
Abstract
Inflammasomes are cytoplasmic multiprotein complexes comprising a sensor protein, inflammatory caspases, and in some but not all cases an adapter protein connecting the two. They can be activated by a repertoire of endogenous and exogenous stimuli, leading to enzymatic activation of canonical caspase-1, noncanonical caspase-11 (or the equivalent caspase-4 and caspase-5 in humans) or caspase-8, resulting in secretion of IL-1β and IL-18, as well as apoptotic and pyroptotic cell death. Appropriate inflammasome activation is vital for the host to cope with foreign pathogens or tissue damage, while aberrant inflammasome activation can cause uncontrolled tissue responses that may contribute to various diseases, including autoinflammatory disorders, cardiometabolic diseases, cancer and neurodegenerative diseases. Therefore, it is imperative to maintain a fine balance between inflammasome activation and inhibition, which requires a fine-tuned regulation of inflammasome assembly and effector function. Recently, a growing body of studies have been focusing on delineating the structural and molecular mechanisms underlying the regulation of inflammasome signaling. In the present review, we summarize the most recent advances and remaining challenges in understanding the ordered inflammasome assembly and activation upon sensing of diverse stimuli, as well as the tight regulations of these processes. Furthermore, we review recent progress and challenges in translating inflammasome research into therapeutic tools, aimed at modifying inflammasome-regulated human diseases.
Similar content being viewed by others
Introduction
Inflammation is a vital physiological response triggered by noxious agents in all metazoan organisms. Virtually any challenge to the body’s homeostasis may elicit an inflammatory response at the local or systemic levels1. The innate immune system’s task is to generate a protective response against signals of danger including both pathogenic microorganisms and sterile incursions such as trauma, cancer, ischemia, and metabolic perturbations. Technically, factors eliciting an innate inflammatory response can be classified as pathogen-associated molecular patterns (PAMPs), conserved compounds of infectious agents, and damage-associated molecular patterns (DAMPs), which are signals of host cellular distress. PAMPs and DAMPs are sensed by an increasingly appreciated variety of pattern recognition receptors (PRRs) and cells of innate and adaptive immunity2,3.
Upon encounter of a pathogenic agent or tissue injury, the innate immune system is challenged to integrate a wealth of signals to initiate a proper response. In addition to Toll-like receptors (TLR)4, Lectin receptors5, RIG-I-like receptors6, and oligoadenylate synthase (OAS)-like receptor7, inflammasomes emerged in the last decade to constitute fundamental processing units contributing to PAMP and DAMP sensing, which actively participate in integration of their downstream signaling3,8. The term “inflammasome” has originally been coined by Martinon et al.8 in a seminal report in 2002 that described the assembly of these supramolecular structures in the cytoplasm of activated immune cells, thereby leading to proteolytic activation of proinflammatory caspases, which drives subsequent systemic immune responses and inflammation. Importantly, while inflammasome signaling has been shown to be critical to host defense, the elicited immune response needs to be tightly regulated in order to limit collateral damage to the host. This implies that appropriate regulation of inflammasomes is intrinsic to the control circuit of the associated inflammatory processes.
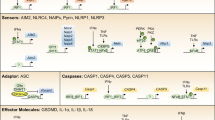
Several cytoplasmic PRRs are able to assemble into an inflammasome complex and are classified by their protein domain structures. For example, the NBD leucine-rich repeat-containing receptor (NLR) family implicates subfamilies distinguishable by their N-terminal effector domains. There are four recognizable NLR N-terminal domains: the acidic transactivation domain, pyrin domain, caspase recruitment domain (CARD), and baculoviral inhibitory repeat (BIR)-like domains9. Another class of inflammasome assembling PRRs is represented by PYHIN protein family members, such as absent in melanoma 2 (AIM2), which contain HIN200 and pyrin domains10. Inflammasome-assembling PRRs are expressed in many cell types, including macrophages, dendritic cells (DCs), neutrophils, and epithelial cells11. The final common pathway of inflammasome signaling is inflammatory caspase-activation. This task is achieved by the assembly of a hetero-oligomeric complex based on a scaffold protein, such as NOD-like receptor-pyrin-containing proteins (NLRP) or AIM2 protein, and in some inflammasomes the additional recruitment of adapter and effector partners, such as apoptosis-associated speck-like protein containing a CARD (ASC)12. The activation of caspases results in the proteolytic activation of the proinflammatory cytokines interleukin-1β (IL-1β) and/or interleukin-18 (IL-18). In particular, IL-1β is considered a gatekeeper cytokine which is critically involved in many events related to activation and regulation of inflammation13.
Considering the potency of the inflammasome-dependent immune responses, it is not surprising that dysregulated inflammasome activity is associated with a number of inflammatory disorders or of multi-factorial diseases involving an inflammatory component, including autoinflammatory disorders, cardiometabolic diseases, infection, cancer and neurological disorders12,14,15,16. Therefore, regulation of inflammasome activity and therapeutic interventions targeting structures related to inflammasome signaling constitute promising areas of basic and translational research. As it is impossible for a single review to fully cover the large and rapidly growing body of high-quality research in inflammasome biology, we aim to provide an introduction to key concepts and an update on recent evidence highlighting new aspects of inflammasome signaling regulation and its implications in health and disease, while referring to reviews dedicated to more specific features of inflammasome research throughout the text.
Inflammasome activation and assembly
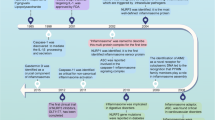
As an important arm of innate immunity, one of the most outstanding functions of inflammasomes is to detect and sense a variety of endogenous or exogenous, sterile or infectious stimuli that are encountered within the cell, and to induce cellular responses and effector mechanisms. The assembly of the inflammasome platform is a critical and well-organized process involving several core parts: the upstream sensors recognizing activating signals, the adapters and the downstream effectors (Fig. 1).

Inflammasomes can be activated by a multitude of infectious and sterile stimuli, including microbiome-derived signals (e.g., bacteria, fungi, parasites, and viruses) and host-derived signals (e.g., ion flux, mitochondrial dysfunction, ROS, and metabolic factors). Upon activation, the inflammasome sensors NAIP/NLRC4, NLRP3/6/7, AIM2/IHI16 initiate the canonical inflammasome assembly by recruiting and forming pro-caspase-1 filaments, with or without the ASC adapter. The assembly of non-canonical inflammasomes involves the pro-caspase-11 (−4/5 in humans) or pro-caspase-8. Consequently, the active caspase-1 or caspase-8 leads to the maturation and secretion of inflammatory cytokines IL-1β and IL-18. The active capsase-1 or caspase-11/4/5 triggers the cleavage of GSDMD, which can either cause pyroptosis or activate the NLRP3 inflammasome complex. In addition, the active caspase-8 mediates another effector function, apoptosis.
