of the MCH‒MCHR1‒Gi and MCH‒MCHR2‒Gq complexes were resolved by cryo-EM to 3.01 and 2.40 Å, respectively (Fig. 1b‒e; Supplementary Figs. S1, S2 and Table S1). The clarity of the cryo-EM density maps allowed for the accurate modeling of most side chains, encompassing MCH, the receptors, the Gαβγ heterotrimer, and both scFv16 and Nb35 (Fig. 1c, e; Supplementary Fig. S3). In terms of receptor composition in the final structure models, MCHR1 spans residues 107–393, and MCHR2 covers residues 31–317. Regarding the ligand, the residues 2‒18 of MCH in the MCH‒MCHR1‒Gi complex are well-defined. However, in the MCH‒MCHR2‒Gq complex, the N-terminal 4 amino acids of MCH are absent from the EM map.
The overall structures of MCHR1 and MCHR2 in both complexes are highly similar, adopting a canonical seven transmembrane helix domain (TMD), with root mean square deviation (RMSD) values of 1.009 Å over 226 Cα atoms for the receptors and 1.874 Å over 778 Cα atoms for the complete complexes. The ligand, MCH, also adopts a similar γ-shaped structure in both receptor complexes, with the RMSD value of 0.51 Å for the central 10 residues between the two disulfate-bond cysteines (C7 and C16). The binding site of MCH in both receptors is at the orthosteric pocket formed by transmembrane helices TM2, TM3, and TM5‒7, as well as three extracellular loops (ECL1‒3) (Supplementary Tables S2‒S4). The overall binding mode of MCH to MCHR1 and MCHR2 is similar to the binding mode of other cyclic peptides, including somatostatin-14 (SST14) to somatostatin receptor 2 (SSTR2) and arginine vasopressin (AVP) to vasopressin type 2 receptor (V2R).
MCH recognition by MCHR1
MCH in the MCH‒MCHR1 complex adopts a γ-shaped configuration, with the central 10 residues forming a cyclic loop with a disulfate bond by cysteine residues at positions 7 and 16. The central loop residues are buried in the TMD pocket, with the central arginine from the conserved LGRVY core motif (residues 9‒13) inserted deeply into the bottom of the pocket. The N-terminal portion of MCH (residues 2‒6) runs toward TM2 and the extracellular loop 1 (ECL1), where the C-terminal portion of MCH (residues 17 and 18) runs towards TM5 and packs along with the extracellular loop 2 (ECL2) (Fig. 2a).

a Cross-section of the MCH-binding pocket in MCHR1. b‒d Detailed molecular interactions between MCH (depicted in medium purple) and MCHR1 (shown in light salmon). Hydrogen bonds are presented with gray dashed lines. e Functional implications of mutations within the MCHR1 binding pocket, represented as ΔpEC50 values. ΔpEC50 represents the difference in pEC50 values between the WT and the mutants of MCHR1. Data from three independent experiments, each of which was performed in triplicate, are presented as mean ± SEM. Statistical differences were determined by two-sided one-way ANOVA with Tukey’s test compared with WT. NA no activity; NS no significant difference; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
The binding mode of MCH in MCHR1 is stabilized by extensive interactions of MCH with specific residues in the receptor binding pocket, as summarized in Supplementary Table S2. The most prominent interaction is mediated by R11 of MCH, the side chain of which is docked near the bottom of the TMD pocket and forms a salt bridge with D1923.32, a hydrogen bond with Q1963.36 and cation–π interaction with Y3707.43 (Fig. 2b). Additionally, D1923.32 forms a hydrogen bond with Y3707.43. Surrounding the central arginine are several hydrophobic residues (the LGRVY motif), which form extensive hydrophobic interactions with residues F1612.53, L1722.64, I265ECL2, Y3416.51, Y3627.35 in the TMD pocket (Fig. 2b, c). Besides, the main chain of Y13 forms a hydrogen bond with Q1712.63 (Fig. 2c). Other hydrophobic residues within the cyclic loop (M8 and P15) also make hydrophobic interactions with receptor residues (Y3627.35, I3667.39, and I254ECL2) in the upper part of the TMD pocket (Fig. 2c, d). In addition, the hydrophobic residue of L5 and the hydrophobic portion of R6 contact residues from ECL2 and ECL3, including F256ECL2 and P354ECL3 (Fig. 2d). Alanine mutation at key pocket residues, led to varied degrees of reduced receptor activation, with the Y3707.43 mutation abolishing activity completely, highlighting the importance of these interactions (Fig. 2e; Supplementary Fig. S4 and Table S5).
MCH recognition by MCHR2
The mode of MCH recognition by MCHR2 closely resembles that by MCHR1, with the MCH displaying a similar binding pose. The central arginine from the conserved LGRVY core motif is also deeply buried into the bottom of the orthosteric pocket of MCHR2 (Fig. 3a). Overall, MCH engages in similar interactions with both MCHR1 and MCHR2, as summarized in Fig. 3b‒f; Supplementary Fig. S5 and Tables S2‒S4. However, MCH makes more extensive and tighter interactions with the residues in MCHR2 than in MCHR1, especially with ECL2 of MCHR2 (Fig. 3c‒f).