Pathogen-derived activating signals
A plethora of PAMPs play key roles in initiating activation of various inflammasomes, among which the most well-studied are bacteria-associated signals. Pathogenic activators of the NLRC4 inflammasome are mainly derived from Gram-negative bacteria namely Salmonella, Legionella, Shigella, and Pseudomonas spp. These bacteria possess flagellin, or a type III (T3SS) or type IV (T4SS) secretion system rod proteins that are recognized by the NAIP proteins, constituting unique binding partners of NLRC417. The murine NLRP1b inflammasome detects Bacillus anthracis lethal toxin in the cytoplasm18. The NLRP3 inflammasome can be activated by the pore-forming activity of a wide array of Gram-positive and Gram-negative bacteria, including Staphylococcus aureus, Streptococcus pneumoniae, enterohemorrhagic Escherichia coli and others, mainly through triggering potassium (K+) eflux19,20,21. NLRP6 inflammasome can sense lipoteichoic acid derived from Gram-positive pathogens like Listeria monocytogenes22. In human macrophages, the NLRP7 inflammasome recognizes acylated lipopeptides, a microbial cell wall components23. Free cytosolic DNA released from a variety of bacteria species, including but not limited to Francisella novicida, is required to activate the inflammasome-forming DNA sensor AIM224. Modification and inactivation of the Rho GTPases by bacterial toxins, for example the Clostridium difficile cytotoxin TcdB, and Clostridium botulinum ADP-ribosylating C3 toxin, are important to activate the Pyrin inflammasome, dependent on their enzymatic activities25. In addition, intracellular lipopolysaccharide (LPS) from Gram-negative bacteria is known to be recognized by the mouse caspase-11, or human caspase-4/5, in activating non-canonical inflammasomes26. Notably, although the inflammasome activation facilitates host defense against intracellular pathogenic infections, some bacteria have developed effective strategies to dampen or evade inflammasome activation, which has been recently reviewed in detail elsewhere27. For example, bacteria such as Streptococcus spp. can restrain inflammasome activation by producing hydrogen peroxide, which in turn dampens bacterial clearance from host cells28.
Moreover, specific factors from viruses, fungi and parasites have been recently shown to also activate inflammasomes. For example, the NLRP3 inflammasome in macrophages can be activated by a multitude of viruses and viral proteins, such as the hepatitis C virus core protein29, severe acute respiratory syndrome coronavirus (SARS-CoV) viroporin30, Influenza virus M231, and Encephalomyocarditis virus viroporin 2B32. NLRP9b, mainly expressed in intestinal epithelial cells (IECs), is capable of recognizing the short double-stranded RNA of rotavirus through host RNA helicase Dhx933. Major fungal PAMPs such as β-glucan upon Aspergillus fumigatus infection34, fungal CPG35, Candidalysin secreted by Candida albicans36,37 are direct inducers of inflammasome assembly, mainly for the NLRP3 inflammasome. Moreover, activation of both canonical and noncanonical inflammasomes (mainly involving mouse caspase-11 or human caspase-4/5) by diverse parasitic stimuli, such as Leishmania and its lipophosphoglycan38,39 and Fasciola hepatica-derived molecule cathepsin L340 have recently been discovered as an important strategy for the restriction and control of parasitic invasion.
The mammalian body is inhabited by a complex community of microorganisms, collectively termed the microbiome. Selective commensal bacteria can not only induce IL-18 secretion in IECs, which is mediated by NLRP6 inflammasome41, but also induce IL-1β maturation mediated by NLRP3 inflammasome signaling in intestinal monocytes, which subsequently promotes intestinal inflammation42. NLRP3 inflammasome activation induced by commensal bacteria is K+ efflux-dependent, but does not depend on bacterial viability or phagocytosis43. Sensing of commensal gut fungi through the Card9–Syk signaling axis promotes inflammasome activation and maturation of IL-18, which plays a protective role in colitis and colitis-associated carcinogenesis44.
Host-derived activating signals
In addition to PAMPs, endogenous DAMPs (or host danger signals), which are released upon tissue injury, emerged as another major source for inflammasome activation. For example, NLRP3 inflammasome which is activated by a diverse numbers and structures of exogenous stimuli ranging from particulate matter (such as silica) to bacterial-derived toxins45 can also sense other host-derived signals downstream of all these exogenous stimuli. Some of these endogenous signals, including ion efflux, mitochondrial dysfunction and reactive oxygen species (ROS) are well-characterized (see below), while others remain uncharacterized to date. An elegant example of a new host-related activator was demonstrated in a recent study showing that diverse NLRP3 stimuli can trigger the disassembly of trans-Golgi network (TGN), and the dispersed TGN acts, in turn, as a scaffold for NLRP3 recruitment through ionic bonding and subsequent inflammasome assembly and activation46.
Cytosolic K+ efflux is a common trigger involved in both canonical NLRP3 inflammasome activation21 and caspase-11-mediated noncanonical activation47. NEK7, a mitosis-related serine-threonine kinase, is essential to regulate NLRP3 oligomerization and activation downstream of K+ efflux48,49. Intracellular chloride efflux, another event downstream of K+ efflux, is a critical upstream event for NLRP3 activation50. The role of calcium (Ca2+) signaling in activating NLRP3 inflammasome remains debatable. Although some studies showed that inflammasome activation essentially requires elevation of intracellular and extracellular Ca2+51,52, other studies argued that Ca2+ is not strictly required for initiation of NLRP3 signaling53,54. The roles and mechanisms of other upstream ion flux disturbances, including Na+ and Zn2+ in orchestrating NLRP3 inflammasome activation have been extensively reviewed elsewhere55. Although it remains to be answered whether these ions directly bind to NLRP3 and trigger downstream processes, such advances provide potential ion-associated targets for modulating NLRP3-driven disorders.
Mitochondrial dysfunction is another emerging elicitor of NLRP3 activation, operating via mitochondrial DNA (mtDNA) release, mitophagy and apoptosis. For example, in aging hematopoietic stem cells, increased mitochondrial stress leads to aberrant NLRP3 inflammasome activation and contributes to characteristic aging-associated defects56. Recently, it was found that mtDNA synthesis and oxidized mtDNA release into the cytosol driven by TLR signaling is crucial to prime NLRP3 activation, while inhibition of mtDNA synthesis via TFAM or IRF1 ablation prevents NLRP3 inflammasome activation57. However, other studies show that TFAM depletion leads to increased rather than decreased cytosolic mtDNA to activate antiviral immune responses through the cGAS–STING pathway58, and that IRF1 is dispensable for the NLRP3 inflammasome activation59. Furthermore, cholesterol-dependent mtDNA accumulation in macrophages results in AIM2, but not necessarily NLRP3 inflammasome activation60. Cytosolic mtDNA activates the NLRP3 inflammasome and promotes the release of IL-1β and IL-18, while the translocation of mtDNA into cytosol, in turn, requires NLRP3, indicating that NLRP3 might act both upstream and downstream of mtDNA release61. On the other hand, damaged mitochondria, stimulated by NLRP3 activators, initiate an intrinsic “NF-κB-p62-mitophagy” pathway through which NF-κB limits NLRP3 inflammasome activation62. These results highlight a regulatory loop existing between the NLRP3 inflammasome and mitochondria. In addition, oxidized mtDNA released into the cytosol during apoptosis binds to the NLRP3 inflammasome, highlighting a link between apoptosis and inflammasome activation63. However, this conclusion has been challenged by another study showing that genetic deletion of important executioners of mitochondrial apoptosis does not affect the activation of NLRP3 inflammasome64. Indeed, besides mtDNA, intrinsic apoptosis resulting from mitochondrial membrane damage in macrophages activates Caspase-3 and -7 to drive IL-1β secretion downstream of both NLRP3 inflammasome and caspase-8 signaling65.
Emerging evidence suggests that ROS constitute another central signaling hub among a diversity of NLRP3-related stimuli45. ROS induce the binding of thioredoxin-interacting protein to NLRP3, which is essential for NLRP3 inflammasome activation66. Although some studies suggest that NLRP3 inflammasome activation may be triggered by ROS generated by an NADPH oxidase67, other studies show that activation of the NLRP3 inflammasome is independent upon NADPH oxidase-generating ROS68 or its oxidative activity69, challenging the proposed indispensable role of ROS in NLRP3 inflammasome activation. Of note, ROS generation is frequently accompanied by mitochondrial dysfunction and ion efflux. It would be worth investigating the interplay among these triggers in contributing to inflammasome activation.
Growing evidence suggests that dysregulated metabolic factors may constitute novel stimuli of inflammasome activation. These include, as an example, alterations in sterol biosynthesis70 and glycolysis metabolism71. Cholesterol overload leads to activation of the AIM2 inflammasome through impairing mitochondrial metabolism and eliciting mtDNA release60. Increased choline uptake and metabolism in macrophages keeps the NLRP3 inflammasome in an active state by maintaining functional mitochondria72. In the central nervous system, cholesterol accumulation in myelin debris in aged mice results in NLRP3 inflammasome activation, which hampers remyelination and nerve repair73. Collectively, the discovery of multiple metabolic activators of inflammasomes necessitates the investigation of the underlying mechanisms by which metabolic dysfunction impacts on inflammasome biology.
Assembly of inflammasomes
Upon activation, assembly of inflammasomes requires interactions between the inflammasome sensor and inflammatory caspase-1 or noncanonical caspase-11, with or without co-binding of the adapter protein ASC. With the help of advanced techniques such as cryo-electron microscopy, rapidly growing evidence begins to disentangle the structural mechanisms of inflammasome assembly.
NLRC4 inflammasome
The interaction between the nucleotide-binding domain (NBD) and winged-helix domain (WHD) is essential to keep NLRC4 in an auto-inhibited state74. Studies using a purified PrgJ–NAIP2–NLRC4 inflammasome revealed that one single PrgJ-bound NAIP2 molecule with a catalytic surface is sufficient to activate NLRC4 by matching its oligomerization surface, which comprises a large portion of NBD and a small part of LRR. Activated NLRC4 undergoes substantial structural reorganization, interacts, and activates another quiescent NLRC4 molecule in a self-propagating approach to ultimately form a 10- to 12-spoke wheel- or disk-like architecture75,76. Similarly, studies on the assembled flagellin–NAIP5–NLRC4 inflammasome showed that the conserved regions of the flagellin ligand recognize multiple domains of the NAIP5 molecule, resulting in NAIP5 activation77 and mediating progressive NLRC4 oligomerization78. More importantly, the extreme C-terminal side of different bacterial flagellins forms a key structural epitope for their specific detection by NAIP5, potentially explaining why different bacteria possess different potency in inducing the NLRC4 inflammasome79. How different NAIPs precisely interact with their respective ligands provides an intriguing question for further investigations.
NLRP3 inflammasome
Unlike NLRC4, the assembly of the NLRP3 inflammasome requires the presence of NEK7, a serine and threonine kinase that is critically involved in mitotic cell cycle progression48,80. NEK7 directly binds to the LRR domain of NLRP3 to promote inflammasome assembly during cell interphase49. A recent cryo-electron microscopic study focusing on the human NLRP3-NEK7 complex found that NEK7 is indispensable to bridge the gaps between adjacent NLRP3 subunits and mediate NLRP3 oligomerization, thereby clarifying the structural basis of NEK7-mediated NLRP3 inflammasome activation81. Despite these advances, it remains to be investigated whether NEK7 serves as a common sensor for multiple stimuli-induced NLRP3 activation states, and whether NEK7 is sufficient to trigger a nucleated oligomerization.
Other NLRP inflammasomes
In vitro reconstitution of the human NLRP1 inflammasome by purified recombinant proteins has characterized that NLRP1 oligomerization can directly recruit caspase-1 via its CARD domain, and can be further enhanced by binding to ASC via the pyrin domain (PYD)82. Crystal structure analysis suggests that interaction between human NLRP1 and procaspase-1 CARDs is potentially mediated by electrostatic attractions83. The role of enzymatic cleavage of murine NLRP1b in its activation is discussed in detail below. Another NLRP subfamily member, NLRP6, is capable of forming filamentous structures though the PYD by self-assembly. Remarkable conformational changes following this step, enable subsequent recruitment of the ASC adapter through PYD–PYD interaction, while the NBD of NLRP6 features a synergistic role in enhancing the assembly process84. NLRP7 can self-associate to form an oligomer through NACHT domain interaction upon activation85, which essentially requires the ATP-binding and hydrolysis activities of the NLRP7 NBD86. In a recent study, a previously uncharacterized member of the NLR family, NLRP9b, has been shown to assemble an inflammasome in the intestine upon murine enteric rotavirus infection33. The authors demonstrated that NLRP9b recognizes short dsRNA of rotavirus to form inflammasome complexes with ASC and caspase-1 to promote maturation of IL-18 and GSDMA-induced cell death33.
AIM2 and IFI16 inflammasomes
Assemblies of the AIM2 and IFI16 inflammasomes are considered different from the aforementioned NLR-dependent inflammasomes, since both the AIM2 and IFI16 sensors specifically recognize cytosolic DNA through their hematopoietic interferon-inducible nuclear (HIN) domain, while lacking the NOD for self-oligomerization. Electrostatic binding of double-stranded DNA backbone to the positively charged HIN domain liberates AIM2 autoinhibition87,88. Following this event, the helical assemblies of AIM2PYD generate a polymerization platform to nucleate downstream ASCPYD filaments, underpinning the assembly of an inflammasome89,90. Unlike AIM2, the isolated IFI16 HIN domain possesses relatively weaker DNA-binding affinity87, which can be strengthened by the presence of a full length IFI16 protein91. Furthermore, the non-DNA-binding PYD of IFI16 is necessary for the cooperative assembly of IFI16 filaments on dsDNA91.
Recruitment of ASC and caspase-1
In CARD-absent NLRs including AIM2, IFI16, NLRP3, NLRP6, and NLRP7, PYD–PYD interactions between NLRs and ASC nucleate the PYD filaments of ASC, which in turn nucleates CARD filaments of caspase-1 through CARD–CARD interactions92. This brings monomers of pro-caspase-1 into close proximity and initiates caspase-1 self-cleavage and activation. Another innate immune sensor encoded by MEFV gene, pyrin, also forms an inflammasome complex in an ASC-dependent approach93. Multiple binding sites on both PYDs of pyrin and ASC identified by spectrometry provide a molecular basis for their interactions94. In NLRC4, which contains a CARD, the nucleation and activation of downstream caspase-1 can occur through the homotypic CARD–CARD binding, and this process can be independent of ASC95. The orientation of the CARD of NLRC4 and the ability to integrate ASC into the structure is currently unknown and warrants further studies.
Noncanonical inflammasome activation
While the final common pathway of canonical inflammasome activation involves recruitment of caspase-1 in response to multiple microbial or danger signals, the newly discovered “noncanonical inflammasome” signals in a caspase-1-independent manner through direct recognition of the cytosolic LPS by the CARDs of caspase-4 and caspase-5 (in humans) and caspase-11 (in mice). This elicits caspase-dimerization and activation, resulting in cleavage of GSDMD96,97. Activation and regulation of non-canonical inflammasomes is described in detail below.