a Cross-section of the MCH-binding pocket in MCHR2. b‒f Detailed molecular interactions between MCH (depicted in medium violet red) and MCHR2 (shown in teal). Hydrogen bonds are highlighted with gray dashed lines. g Functional data derived from alanine mutations of residues within the MCHR2 binding pocket, represented as ΔpEC50 values. ΔpEC50 represents the difference in pEC50 values between the WT and the mutants of MCHR2. Data are presented as mean ± SEM. from three independent experiments performed in triplicate and analyzed by two-sided one-way ANOVA with Tukey’s test compared with WT. NA no activity; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
In addition to various similar interactions, MCH and MCHR2 engage in additional polar interactions. The central arginine of MCH can form hydrogen bonds with W932.64 and S2957.42 of MCHR2 (Fig. 3b). The corresponding positions of MCHR1 are substituted by L1722.64 and G3697.42, which cannot form hydrogen bonds with R11. Among the other residues of the core motif, the backbone amine group of G10 and the backbone carbonyl group of Y13 can form hydrogen bonds with Q2716.55 and the main chain amine group of F186ECL2, respectively, and the hydroxyl group of Y13 and Y2857.32 can form a hydrogen bond (Fig. 3c, d). The sidechain of R6 forms electrostatic interaction with D179ECL2 and a hydrogen bond with the backbone carbonyl of G96ECL1 (Fig. 3e). In addition, ECL2 of MCHR2 forms a hydrophobic cap composed of F177ECL2, V181ECL2, S183ECL2, C184ECL2, A185ECL2, F186ECL2, and L188ECL2, which makes intensive hydrophobic interactions with MCH residues L5, L9 and V12‒P15 (Fig. 3c‒f).
Consistently, alanine mutations of the aforementioned receptor pocket residues significantly decrease or even abolish receptor activity, highlighting the essential role of these amino acids in the MCH binding to MCHR2 (Fig. 3g; Supplementary Fig. S4 and Table S6). In addition, MCH is closely packed against ECL2 of MCHR2 (Fig. 3c‒f). The A185ECL2F mutation results in a decrease in activity by more than 1000-fold, while the A185ECL2W mutation completely abolishes activity (Fig. 3g). This suggests that increased steric hindrance of ECL2 may hinder MCH from effectively accessing the ligand-binding pocket.
Differences in MCH binding between MCHR1 and MCHR2
While MCHR1 and MCHR2 share highly similar overall structures and MCH binding modes, subtle differences in their binding pockets confer differences in MCH binding (Fig. 4a‒c). Notably, residues deep within the binding pocket of MCHR2 engage in extensive interactions with the critical R11 of MCH. R11 forms hydrogen bonds with S7.42 and W2.64 in MCHR2, enabling a unique interaction pattern (Fig. 4d). In contrast, in MCHR1, R11 shifts away from TM6 due to a potential steric clash with W6.48 and the absence of a hydrogen bond with G7.42 (corresponding to A6.48 and S7.42 in MCHR2) (Fig. 4d).

a Structural superposition of MCH-bound MCHRs, presented from both side-view (the left) and top view (the right). b, c Mesh representations of MCH within the binding pockets of MCHR1 and MCHR2. d, e The differential interactions between MCH and MCHRs, focusing on R11 (d) and the C7, M8, and R14 residues of MCH (e).
Additionally, residues 2.64 and 7.32 confer other differences. MCHR1 has L2.64 and V7.32, while MCHR2 has W2.64 and Y7.32. The bulky tryptophan and tyrosine residues introduce steric hindrance that brings C7 and M8 of MCH closer to TM4 and TM5 in MCHR2 (Fig. 4e). This enhances interactions between MCH and ECL2 of MCHR2, which dips slightly towards the binding pocket. Furthermore, R14 of MCH clashes with Q6.58 in MCHR1, causing it to shift downwards compared to N6.58 in MCHR2 (Fig. 4e). Collectively, these distinctions display unique differences between MCH binding to MCHR1 and MCHR2.
Activation mechanism of MCHRs
To elucidate the activation mechanisms of MCHR1 and MCHR2, we compared their active structures to the AlphaFold2-generated inactive models from GPCR databank (Fig. 5). Structural alignment showed that upon MCH binding and activation, the cytoplasmic ends of TM6 in MCHR1 and MCHR2 moved outward by approximately 7.50 Å and 9.01 Å respectively, as measured at the Cα of residue 6.31 (Fig. 5a, b). This outward movement of TM6 is a hallmark of GPCR activation and enables coupling to downstream G proteins. Additionally, TM7 underwent an inward shift of 2.37 Å in MCHR1 and 1.03 Å in MCHR2 at the Cα of residue 7.53 (Fig. 5a, b). The combination of TM6 and TM7 movements opens a cleft on the intracellular side to accommodate G protein binding.

a, b Structural superposition between cryo-EM structures of MCH-bound MCHR complexes and the AlphaFold2-predicted inactive models from the GPCR databank is shown from the side view and the cytoplasmic view. c Structural alignment highlighting the toggle switch mechanism. d Depiction of shifts in H/F2.53, D3.32, and Y7.43 at the base of the binding pocket. e‒g Illustration of the conserved motifs in MCH-activated MCHRs: DRY motif (e); NPxxY motif (f); PIF motif (g).
The activation of MCHR1 and MCHR2 is intricately regulated by MCH binding. This interaction occurs within the orthosteric binding pocket, leading to conformational changes that extend through the helices. However, contrary to the commonly observed shift of the conserved toggle switch W6.48 in most class A GPCRs, we observed minimal movement of the tryptophan in MCHR1 and an alanine substitution at this position in MCHR2 (Fig. 5c). Instead, our study reveals a novel mechanism of activation for these receptors. MCH disrupts the polar interaction between Q3.36 and H/Y6.52, leading to the displacement of these residues, a critical event for receptor activation (Fig. 5c). This was further substantiated by our mutagenesis experiments. In MCHR1, mutations at Q1963.36 and Y3426.52 increased basal activity by approximately 20%, confirming the significance of disrupting this polar interaction for receptor activation (Supplementary Fig. S4a‒d). In contrast, similar mutations in MCHR2 resulted in distinct outcomes: the H2686.52 to F mutation led to receptor inactivity, and the Q1173.36A mutation had a minimal impact (Supplementary Fig. S4b‒e). These findings highlight a unique activation mechanism for MCHR2, divergent from that of MCHR1. The differential effects observed upon mutating Q3.36 and H/Y6.52 between MCHR1 and MCHR2 provide a deeper understanding of the activation mechanisms in these receptors, emphasizing the complexity and diversity of GPCR activation.
Furthermore, R11 of MCH forms critical interactions with D3.32 and Y7.43 at the base of the pocket. This results in downward shifts of D3.32 and Y7.43, which in turn induces the upward movement of R3.50 in the DRY motif and subsequent downward shift of Y7.53 in the NPxxY motif (Fig. 5d‒f). The H/Y6.52 displacement also shifts F6.44 in the PIF motif (Fig. 5g). Collectively, these rearrangements within key structural motifs facilitate the activated state.
G-protein coupling of MCHR1 and MCHR2
MCHR1 and MCHR2 exhibit distinct G-protein coupling profiles. Specifically, MCHR1 is capable of associating with a diverse set of G-proteins, namely Gαi/o and Gαq/11. Conversely, MCHR2 demonstrates specificity by predominantly coupling to Gαq/11. Experimental analyses further elucidated that MCHR1 manifests a heightened efficacy in its coupling to Gαi as opposed to Gαq, as depicted in Supplementary Fig. S4c.
Structural alignments between Gαi and Gαq revealed notable conformational differences (Fig. 6a). The α5 helix of both Gαi and Gαq exhibits a deflection of ~7°, and a discernible spatial separation of ~10.3 Å is observed in their respective αN regions (Fig. 6b, c). Intriguingly, both MCHR1 and MCHR2 engage with Gαi or Gαq through transmembrane domains 2–7 (TM2‒7) and intracellular loops 2 and 3 (ICL2 and ICL3) (Fig. 6d). Additional interactions via TM1 and Helix8 are uniquely observed in MCHR2 (Fig. 6d).

a Structural alignment between MCHR1‒Gi and MCHR2‒Gq complexes. b, c Comparative analysis of the Gα conformation in the α5 (b) and αN (c) domains. d Key residues in MCHR1 and MCHR2 that interact with the downstream Gαi or Gαq proteins. e Detailed interactions between the ICL2 of MCHR1 and MCHR2 with the αN domain of Gαi or Gαq. f IP-one assay data for wild-type and T22134.54R of MCHR1, with data presented as mean values ± SEM from three independent experiments performed in triplicate.
A critical feature of Gαq-coupled receptors is the presence of a basic residue, typically arginine or lysine, at positions 34.54 or 34.55 within ICL240. In MCHR1, the threonine residue at position 34.54 (T221) engages in a polar interaction with the αN domain of Gαq (Fig. 6e). Interestingly, when this threonine was mutated to arginine, there was a significant tenfold increase in the coupling efficiency of MCHR1 to Gαq, underscoring the importance of this residue in G-protein coupling specificity (Fig. 6f). In contrast, a similar mutation in MCHR2 (T14234.54 to R) did not enhance the coupling efficiency to Gαq (Supplementary Fig. S4e). This disparity in functional outcomes between MCHR1 and MCHR2 suggests that the determinants of G-protein coupling specificity extend beyond a single residue alteration. The lack of a similar effect in MCHR2 indicates the complexity of GPCR‒G protein interactions, where a broader network of interactions and structural conformations plays a crucial role. Thus, while T34.54 is a pivotal element in modulating Gq coupling in MCHR1, its role in MCHR2 appears to be distinct, likely influenced by other structural factors and residues. These findings highlight the intricate and receptor-specific nature of GPCR‒G protein interactions, emphasizing that insights derived from one receptor subtype cannot be directly transferable to another, even within the same receptor family. Our study provides an understanding of G-protein coupling specificities in MCHR1 and MCHR2, revealing the complex interplay of residues and structural elements that govern these interactions.
Comparison of ligand-activated SSTR2 and V2R
MCH, SST14, and AVP are cyclic polypeptides, each stabilized by disulfide bonds. These peptides adopt analogous conformations, with their cyclic segments deeply embedded within their respective receptor binding pockets (SST14 in SSTR2; AVP in V2R; Supplementary Fig. S6). Distinctively, MCH adopts a twisted γ-shaped ring conformation, diverging from the near-planar cyclic amino acid backbones exhibited by SST14 and AVP (Fig. 7).

a Binding conformation of MCH in MCHR1. b Binding conformation of MCH in MCHR2. c Binding conformation of SST14 in SSTR2 (PDB: 7Y27). d Binding conformation of AVP in V2R (PDB: 7DW9).
SST14, characterized by its expansive loop encompassing 12 amino acids, positions its disulfide bond above the receptor pocket. Central residues, W8 and K9, from its FWKT motif, play pivotal roles in receptor engagement (Fig. 7c). In contrast, AVP, with its compact 6-amino acid ring, situates its disulfide bond intrinsically within the receptor pocket (Fig. 7d). Key residues, Y2 and F3, delve into the pocket’s depths, with their side chains docked horizontally into the pocket, which contrasts with the vertical docking of the sidechain of R11 from MCH into both MCHR1 and MCHR2 pocket (Fig. 7a, b and d). The profound penetration of MCH into the receptor pocket surpasses that of both SST14 and AVP.