Inflammasome effector functions
Inflammasome-mediated maturation of IL-1 family cytokines IL-1β and IL-18
Secretion of the proinflammatory cytokines IL-1β and IL-18, together with induction of pyroptotic cell death (see below), represent the main outcomes of inflammasome activation upon caspase-1 activation98. Akin to caspase-1, IL-1β, and IL-18 accumulate in the cytoplasm as inactive precursors (pro-IL-1β and pro-IL-18, respectively). Caspase-1 cleaves pro-IL-1β into a 17 kDa mature fragment99 and proIL-18 into a 17.2 kDa mature protein100. IL-1β stimulates the release of other cytokines such as IL-6, tumor necrosis factor (TNF)-α and IL-1α, as well as other crucial factors responsible for growth and differentiation of immune cells13. Both IL-1α and IL-1β bind to IL-1R1, thereby enabling recruitment of its co-receptor IL-1RAc. Similarly, upon binding of IL-18 to IL-18Rα, the latter heterodimerizes with IL-18Rβ. Approximation of the intracellular TIR domains of the IL-1R or IL-18R complex results in recruitment of MyD88 followed by a cascade of downstream events, which ultimately results in the activation of important signaling proteins and transcription factors, such as NF-κB, regulating inflammation101.
Pyroptosis
Both canonical inflammasome signaling engaging caspase-1 and noncanonical inflammasome activation recruiting caspase-4, caspase-5 (in humans), and caspase-11 (in mice) elicit an inflammatory type of cell death termed “pyroptosis”102. Pyroptosis is a lytic form of programmed cell death in response to sensing of pathogens or host-derived danger signals. It is morphologically distinct from apoptosis and is characterized by cell swelling, membrane rupture, and subsequent release of inflammatory compounds into the extracellular space, such as IL‐1β, IL-6, and IL‐18103,104. A single protein from the gasdermin family, gasdermin-D (GSDMD) represents the key pyroptotic substrate of inflammatory caspases105. GSDMD is expressed in various cell types, with a particularly high density in intestinal epithelia. Pyroptosis plays a critical role in the clearance of intracellular bacteria, effectively resulting in the clearance of the pathogen’s niches106. Studies utilizing engineered bacteria showed that pyroptosis of the host cell does not directly cause bacterial death. Instead, the pathogen remains trapped within the cell remnant which is termed “pore-induced intracellular trap (PIT)”. PITs arrest bacteria during simultaneous recruitment of neutrophils and macrophages, which ultimately results in subsequent killing of the pathogen107,108.
Apoptosis
Caspase-8-driven apoptosis, also regarded as “secondary pyroptosis”, is increasingly studied as another effector function of inflammasome activation. Studies have confirmed the existence of a caspase-8 dependent apoptotic death pathway activated by AIM2, NLRP3, and NAIP–NLRC4 inflammasomes, which is parallel to, but distinct from the canonical caspase-1 dependent pyroptosis, and is present in various cell types109,110,111. In the absence of caspase-1, these inflammasomes are capable of triggering caspase-8-dependent apoptosis upon sensing of diverse stimuli112,113. Importantly the presence of caspase-1 protease activity suppresses activation and induction of caspase-8-mediated apoptosis by the sensor NLRP1b and NLRC4114. Procaspase-8 binds to the ASC through the adapter protein FADD, and co-localizes to ASC inflammasome specks, adding another layer of complexity to the inflammasome structure109,114. Interestingly, a recent study shows that apoptosis induced by inflammasome stimuli can be initiated by caspase-1 itself, in the absence of GSDMD, through the Bid–caspase-9–caspase-3 axis115. The molecular mechanisms regulating inflammasome-induced apoptosis, and the interaction between apoptosis and pyroptosis, remain to be further investigated.
Regulatory mechanisms of inflammasome activation
In general, in a well-timed and carefully controlled inflammatory response, it is critical to seek the optimal balance between inflammasome activation and inhibition. This depends on complex and highly organized regulatory mechanisms. Due to distinctions in the structures and expression patterns of inflammasome sensors, different inflammasomes are activated and regulated by different mechanisms.
NLRP1 inflammasome
Unlike other NLR family members, human NLRP1 and murine NLRP1b contains a CARD domain at its C-terminus, and a unique function-to-find domain (FIIND). NLRP1 undergoes autolytic proteolysis within the FIIND, which generates N-terminal and C-terminal fragments that remain in an auto-inhibited state, while NLRP1 inflammasome activation can be blocked by abolishing FIIND auto-processing activity18,116. Mouse NLRP1b and its rat paralog activate caspase-1 in response to a lethal toxin secreted by B. anthracis18,117. Both rodent and human NLRP1 molecules are directly proteolytically cleaved by the anthrax lethal factor, which is necessary and sufficient to activate the NLRP1b inflammasome118,119. A “functional degradation” model was recently proposed, providing a universal molecular mechanism for murine NLRP1b inflammasome activation induced by diverse pathogen enzymes. Direct cleavage of NLRP1b by pathogen-encoded proteases like lethal factor or Shigella effector IpaH7.8 induces degradation and instability of the NLRP1b N-terminus, thereby liberating the C-terminal fragment to activate caspase-1120,121. The N-terminus degradation of NLRP1b is mediated by the N-end rule ubiquitin ligase UBR2, accompanied with an E2 ubiquitin-conjugating enzyme UBE2O, as revealed by genome-wide CRISPR/Cas9 knockout screens121,122. Release of the C-terminal FIIND(UPA)–CARD fragment is required to recruit CASP1 and initiate NLRP1b inflammasome assembly120.
NLRP1b also acts as an inflammasome sensor for L. monocytogenes123 and the intracellular parasite Toxoplasma gondii124, as well as secondary to depletion of cytosolic ATP125. It remains to be investigated how human NLRP1 or murine NLRP1b senses specific pathogens, whether these pathogens also activate NLRP1 via “functional degradation”, and whether ATP depletion acts as a unifying event upstream of NLRP1 or NLRP1b inflammasome activation. Furthermore, the role of the NBD and LRR sites of NLRP1 remains elusive. A previous study showed that the PYD and LRR domains of NLRP1 inhibit its self-organization, while disruption of these two domains by NLRP1 mutation leads to inflammasome activation, and contributes to skin inflammation and skin cancer in humans126. This provides a novel mechanism for NLRP1 PYD- and LRR-mediated inflammasome regulation, which is also dependent on the auto-proteolytic cleavage within the FIIND domain. However, whether these domains also contribute to the identification of pathogen-encoded effectors warrants further studies.
Additionally, recent evidence shows that inhibition of the cytosolic serine proteases Dpp8 and Dpp9 activates the murine NLRP1b inflammasome to induce pyroptosis, which is dependent on proteasome activity, but does not require proteolysis127. Direct binding of human DPP9 to the FIIND and its catalytic activity synergize in keeping NLRP1 in an auto-inhibitory state128, while DPP8/9 inhibitors activate murine NLRP1b through an endogenous proteasomal degradation pathway122. The substrates of Dpp8/Dpp9 and the molecular mechanisms for this pathway require further investigation.
NLRP3 inflammasome
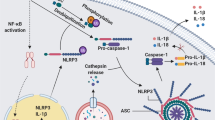
The NLRP3 inflammasome best demonstrates the complexity of inflammasome regulation in both the priming and activation steps (Fig. 2). NLRP3 inflammasome activity is tightly regulated to prevent uncontrolled activation, since aberrant signaling is associated with various autoimmune and metabolic consequences, including exacerbation of gouty arthritis, induction of cryopyrin-associated periodic syndromes (CAPS) in susceptible individuals, and possible contribution to obesity, type 2 diabetes, and Alzheimer’s disease (AD)14. It is tempting to assume that the proposed link of the NLRP3 IL-1/IL-18 axis with this multitude of metabolic and degenerative diseases may hold therapeutic potential in treating these diseases, via NLRP3 inhibition. However, recent translational studies suggest a more complex disease specific function of NLRP3 inflammasome inhibition. Notably, the results of the CANTOS trial showed that, although IL-1β inhibition with canakinumab reduced cardiovascular events in patients with a history of myocardial infarction, it did not reduce the incidence of new-onset diabetes129.