Analysis of AlphaFold2 predictions and cryo-EM structures
In our endeavor to comprehensively understand the structural aspects of MCHR1 and MCHR2, we employed AlphaFold2 to predict the structures of the MCHR1‒Gi and MCHR2‒Gq complexes. This comparison aimed to assess the accuracy and limitations of computational predictions in contrast to experimental cryo-EM data. The AlphaFold2 models exhibited remarkable similarity to our cryo-EM structures (Supplementary Fig. S7a), with RMSDs of 0.988 Å over 229 Cα atoms for MCHR1 and 1.249 Å over 242 Cα atoms for MCHR2. These low RMSDs underscore the impressive accuracy of AlphaFold2 in predicting the overall architecture of these GPCRs.
Despite the overall structural congruence, we identified several key differences between cryo-EM structures and computational predictions. In the AlphaFold2 models, the MCH ligand exhibited a short alpha-helical conformation for residues 2‒7, diverging significantly from the short beta-strand configuration observed in our cryo-EM structures (Supplementary Fig. S7). This distinction critically alters the ligand‒receptor interaction and the shape of the binding pocket. The altered conformation of MCH in the AI models resulted in an expanded binding pocket, with observable outward movements in ECL2, ECL3, TM1, and TM2 (Supplementary Fig. S7b, d). These structural adjustments in response to the ligand’s conformation highlight the sensitivity of receptor architecture to ligand shape. We also noted significant outward movements in the cytoplasmic end of TM6 in the AI models, with a corresponding 2‒3 Å displacement of the G protein heterotrimer (Supplementary Fig. S7a). These deviations suggest differences in the predicted vs actual conformational states of the receptor‒G protein interface.
This comparative analysis reveals the strengths and limitations of current AI/ML tools in structural predictions of GPCRs. While the overall structural predictions were highly accurate, the finer details of the ligand conformation and receptor‒G protein interactions were not completely captured by the AI models. These differences are crucial for understanding the functional dynamics of receptors and have significant implications for drug design.
Discussion
In this study, we have advanced our understanding of the structural mechanisms underlying the activation of MCHR1‒Gi and MCHR2‒Gq by the endogenous ligand MCH. Our approach, integrating high-resolution structural determination, mutagenesis, and functional assays, has provided a detailed view of the ligand-binding sites in MCHR1 and MCHR2. We highlight the crucial role of the ring structure formed by Cys6 and Cys17 in ligand‒receptor interactions.
Our comparative analysis of the ligand-binding pockets in MCHR1 and MCHR2 unveils unique molecular interactions. Notably, the absence of a hydrogen bond between S7.42 and R11 in MCHR1, due to the presence of G7.42, contrasts with the hydrogen bond formation in MCHR2. Additionally, the differences in steric hindrance between W6.48 in MCHR1 and A6.48 in MCHR2 lead to distinct displacement and interaction patterns with the ligand. These differences are further illustrated by the unique contacts MCH makes with MCHR2, facilitated by the spatial arrangement of W2.64 and Y7.32.
The activation process in both MCHR1 and MCHR2 involves significant conformational changes in the canonical motifs DRY, NPxxY, and PIF, indicative of receptor activation. However, we observe a divergence from the typical toggle switch mechanism seen in class A GPCRs. In MCHR1, W6.48 shows minimal movement after activation, whereas MCHR2 has an alanine at this key position. Importantly, the disruption of the polar interaction between Q3.36 and H/Y6.52 in MCHR1, as evidenced by our mutagenesis studies, plays a pivotal role in receptor activation. These findings are complemented by the differential effects of mutations at these positions in MCHR1 and MCHR2, underlining the unique activation mechanisms in these receptors.
Furthermore, our structural insights extend to the interactions between MCHRs and G proteins. The T34.54R mutation in the ICL2 of MCHR1, identified through our mutagenesis studies, significantly enhances Gαq coupling efficiency. This observation emphasizes the role of specific residues in determining G protein coupling specificity. However, a similar mutation in MCHR2 does not yield an increase in Gαq coupling efficiency, highlighting the complexity of GPCR‒G protein interactions and the necessity to consider receptor-specific structural and functional nuances.
Additionally, our comparative analysis with AlphaFold2 predictions has demonstrated both the capabilities and limitations of AI/ML tools in structural biology. While AlphaFold2 accurately predicted the overall receptor architectures, it fell short in capturing the precise conformation of ligands and the subtle yet functionally significant movements in receptor domains and associated proteins. This comparison underscores the importance of combining computational predictions with experimental methods to achieve a comprehensive understanding of complex biological systems like GPCRs.
Overall, our study provides a rich and comprehensive understanding of the molecular dynamics in ligand binding, receptor activation, and G protein coupling in MCHR1 and MCHR2. These findings pave the way for targeted therapeutic interventions, leveraging the distinct characteristics of these receptors.