Activation of the NLRP3 inflammasome involves both the priming and activation steps. In the priming step (Signal 1), inflammatory triggers such as TLR4 agonist LPS induces the NF-κB-mediated expression of NLRP3, pro-IL-1β and pro-caspase-1. In the activation step, diverse PAMPs and DAMPs trigger the NLRP3 inflammasome assembly though unifying events such as ion flux, mitochondrial dysfunction and ROS formation. Regulation of the NLRP3 inflammasome activation can occur at the post-transcriptional and post-translational levels. MicroRNAs (e.g., MiR-223, MiR-22, MiR-7, and MiR-30e) inhibit the NLRP3 activity by targeting its UTR binding sites, and long non-coding RNAs (e.g., ANRIL, MALAT1, Neat 1, and Gm15441) can either promote or inhibit the inflammasome signaling. The post-translational modifications at different sites and domains of NLRP3 protein include phosphorylation, dephosphorylation, ubiquitination, de-ubiquitination, and S-nitrosylation. Many molecules such as BCAP, IRGM, and DDX3X can interfere the assembly of NLRP3, ASC, and procaspase-1. Other negative regulatory molecules inhibit the inflammasome activation by targeting the K+ efflux (e.g., BHB), cytosolic Ca2+ (e.g., TMEM176B), mitochondrial function (e.g., macrophage CGI-58, IL-10, and NO).
Regulation in the priming step
The priming step of NLRP3 inflammasome, or “signal 1”, typically involves an NF-κB-dependent upregulation of cellular NLRP3, pro-IL-1β transcription, and de novo protein synthesis upon recognition of pro-inflammatory stimuli such as ligands for TLRs130. This transcription-dependent priming to activate the nuclear factor-κB is mediated by the Fas-associated death domain (FADD) and later by caspase-8131. Other evidence uncovered more rapid mechanisms in priming the NLRP3 inflammasome, which are transcription-independent. For example, LPS–TLR4–MyD88 signaling in macrophages can non-transcriptionally prime NLRP3 by stimulating its deubiquitination as early as after 10 min132. Beyond TLR4, binding of TLR3 to dsRNA promotes post-translational NLRP3 inflammasome priming through TRIF/RIPK1/FADD-dependent pathways in intermediate and late phases, both of which require the FADD/caspase-8 scaffolding function133. Notably, simultaneous activation of TLRs and NLRP3 by sensing various microbial ligands triggers a rapid TLR-, IRAK-1-, and IRAK-4-dependent assembly of NLRP3 inflammasome components, which can bypass the priming step and is critical for immediate innate defense134.
Post-transcriptional regulation
Emerging evidence shows that the NLRP3 inflammasome can also be regulated at the post-transcriptional level. Epigenetic factors including DNA methylation and histone acetylation have been demonstrated to regulate NLRP3 mRNA expression in response to Mycobacterium tuberculosis infection135 and bortezomib-induced painful neuropathy136. So far, MicroRNAs are the most studied post-transcriptional regulators of NLRP3 inflammasomes. Among these, miR-223 negatively regulates the activity of NLRP3 inflammasome by targeting the UTR-binding sites of NLRP3 in myeloid cells137 and was proposed to modulate innate immune responses during intestinal inflammation138 and DAMPs-induced acute lung injury138. Aberrant expression of other microRNAs also targets and inhibits the NLRP3 inflammasome in various diseased states, e.g., microRNA-7 in neuroinflammation139, microRNA-30e in Parkinson’s disease140, and microRNA-22 in coronary heart disease141 and oral squamous cell carcinoma142. More recent studies begin to unravel a previously unanticipated role of long noncoding RNAs (lncRNAs) in orchestrating inflammasome activation. Among these, antisense noncoding RNA in the INK4 locus (ANRIL) can sponge miR-122-5p to enhance NLRP3 inflammasome activation in uric acid nephropathy143, and MALAT1 has been implicated to sponge miR-133 in ischemia–reperfusion injured myocardium144. Other lncRNAs were reported to either promote or attenuate inflammasome signaling, including nuclear enriched abundant transcript 1 (Neat1)145 and Gm15441146. These promising data provide potential for miRNA- and lncRNA-targeted therapies of diseases linked to NLRP3 dysfunction. More studies are required to clarify the roles of the complex miRNA networks in fine‐tuning NLRP3 inflammasome activation to facilitate translation into clinical applications.
Post-translational modulation
Regulation of the NLRP3 inflammasome by post-translational modifications, including phosphorylation and deubiquitination, is increasingly studied. JNK1-mediated phosphorylation of NLRP3 at Ser194 (corresponding to human Ser198) is a key non-transcriptional priming event eliciting NLRP3 self-association and subsequent inflammasome assembly147. Furthermore, E3 ubiquitin ligase Pellino2 promotes the ubiquitination of NLRP3 in the non-transcriptional priming step and facilitates the activation of the NLRP3 inflammasome148, while FBXL2 E3 ligase-mediated ubiquitination and degradation of NLRP3 plays a negative regulatory role in this inflammasome activation148.
During the activation step, phosphorylation of NLRP3 by Golgi-mediated protein kinase D (PKD) at Ser293 (or human Ser295) is sufficient to trigger inflammasome assembly149. In contrast, PKA-induced phosphorylation of NLRP3 at mouse Ser 291 mediates negative regulation of NLRP3 inflammasome induced by bile acids150. Dephosphorylation of NLRP3 PYD by Phosphatase 2A (PP2A)151, or NLRP3 Tyr861 by protein tyrosine phosphatase non-receptor 22 (PTPN22)152 also promotes efficient NLRP3 activation. Besides phosphorylation, the deubiquitinating enzyme BRCA1/BRCA2-containing complex subunit 3 (BRCC3) promotes an inflammasome activation by deubiquitinating NLRP3 at its LRR domain153. ABRO1, a component of the BRCC3 complex, enhances NLRP3 inflammasome activation by regulating NLRP3 deubiquitination after LPS priming154. Other examples of ubiquitination-mediated negative regulation of the NLRP3 inflammasome include the dopamine-induced E3 ligase MARCH7155, Ariadne homolog 2 (ARIH2)156 and TRIM31157. Interestingly, another small molecule, nitric oxide (NO), attenuates NLRP3 inflammasome activity at the post-translational level by direct S-nitrosylation158.
Negative regulation of NLRP3 inflammasome activation
In addition to post-translational and post-transcriptional regulation, a number of molecules can directly or indirectly interact with different components of the NLRP3 inflammasome, through which they impede the assembly of NLRP3 with ASC and caspase-1. For example, the B-cell adapter for phosphoinositide 3-kinase (BCAP) interacts with the caspase-1 pseudosubstrate inhibitor Flightless-1, thereby delaying the recruitment and activation of pro-caspase-1 within the “pre-inflammasome” containing only NLRP3 and ASC, and contributing to delayed clearance of pathogens159. The Crohn’s disease risk factor IRGM, directly interacts with NLRP3 and ASC, blocks their oligomerization and impedes inflammasome assembly, and alternatively mediates selective autophagy of NLRP3 and ASC, thus providing protection in gut inflammatory disorders160. Pyrin-only protein (POP) family members including POP1161 and POP2162 block inflammasome assembly by binding to ASC and inhibiting the recruitment of ASC to NLRP3. A stress granule protein DDX3X, which is involved in both NLRP3 inflammasome and stress granules assembly, regulates NLRP3 inflammasome, thereby constituting a critical checkpoint in stressed cells163. Other molecules inhibiting NLRP3–ASC interaction include the orphan nuclear receptor small heterodimer partner164, heat shock protein 70165, and NLR family CARD-containing 3 (NLRC3) protein166.