Materials and methods
Constructs cloning
The gene sequences encoding human MCHR1 (residues 70‒405) and MCHR2 (residues 1‒331, with L2566.40Y mutation) were subcloned into pFastBac vector (Invitrogen) with an N-terminal haemagglutinin signal peptide (HA) and a thermostabilized BRIL fusion, followed by a C-terminal 15-amino acid linker, a LgBiT subunit and two MBPs to facilitate receptor expression and stability. The Gαi construct was designed with dominant-negative mutations S47N, G203A, A326S, and E245A to decrease the affinity of nucleotide-binding and stable Gαβγ complex. The mini-Gαq construct was designed with the C-terminal α-helix (αH5) of Gαq, which is critical for G protein coupling selectivity. The core of the construct, known as the Ras domain, is derived from Gαs. This design choice was made to enable the binding of the nanobody Nb35, which stabilizes the complex for cryo-EM analysis. Additionally, the N-terminal helix from Gαi was incorporated into the construct to facilitate the binding of the single-chain variable fragment (scFv16), aiding in cryo-EM studies. Rat Gβ1 was constructed with a N-terminal 16× His-tag for purification and a C-terminal HiBiT to form a NanoBiT with LgBiT. Gαi, mini-Gαq, Gβ1, Gγ2, scFv16, and Nb35 were all cloned into the pFastBac vector separately. The wild-type and mutants of MCHR1 and MCHR2 were constructed into the pcDNA6.0 vector (Promega) for functional assays.
Complex expression and purification
MCHRs and G proteins were co-expressed in High Five insect cells (Thermo Fisher Scientific) using Bac-to-Bac baculovirus system (Thermo Fisher Scientific). The cells were cultured in ESF 921 serum-free medium (Expression Systems) to a density of 3 × 106 cells/mL. The cells were infected with MCHR1, Gαi, Gβ1, Gγ2, scFv16, and Ric8B at the ratio of 1:2:2:2:2:2 or infected with MCHR2, Gαq, Gβ1, Gγ2, and Ric8A at the ratio of 1:2:2:2:2. After 48 h of infection at 27 °C; the cells were harvested by centrifugation at 2000 rpm and then stored at ‒80 °C for future use.
The frozen cells were thawed at room temperature (RT) and resuspended in a buffer containing 20 mM HEPES, pH 7.4, 100 mM NaCl, 10% (v/v) glycerol, 10 mM MgCl2, 10 mM CaCl2, 100 μM TCEP, 25 mU/mL apyrase, 1× protease inhibitor cocktail (TargetMol, 1 mL/100 mL suspension), 10 μM MCH and 10 μg/mL Nb35 (for MCHR2 only). The suspension was incubated at RT for 1 h and then solubilized by 0.5% (w/v) lauryl maltose neopentyl glycol (LMNG, Anatrace) and 0.1% (w/v) cholesteryl hemisuccinate TRIS salt (CHS, Anatrace) at 4 °C for 3 h. The supernatant was separated by centrifugation at 30,000 rpm for 35 min and then incubated with re-equilibrated dextrin beads (Smart-Lifesciences) at 4 °C for 3 h. The resin was collected by centrifugation at 500× g for 5 min, loaded onto a gravity-flow column and washed with 20 column volumes of wash buffer containing 20 mM HEPES, pH 7.4, 100 mM NaCl, 10% (v/v) glycerol, 5 mM MgCl2, 5 mM CaCl2, 25 μM TCEP, 10 μM MCH, 0.01% (w/v) LMNG, 0.01% (w/v) glyco-diosgenin (GDN, Anatrace) and 0.004% (w/v) CHS. The protein was eluted with wash buffer adding 10 mM maltose, concentrated using a 100-kDa Amicon Ultra Centrifugal Filter (Millipore), and loaded onto Superose 6 Increase 10/300 GL column (GE Healthcare) pre-equilibrated with buffer including 20 mM HEPES, pH 7.4, 100 mM NaCl, 2 mM MgCl2, 2 mM CaCl2, 100 μM TCEP, 0.0005% (w/v) digitonin (Biosynth), 0.00075% (w/v) LMNG, 0.00025% (w/v) GDN, and 0.0002% (w/v) CHS. The receptor and G protein complexes were collected and concentrated for electron microscopy experiments.
Expression and purification of Nb35
Nanobody-35 (Nb35) was expressed in E. coli BL21 cells which were cultured in TB medium with 100 μg/mL ampicillin at 37 °C for about 3 h to OD600 of 1.0 and then induced with 1 mM IPTG at 25 °C for 16 h. Each liter of harvested cells was resuspended with 15 mL TES (0.2 M Tris, pH 8.0, 0.5 mM EDTA, 0.5 M sucrose), stirred at 4 °C for 1 h and another 45 min with 30 mL TES/4. The supernatant was collected by centrifugation at 30,000 rpm for 30 min and incubated with pre-equilibrated Nickel resin at 4 °C for 1 h. After washing with 20 column volumes of wash buffer containing 20 mM HEPES, pH 7.4, 100 mM NaCl, and 10% (v/v) glycerol, Nb35 was eluted with wash buffer, adding 300 mM imidazole. The protein was further purified by HiLoad 16/600 Superdex 75 column with 20 mM HEPES pH 7.4, and 100 mM NaCl. The targeted fractions were collected and concentrated to 5 mg/mL and then flash-frozen in liquid nitrogen before storing at −80 °C.
Cryo-EM grid preparation and data collection
For cryo-EM grid preparation of the MCH‒MCHR1‒Gi complex, 3 μL of the purified complex at a concentration of 5.72 mg/mL was applied to glow-discharged holey carbon grids (Quantifoil R1.2/1.3, Au/C 300 mesh) that were subsequently vitrified by plunging into liquid ethane using a Vitrobot Mark IV (Thermo Fisher Scientific) at 4 °C. A Titan Krios G4 equipped with a Gatan K3 direct electron detector with super-resolution mode and EPU was used to acquire cryo-EM movies at the Advanced Center for Electron Microscopy at Shanghai Institute of Materia Medica, Chinese Academy of Sciences. A total of 14,952 movies were recorded with a pixel size of 0.824 Å at a dose of 50 electrons per Å2 for 32 frames. The defocus range of this dataset was ‒0.8 to ‒1.8 μm.