Negative regulatory molecules targeting ion efflux, mitochondrial function, and ROS signaling likewise block NLRP3 inflammasome activation. The ketone body β-hydroxybutyrate (BHB)167 inhibits NLRP3 inflammasome-mediated inflammation by preventing K+ efflux. Transmembrane protein 176B (TMEM176B) inhibits the NLRP3 inflammasome by controlling cytosolic Ca2+, while targeting TMEM176B can improve antitumor activity through enhanced inflammasome activation168. G protein-coupled receptors, which encompass a diverse class of membrane receptors, not only initiate but also suppress NLRP3 inflammasome activation by regulating ion fluxes and mtROS production, which has been reviewed recently elsewhere169. NO is also regarded as a crucial negative regulator of NLRP3 inflammasome by inducing stabilization of mitochondria158. The inflammatory cytokine IL10 inhibits NLRP3 inflammasome activation by reducing mitochondrial ROS production170. Other examples of molecules inhibiting NLRP3 inflammasome activity by targeting mitochondrial function and ROS include macrophage CGI-58 modulating insulin resistance171, and sestrin suppressing sepsis172.
Negative regulation targeting other NLRP3-related signaling pathways includes, for example, myeloid β-catenin, which is required for heat shock transcription factor 1-mediated immune regulation of NLRP3 functions in murine models of hepatic ischemia/reperfusion injury173. Blockade of IRE1α signaling, an important pathway involved in NLRP3 inflammasome assembly, dampens its assembly and subsequent caspase-1 activation174.
NAIP–NLRC4 inflammasome
The NLRC4 inflammasome is generally activated by PAMPs including bacterial flagellin and type III secretion system (T3SS) components. Unlike other NLRPs, NLRC4 partners with another NLR family member, NAIP, which directly binds to bacterial ligands. NAIPs are initially recognized and activated by PAMPs, and subsequently bind to NLRC4, releasing autoinhibition of NLRC4 and inducing inflammasome formation175. At the molecular level, ligand specificity is mediated by NBD-associated helical domains of mouse NAIPs176. A recent study revealed that caspase-7 and GSDMD are critical mediators downstream of the NAIP5/NLRC4 inflammasome for restriction of pathogenic infection177.
The role of post-translational modulation in regulating the activity of the NLRC4 inflammasome remains controversial. For example, phosphorylation of NLRC4 Ser533 by protein kinase Cδ (PKCδ) necessitates NLRC4 inflammasome activity against Salmonella typhimurium infection178. NLRC4 phosphorylation induced by flagellin can also prime the inflammasome for activation by NAIP5, suggesting a biphasic activation mechanism for the NLRC4 inflammasome179. However, another study showed that NLRC4 phosphorylation by PKCδ kinase is dispensable upon recognition of Shigella T3SS inner rod protein MxiI by NAIP2180. Interestingly, NLRC4 S533A interacts directly with infection-induced NLRP3, which further recruits ASC and activates caspase-1181. The association between NLRC4 and NLRP3 is not only noted to occur in the NLRC4 S533A mutant, but also wildtype NLRC4 and NLRP3 both can be recruited to a macromolecular complex containing ASC and caspase-1182. The involvement of NAIPs in NLRC4–NLRP3 interaction remains to be investigated. Besides phosphorylation, a 26S proteasome-associated protein, surg1, binds to the 91–253 residues of NLRC4 and activates NLRC4 by ubiquitination. Future studies are required to disentangle the types and sites of NLRC4 phosphorylation and ubiquitination, and the molecular mechanisms underlying positive and negative regulation of the NLRC4 inflammasome. It should be noted that regulatory mechanisms governing NAIPs are equally important. A recent study uncovered interferon regulatory factor 8 (IRF8) as a critical transcriptional regulator of different murine NAIPs, which act in concert as a prerequisite for optimal NLRC4 inflammasome activation serving defense against infectious pathogens183.
NLRP6 inflammasome
NLRP6, previously called PYPAF5, has initially been identified as an activator of NF-κB and caspase-1 in different immune cells184. NLRP6 is characterized by high expression in murine IEC, including enterocytes185 and goblet cells186. Indeed, emerging evidence shows that NLRP6 is a central regulator of host-microbiome interactions through both inflammasome-dependent and -independent mechanisms. NLRP6 has been shown to regulate intestinal innate immune defense against enteric viral infections187 and diverse bacterial pathogens188 in an inflammasome-independent manner. Importantly, NLRP6 deficiency in IEC is associated with impaired IL-18 production185 and depleted caspase-1 activation41. The NLRP6 inflammasome in IEC has a particular importance in protection against intestinal inflammation185, colitis-associated tumorigenesis189,190 persistent enteropathogenic infection and regulation of goblet cell mucus secretion186. Mechanistically, commensal bacteria and microbiota-associated metabolites, such as taurine, regulate NLRP6 inflammasome signaling, IL-18 secretion, and downstream antimicrobial peptides in the intestine to shape the host–microbiome interface, via direct or indirect mechanisms not elucidated to date41. The NLRP6 inflammasome can also be activated by bacterial TLR ligands in specific sentinel goblet cell through TLR-Myd88 signaling, which subsequently triggers muc2 production191. Furthermore, lipoteichoic acid, produced by gram-positive pathogens, binds and activates the NLRP6 inflammasome through recruitment of ASC, leading to both caspase-1 and caspase-11 mediated IL-18 secretion, and ultimately exacerbating systemic infection22. Interestingly, stress-induced corticotropin-releasing hormone specifically inhibits NLRP6 inflammasome activation, leading to gut microbiome alterations and intestinal inflammation192. Future studies will need to identify more pathogenic activators and negative regulators of NLRP6 inflammasome and the mechanisms by which regulation of NLRP6 contributes to modulation of intestinal inflammasome-associated diseases.
Notably, the NLRP6 inflammasome is one the first host factors suggested to regulate the composition and function of the intestinal microbiome. Susceptibility of mice with NLRP6 deficiency to inflammation and carcinogenesis is transmissible to wild-type mice via fecal microbial transfer185,193. More specifically, NLRP6 has been proposed to limit intestinal colonization by specific colitogenic species. Specific-pathogen free NLRP6-IL-10 double knockout mice are prone to colitis resulting from expansion of the mucin degrading species Akkermansia muciniphila, mediated by the associated under-expression of IL-18194. This diseased phenotype is transmissible to single IL-10−/− knockout mice by oral gavage of A. muciniphila194. Importantly, these results have been shown to be vivarium and microbiome-specific while lesser or significant changes in gut microbial composition are shown to occur in mice lacking NLRP6 in some vivaria compared to their WT littermates195,196. Likewise, NLRP6 inflammasome baseline regulation of goblet cell function and mucus secretion appears to depend on the microbiome context, differentially driven by “sentinel goblet cell” activation by defined commensal signals197. Collectively, these results likely are explained by differences in the experimental design and facility-specific microbiota characteristics, leading to differential effects on inflammasome signaling198. Distinct community structures and, in particular, exposure to specific potentially pathogenic symbionts (pathobionts), such as members of the Helicobacteraceae family, are likely required for dysbiosis to manifest biological effects in the context of NLRP6 deficiency199.