For the MCH‒MCHR2‒Gq complex, 20.47 mg/mL of purified protein was used for cryo-EM grid preparation. Cryo-EM movies were collected by a Titan Krios G4 CEFG at 300 kV accelerating voltage equipped with Falcon4 direct electron detector and Selectris X (Thermo Fisher Scientific). A total of 8789 EER movies were recorded with a pixel size of 0.73 Å at a dose of 50 electrons per Å2. The defocus range of this dataset was ‒0.8 to ‒1.8 μm.
Cryo-EM data processing and three-dimensional reconstruction
For the MCH‒MCHR1‒Gi complex, all dose-fractionated image stacks were subjected to beam-induced motion correction by Relion4.041. The defocus parameters were estimated by CTFFIND4.142. Blob picking and Template picking yielded 32,120,445 particles, which were processed by reference-free 2D classification using Cryosparc43. With the initial model from Cryosparc, after several rounds of 3D classification using Relion, 311,033 particles were used to further CTF Refinement and polishing, yielding a reconstruction with a global resolution of 3.01 Å at a Fourier shell correlation (FSC) of 0.143, and subsequently post-processed by DeepEMhancer44.
For the MCH‒MCHR2‒Gq complex, EER movies were aligned with Relion4.041. Initial contrast transfer function (CTF) fitting was performed with CTFFIND4.142 from Cryosparc43. Blob picking and Template picking yielded 14,206,080 particles, which were processed by reference-free 2D classification using Cryosparc, 2D Classification was processed using Cryosparc, producing 1,498,863 particles for further processing. With the initial model, two rounds of 3D classifications were carried out with Relion4.0, in which 879,702 particles were subjected to 3D auto-refinement, CTF refinement, and polishing. A map with an indicated global resolution of 2.4 Å at FSC of 0.143 was generated from the final 3D refinement and subsequently post-processed by DeepEMhancer44.
Model building and refinement
All PDB coordinates using alphafold245 were served as a starting model for building the atomic model. All model were fitted into cryo-EM density map using chimera46 followed by a manual adjustment in Coot47. The model was refined by Phenix48.
GloSensor cAMP assay
GloSensor cAMP assay was used to detect the downstream Gαi signal of MCHR1 using GloSensor cAMP Reagent (Promega). HEK293 cells were cultured in DMEM/high glucose supplemented with 10% (v/v) fetal bovine serum and 1% (v/v) penicillin–streptomycin at 37 °C in 5% CO2. Cells were grown at 12-well plates for 18 h and transfected with MCHR1 constructs and the cAMP biosensor GloSensor-22F (Promega) at a ratio of 3:2. After transfection for 24 h, cells were harvested, resuspended with CO2-independent media containing 2% GloSensor cAMP Reagent (Promega), and then distributed to 384-well plates at a density of 3 × 105 cells/mL (20 μL/well). The cells were incubated at 37 °C for 1.5 h. Ligand of different concentrations was mixed with forskolin at the final concentration of 1 μM (Sigma), and the mixture was added to each well (10 μL/well). The sample mixtures were immediately measured with an EnVision multi-plate reader (PerkinElmer).
Inositol phosphate accumulation assay
IP1 accumulation assay was applied for the detection of the downstream Gαq signal of MCHR2 using the IP-One HTRF kit (Cisbio). AD293 cells were cultured in DMEM/high glucose supplemented with 10% (v/v) fetal bovine serum and 1% (v/v) penicillin–streptomycin at 37 °C in 5% CO2. After transfection for 24 h, cells were harvested, resuspended with 1× stimulation buffer to a density of 8 × 105 cells/mL, and then seeded to 384-well plates for 7 μL/well. After dispensing 7 μL different concentrations of ligand diluted with 1× stimulation buffer, the mixture was incubated at 37 °C for 1 h. IP1-d2 and anti-IP1 cryptate were dissolved in 1× lysis buffer and sequentially added to the plates for 3 μL/well. Before measurement, the samples were incubated at RT for 30 min and measured with EnVision multi-plate reader (PerkinElmer).
Cell surface expression assay
MCHR1/2 wild type and their mutants were cloned into pcDNA6.0 (Invitrogen) with an N-terminal 3× Flag tag, and the cell surface expression was determined by flow cytometry. The expression of MCHR1 was tested by HEK293, and the expression of MCHR2 was measured by AD293. After transfection for 24 h, cells were collected and then blocked with 50 μL 5% (w/v) bovine serum albumin (BSA) in PBS at RT for 15 min, followed by incubation with primary mouse anti-Flag antibody (ABclonal) at RT for 1 h. The cells were then washed three times with 1 mL 1% BSA (w/v) and incubated with anti-mouse Alexa-488-conjugated secondary antibody (Invitrogen) at 4 °C in the dark for 1 h. After another three washes with 1 mL 1% BSA (w/v), cells were resuspended with 200 μL 1% BSA (w/v) for detection in Guava EasyCyte™ System at excitation 488 nm and emission 525 nm.
Alphafold2 multimer prediction
The 2.3.2 version of AlphaFold 2 multimer version49,50 was applied for the prediction of MCH‒MCHR1‒Gi and MCH‒MCHR2‒Gq structures. The input sequences and chain IDs are the same as the cryo-EM structures. The template date was 2021-11-01, and the full database present was applied. The prediction was run on a Tesla V100 GPU.