AIM2 inflammasome
The ALRs are cytosolic sensors of a variety of endogenous and exogenous ligands. AIM2 is a non-NLR protein belonging to the ALR family, whose structure is characterized by a HIN200 and an N-terminal pyrin (PYD) domains24,200. In a quiescent state, AIM2 is mostly expressed in the spleen, peripheral blood and the intestine, but may rapidly be induced, predominantly by type I IFN signaling upon NF-κB transcriptional activation (“signal 1”)201.
In eukaryotic organisms, DNA is normally absent from the cytosol. AIM2 is activated via its HIN200 domain by the cytosolic presence of dsDNA, which can be considered a danger signal202. AIM2 recognizes cytosolic dsDNA in a non-sequence-specific manner, however, the sequence is required to be at least 80 base pairs long87. ALRs bind dsDNA of multiple origins, including host, microbial, and synthetic molecules201. Many bacterial organisms activate AIM2, including Francisella tularensis, L. monocytogenes, S. pneumoniae, Mycobacterium species, Legionella pneumophila, and S. aureus203. Bacteriolysis is a prerequisite for pathogen recognition in order to enable cytosolic exposure of free dsDNA. Apart from bacteria, genetic materials from DNA viruses entering the cytoplasm can be recognized by AIM2. This initiates an antiviral immune response against DNA viruses such as mouse cytomegalovirus (MCMV), vaccinia virus, and human papilloma viruses200,204,205. However, AIM2 is not sensed by some DNA-viruses, such as human herpes simplex virus (HSV)-1204. This highlights that some DNA viruses may have evolved escape or inhibition strategies to this inflammasome activation. The mechanisms by which viral DNA is exposed to AIM2 in the cytosol are still unknown. Conversely, AIM2 has been demonstrated to drive anti-viral responses against some RNA viruses206. However, how AIM2 senses RNA remains unclear. In addition to bacteria and viruses, AIM2 participates in defense against eukaryotic pathogens such as the fungus A. fumigatus207 and the protozoan pathogen Plasmodium berghei208. Activation of the AIM2 inflammasome by dsDNA can not only mediate pyroptosis in a caspase-1-dependent manner, but also to instigate apoptosis by engaging caspase-8. The balance between pyroptosis and apoptosis depends on the amount of DNA, with pyroptosis occurring at higher transfected DNA concentrations109.
Beyond the recognition of foreign DNA, the sensing of self-DNA recently emerges as a crucial function of the AIM2 inflammasome. Under physiological conditions, human host DNA is compartmentalized in the nucleus and mitochondria. Alterations of the nuclear envelope integrity and subsequent release of nuclear DNA into the cytosol promote AIM2 inflammasome signaling. Perturbation of nuclear envelope integrity can therefore directly initiate an innate immune response and cell death209. Surprisingly, AIM2 is also shown to sense ionizing-radiation-induced DNA damage within the nucleus, thereby triggering caspase-1-mediated cell death in intestinal epithelial and bone marrow-derived cells210. Together, these findings highlight the regulatory functions of the AIM2 inflammasome in distressed tissues beyond its well-established role as a pathogen sensor.
Some mechanisms regulating AIM2 activity have been recently deciphered (Fig. 3). One way of regulating the AIM2 inflammasome activity involves the engagement of decoy proteins. The PYD-only proteins (POPs) represent a family of inflammasome inhibitors occupying the PYD in ASC and PYD-containing PRRs, thereby blocking PYD–PYD interactions required for inflammasome assembly211,212. Each main branch of inflammasome-activating PRRs in humans may have evolved a corresponding POP regulator. The type I IFN-inducible member of the POP family, POP3, was shown to inhibit the activation of ALR inflammasomes upon encounter of immunogenic DNA by interacting with PYD, thereby preventing the recruitment of ASC213. In addition to POPs, a protein within the ALR family has been identified as a decoy inhibitor of AIM2 in mice. Akin to AIM2, the HIN-200 protein p202 binds transfected dsDNA but lacks a PYD, therefore, preventing inflammasome activation214. Moreover, mouse p202 contains two HIN-domains: HIN1 attracts DNA to its surface while HIN2 mediates tetramerization of p202. The tetramers directly interact with the HIN-domain of AIM2 to prevent clustering with ASC215. Certain DNA sequences such as the TTAGGG repeat, commonly found in mammalian telomeres, can serve to suppress innate immune activation. A suppressive ssDNA oligodeoxynucleotide6 composed of four repeats of TTAGGG (ODN A151) has been shown to competitively inhibit AIM2 signaling by blocking availability of activating DNA ligands216. However, the in vivo role of ODNs in regulating AIM2 remains to be elucidated.

AIM2 is composed of the N-terminal pyrin (PYD) and the C-terminal HIN200 domains. Interaction of both domains renders the molecule inactive. Binding of free cytosolic double-stranded (ds)DNA releases auto-inhibition with subsequent oligomerization, recruitment of adapter ASC via PYD–PYD interaction and polymerization of the ASC protein. The dsDNA instigating the cascade can originate from microorganisms of different kingdoms and from host cellular damage. Through ASC polymerization pro-caspase-1 is recruited to the complex via CARD–CARD interactions. This induces the maturation and secretion of IL-1β and IL-18, and results in pyroptosis. AIM2 inflammasome signaling can be inhibited on several levels by decoy proteins. Decoy ALRs such as P202, which lack a PYD, may interfere with AIM2 binding to dsDNA and may also inhibit AIM2 oligomerization. POPs are decoy ASC proteins inhibit PYD–PYD interactions between ASC and AIM2. Decoy caspase-1 (COPs) lack a caspase domain and inhibit caspase-1 recruitment to the inflammasome by blocking CARD–CARD association.
Pyrin inflammasome
Pyrin is a high molecular mass (86 kDa) protein mostly found in immune cells such as neutrophils, monocytes and DCs217. Human pyrin consists of four functional domains: PYD, a zinc finger domain (bBox), a coiled coil (CC) domain, and a B30.2/SPRY domain217. Pyrin mediates caspase-1 inflammasome assembly in an ASC-dependent way upon recognition of an inactivating modification of the RhoA GTPase by pathogens25,93,218. Pyrin is a unique immune sensor, since it senses bacterial virulence via cytoskeletal remodeling rather than by microbial compounds219. Mouse pyrin has two functional phosphorylation sites, Ser-205 and Ser-241 (corresponding to Ser-208 and Ser 242 in humans), which render pyrin inactive by binding 14-3-3 proteins. Upon toxin stimulation or bacterial infection resulting in Rho modification, Ser-205 and Ser-241 are dephosphorylated leading to 14-3-3 dissociation. This cascade results in activation of Pyrin and formation of an oligomeric pyrin-ASC inflammasome complex (Fig. 4)219. Pathogenic Yersinia spp. secrete a virulence factor, Yersinia outer protein M (YopM), which suppresses pyrin inflammasome activation by employing host kinases PRK1 and PRK2 in order to maintain pyrin in an inactive phosphorylated state220. Pyrin associates with cytoskeletal microtubules and actin filaments221. Drugs interfering with microtubule dynamics, such as vinblastine or colchicine, inhibit pyrin inflammasome formation without affecting the pyrin phosphorylation state. This indicates that microtubule elements control the activation of the pyrin inflammasome, perhaps by impacting on the conformational change of dephosphorylated pyrin and associated ASC recruitment219,222. Together these concepts highlight an interesting new paradigm, in which innate immune components may engage with the cytoskeleton, thereby providing new mechanisms of structural modulation of cellular immunity.