Quantification and statistical analysis
All functional study data were analyzed using Prism 8 (GraphPad) and displayed as means ± SEM from at least three independent experiments. Concentration–response curves were evaluated with a three-parameter logistic equation. The significance was determined with one-way ANOVA with Tukey’s test, and P < 0.05 vs wild type was considered statistically significant.
Data availability
Cryo-EM maps have been deposited in the Electron Microscopy Data Bank under accession codes: EMD-37823 (MCH‒MCHR1‒Gi complex) and EMD-37824 (MCH‒MCHR2‒Gq complex). The atomic coordinates have been deposited in the Protein Data Bank under accession codes: 8WSS (MCH‒MCHR1‒Gi complex) and 8WST (MCH‒MCHR2‒Gq complex).
References
Kawauchi, H., Kawazoe, I., Tsubokawa, M., Kishida, M. & Baker, B. I. Characterization of melanin-concentrating hormone in chum salmon pituitaries. Nature 305, 321–323 (1983).
Skofitsch, G., Jacobowitz, D. M. & Zamir, N. Immunohistochemical localization of a melanin concentrating hormone-like peptide in the rat brain. Brain Res. Bull. 15, 635–649 (1985).
Vaughan, J. M., Fischer, W. H., Hoeger, C., Rivier, J. & Vale, W. Characterization of melanin-concentrating hormone from rat hypothalamus. Endocrinology 125, 1660–1665 (1989).
Bittencourt, J. C. et al. The melanin-concentrating hormone system of the rat brain: an immuno- and hybridization histochemical characterization. J. Comp. Neurol. 319, 218–245 (1992).
Saito, Y., Nothacker, H. P. & Civelli, O. Melanin-concentrating hormone receptor: an orphan receptor fits the key. Trends Endocrinol. Metab. 11, 299–303 (2000).
Nahon, J. L. The melanocortins and melanin-concentrating hormone in the central regulation of feeding behavior and energy homeostasis. C. R. Biol. 329, 623–638 (2006).
Al-Massadi, O. et al. Multifaceted actions of melanin-concentrating hormone on mammalian energy homeostasis. Nat. Rev. Endocrinol. 17, 745–755 (2021).
Jiang, H. & Bruning, J. C. Melanin-concentrating hormone-dependent control of feeding: when volume matters. Cell Metab. 28, 7–8 (2018).
Bandaru, S. S., Khanday, M. A., Ibrahim, N., Naganuma, F. & Vetrivelan, R. Sleep–wake control by melanin-concentrating hormone (MCH) neurons: a review of recent findings. Curr. Neurol. Neurosci. Rep. 20, 55 (2020).
Torterolo, P. et al. Melanin-concentrating hormone (MCH): role in REM sleep and depression. Front. Neurosci. 9, 475 (2015).
Adamantidis, A. et al. Disrupting the melanin-concentrating hormone receptor 1 in mice leads to cognitive deficits and alterations of NMDA receptor function. Eur. J. Neurosci. 21, 2837–2844 (2005).
Tyhon, A. et al. Mice lacking the melanin-concentrating hormone receptor-1 exhibit an atypical psychomotor susceptibility to cocaine and no conditioned cocaine response. Behav. Brain Res. 173, 94–103 (2006).
Schneider, J. E. Energy balance and reproduction. Physiol. Behav. 81, 289–317 (2004).
Naufahu, J., Cunliffe, A. D. & Murray, J. F. The roles of melanin-concentrating hormone in energy balance and reproductive function: are they connected? Reproduction 146, R141–R150 (2013).
Prida, E. et al. Crosstalk between melanin concentrating hormone and endocrine factors: implications for obesity. Int. J. Mol. Sci. 23, 2436 (2022).
Hausen, A. C. et al. Insulin-dependent activation of MCH neurons impairs locomotor activity and insulin sensitivity in obesity. Cell Rep. 17, 2512–2521 (2016).
Roy, M., David, N., Cueva, M. & Giorgetti, M. A study of the involvement of melanin-concentrating hormone receptor 1 (MCHR1) in murine models of depression. Biol. Psychiatry 61, 174–180 (2007).
Roy, M. et al. Genetic inactivation of melanin-concentrating hormone receptor subtype 1 (MCHR1) in mice exerts anxiolytic-like behavioral effects. Neuropsychopharmacology 31, 112–120 (2006).
Potter, L. E. & Burgess, C. R. The melanin-concentrating hormone system as a target for the treatment of sleep disorders. Front. Neurosci. 16, 952275 (2022).
Jeon, J. Y. et al. MCH−/− mice are resistant to aging-associated increases in body weight and insulin resistance. Diabetes 55, 428–434 (2006).
Willie, J. T., Sinton, C. M., Maratos-Flier, E. & Yanagisawa, M. Abnormal response of melanin-concentrating hormone deficient mice to fasting: hyperactivity and rapid eye movement sleep suppression. Neuroscience 156, 819–829, (2008).
Bell, C. G. et al. Association of melanin-concentrating hormone receptor 1 5’ polymorphism with early-onset extreme obesity. Diabetes 54, 3049–3055 (2005).
Ahnaou, A., Dautzenberg, F. M., Huysmans, H., Steckler, T. & Drinkenburg, W. H. Contribution of melanin-concentrating hormone (MCH1) receptor to thermoregulation and sleep stabilization: evidence from MCH1 (−/−) mice. Behav. Brain Res. 218, 42–50 (2011).
Tan, C. P. et al. Melanin-concentrating hormone receptor subtypes 1 and 2: species-specific gene expression. Genomics 79, 785–792 (2002).
Saito, Y. et al. Molecular characterization of the melanin-concentrating-hormone receptor. Nature 400, 265–269 (1999).
Hawes, B. E. et al. The melanin-concentrating hormone receptor couples to multiple G proteins to activate diverse intracellular signaling pathways. Endocrinology 141, 4524–4532 (2000).
Pissios, P., Trombly, D. J., Tzameli, I. & Maratos-Flier, E. Melanin-concentrating hormone receptor 1 activates extracellular signal-regulated kinase and synergizes with G(s)-coupled pathways. Endocrinology 144, 3514–3523 (2003).
Sailer, A. W. et al. Identification and characterization of a second melanin-concentrating hormone receptor, MCH-2R. Proc. Natl. Acad. Sci. USA 98, 7564–7569 (2001).
Baker, B. I. Melanin-concentrating hormone: a general vertebrate neuropeptide. Int. Rev. Cytol. 126, 1–47 (1991).
Chung, S., Parks, G. S., Lee, C. & Civelli, O. Recent updates on the melanin-concentrating hormone (MCH) and its receptor system: lessons from MCH1R antagonists. J. Mol. Neurosci. 43, 115–121 (2011).
Cheon, H. G. Antiobesity effects of melanin-concentrating hormone receptor 1 (MCH-R1) antagonists. Handb. Exp. Pharmacol. 383–403 (2012).
Borowsky, B. et al. Antidepressant, anxiolytic and anorectic effects of a melanin-concentrating hormone-1 receptor antagonist. Nat. Med. 8, 825–830 (2002).
Johansson, A. & Lofberg, C. Novel MCH1 receptor antagonists: a patent review. Expert Opin. Ther. Pat. 25, 193–207 (2015).
Chun, E. et al. Fusion partner toolchest for the stabilization and crystallization of G protein-coupled receptors. Structure 20, 967–976 (2012).
Zhao, L. H. et al. Structure insights into selective coupling of G protein subtypes by a class B G protein-coupled receptor. Nat. Commun. 13, 6670 (2022).
Zhao, L. H. et al. Conserved class B GPCR activation by a biased intracellular agonist. Nature 621, 635–641 (2023).
Zhao, L. H. et al. Molecular recognition of two endogenous hormones by the human parathyroid hormone receptor-1. Acta Pharmacol. Sin. 44, 1227–1237 (2023).
Zhao, L. H. et al. Structure and dynamics of the active human parathyroid hormone receptor-1. Science 364, 148–153 (2019).
Liang, Y. L. et al. Dominant negative G proteins enhance formation and purification of Agonist–GPCR–G protein complexes for structure determination. ACS Pharm. Transl. Sci. 1, 12–20 (2018).
Duan, J. et al. Molecular basis for allosteric agonism and G protein subtype selectivity of galanin receptors. Nat. Commun. 13, 1364 (2022).
Zivanov, J., Nakane, T. & Scheres, S. H. W. Estimation of high-order aberrations and anisotropic magnification from cryo-EM data sets in RELION-3.1. IUCrJ 7, 253–267 (2020).
Rohou, A. & Grigorieff, N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Sanchez-Garcia, R. et al. DeepEMhancer: a deep learning solution for cryo-EM volume post-processing. Commun. Biol. 4, 874 (2021).
Senior, A. W. et al. Improved protein structure prediction using potentials from deep learning. Nature 577, 706–710 (2020).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Evans, R. et al. Protein complex prediction with AlphaFold-Multimer. Preprint at bioRxiv https://doi.org/10.1101/2021.10.04.463034 (2022).
Acknowledgements
The cryo-EM data were collected at the Advanced Center for Electron Microscopy at Shanghai Institute of Materia Medica, Chinese Academy of Sciences. This work was supported by the Natural Science Foundation of Shanghai (23ZR1475200 to L.-H.Z.); the National Natural Science Foundation of China (32071203 to L.-H.Z., 32130022 and 82121005 to H.E.X.); the Young Innovator Association of CAS (Y2022078 to L.-H.Z.); CAS StrateGic Priority Research Program (XDB37030103 to H.E.X.); Shanghai Municipal Science and Technology Major Project (2019SHZDZX02 to H.E.X.); Shanghai Municipal Science and Technology Major Project (H.E.X.); the National Key R&D Program of China (2022YFC2703105 to H.E.X., 2019YFA0904200); the Lingang Laboratory (LG-GG-202204-01 to H.E.X.); State Key Laboratory of Drug Research (SKLDR-2023-TT-04 to H.E.X.).
Author information
Authors and Affiliations
Contributions
Q.H. and L.-H.Z. designed the expression constructs; Q.H. purified the complexes, and prepared the final samples for cryo-EM data collection toward the structure; Q.H. and L.-H.Z. prepared the cryo-EM grids; Q.H. performed function assays and prepared figures and drafted the manuscript; W.H. and K.W. collected the cryo-EM data; Q.Y. performed map calculations; Q.Y. and H.S. built and refined the structure models; C.W., Y.G., Y.Z. and X.H. participated in data analysis; L.-H.Z. and H.E.X. performed structure and function data analysis, and wrote the manuscript; L.-H.Z. and H.E.X. conceived, designed, and supervised the overall project.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
He, Q., Yuan, Q., Shan, H. et al. Mechanisms of ligand recognition and activation of melanin-concentrating hormone receptors. Cell Discov 10, 48 (2024). https://doi.org/10.1038/s41421-024-00679-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41421-024-00679-8