Pyrin is kept autoinhibited by 14-3-3 proteins which are bound to the phosphorylated pyrin. The inactivating pyrin phosphorylation is maintained by the kinases PRK1 and PRK2. The kinases are activated by RhoA GTPase. Inactivation of RhoA by bacterial effector proteins leads to dephosphorylation of PRK1/2. Subsequently, pyrin is de-phosphorylated, leading to dissociation of 14-3-3 and pyrin activation. Activated pyrin recruits ASC via PYD–PYD interactions. The complex then associates with pro-caspase-1 through CARD-CARD interactions. Active caspase-1 mediates IL-1β and IL-18 maturation, as well as pyroptosis. Yersinia outer protein M (YopM) suppresses the pyrin inflammasome activation by employing PRK1/2. Microtubule polymerization inhibition by colchicine leads to release of GEF-H1 which renders RhoA GTPase active, resulting in inactivating phosphorylation of pyrin by PRK1/2.
In contrast to NLRP3 and NLRP6, knowledge regarding specific pyrin inflammasome activators produced by the residing intestinal microbiota remains scarce. In a study employing DSS-induced colitis in mice, pyrin inflammasome signaling prevented dysbiosis, promoted intestinal barrier integrity and ameliorated colonic inflammation and tumorigenesis223. These results shed light on an emerging role of pyrin inflammasome in maintaining intestinal homeostasis. In a recent work using whole-genome pooled CRISPR screen technology, two bile acid analogs (BAA485 and BAA473) have been identified as specific ligands inducing pyrin inflammasome signaling in myeloid and IEC lines224. As the enteric bacterial metagenome is a rich source of bile acid metabolism, similar microbiome-derived pyrin-inflammasome activating ligands may contribute to the regulation of intestinal homeostasis. However, the existence of such pyrin-specific ligands has not been verified in situ yet.
Noncanonical inflammasomes
The term “non-canonical inflammasomes” was first coined in the seminal study by Kayagaki et al.225 to describe the finding that caspase-11 in mice (with the human orthologs caspase-4/5) is activated by mechanisms deviating from the “canonical” NLR-ASC-caspase-1 paradigm of inflammasome activation. Non-canonical inflammasome activation targets caspase-11, while loss of caspase-11 rather than caspase-1 protects mice from lethal LPS exposure225.
In noncanonical inflammasome signaling, caspase-11 acts as a sensor of LPS transfected into the cytosol from Gram-negative bacteria226. Upon recognition of LPS, caspase-11 initiates IL-1β proteolytic maturation and pyroptotic cell death in a GSDMD-dependent manner96. Caspase-11 directly binds to LPS via its CARD domain227. Noncanonical inflammasome activation, therefore, represents an LPS sensing system bypassing membrane-bound TLR4 activation and specifically defending against pathogens invading the cytosol26. In particular, caspase-11 is responsive to the penta- and hexa-acylated lipid A moiety of LPS, whereas tetra-acylated lipid A is not detected, providing a mechanism of evasion by the cytosol-invasive strain Francisella spp228. Caspase-11 signaling engages the NLRP3 inflammasome, thereby cross-recruiting caspase-1 signaling and eliciting maturation of IL-1β and IL-1896.
DCs use PRRs to sense microorganisms or dying cells in order to induce protective immunity. An oxidized phospholipid released from dying cells, oxPAPC, binds to caspase-11 in DCs to induce an inflammatory program. While binding of caspase-11 to oxPAPC and LPS signals to DCs to produce IL-1β and undergo cell death, binding to oxPAPC alone triggers DCs to secrete IL-1β and to induce a strong T-cell mediated immune response229. These results highlight that the outcomes of inflammasome signaling may vary depending on the biological context. Caspase-11 is regulated at both the transcriptional and posttranslational levels. Both NF-κB and signal transducer and activator of transcription (STAT) 1 bind to the promoter region of caspase-11226. TRIF, an adapter molecule responding to activation of TLRs, was identified as a regulator of caspase-11. TRIF is engaged by Gram-negative bacteria to activate caspase-11 via type I IFN signaling20. The NOD2-RIP pathway regulates ROS production and c-JUN N-terminal kinase signaling to control caspase-11 expression and subsequent activation of caspase-11 and NLRP3 inflammasome signaling during infection with Citrobacter rodentium230. Moreover, the complement-related Cbp1–C3a–C3aR axis increases caspase-11 expression by enhancing TLR-4–TRIF–IFNAR signaling via modulation of P38 mitogen-activated protein kinase226.
Similar to caspase-11 in mice, caspase-4/5 in humans are activated by binding hexa-acylated lipid A227. However, the biological significance and the regulation of caspase-4/5 in cytosolic recognition of LPS during bacterial infections in humans requires further investigation.
Recent studies have identified the formation of noncanonical caspase-8-dependent inflammasomes in response to different stimuli. For example, The NLRP3–ASC–caspase-8 noncanonical inflammasome can be activated upon epithelial cell apoptosis231, or Cryptococcus neoformans infection232, as well as by TLR stimulation in microglia233. Another noncanonical inflammasome complex comprising CARD9, Bcl-10, MALT1, caspase-8, and ASC is formed upon exposure to the extracellular pathogen sensor Lectin-1234. Besides the aforementioned caspase-8 mediated apoptosis, activation of the caspase-8-based inflammasome results in processing and maturation of IL-1β in various conditions233,234.
Other inflammasomes
Much of the immunological literature is focused on the aforementioned inflammasomes. Nevertheless, the NLR and PIHYN protein families are large and entail additional molecules capable of assembling inflammasomes, including IFI16 and NLRP7. However, knowledge on their biology, regulatory mechanisms, and medical significance is still limited.
IFI16 inflammasome
IFI16 (interferon-inducible protein 16) is, like AIM2, a member of the PYHIN protein family, and forms an atypical inflammasome. IFI16 is composed of a PYD, HIN-A, and HIN-B domains202. IFI16 is present in humans, but not in mice. In contrast to its cytosolic relative AIM2, inactive IFI16 is predominantly present in the nucleus of human cells235. IFI16 is believed to be a “nucleus-associated inflammasome sensor component” for recognition of viruses replicating within the nucleus235. Upon infection with Kaposi Sarcoma herpesvirus (KSHV), IFI16, ASC, and procaspase-1 interact and are redistributed to the cytoplasm to form a functional inflammasome, leading to caspase-1 activation and IL-1β cleavage235. The existence of nuclear inflammasomes adds another layer of complexity to the innate intracellular immunity. Moreover, IFI16 acts as a DNA sensor that induces cell death of quiescent CD4 T cells in abortive HIV infection236.
NLRP7 inflammasome
The NLRP7 inflammasome serves multiple functions in inflammation and embryonic development237. NLRP7 is only present in humans and no mouse ortholog has been identified to date238. The NLRP7 encoding gene together with its closest relative, NLRP2, is located on the human chromosome 19. The presence of NLRP7 in various immune cells, such as macrophages, mediates the recognition of bacterial lipopeptides to promote ASC-dependent caspase-1 activation, IL-1β and IL-18 maturation, and restriction of intracellular bacterial replication23. Among other nonimmune tissues, NLRP7 is expressed in ovaries and oocytes and exerts its protective role in embryonic development239. A wide array of mutations in the NLRP7 encoding gene have been associated with manifestation of hydatidiform mole, which is a trophoblastic disease resulting in a nonviable pregnancy237,239. However, the mechanisms by which NLRP7 regulates embryonic development are still unclear. Beside its inflammasome forming capabilities, NLRP7 also prevents inflammasome formation238. These apparently contradictory findings are probably best reconciled by assuming that NLRP7 serves as a negative regulator of inflammation in quiescent cells, while in response to a proper stimulus, such as infection, NLRP7 promotes inflammasome assembly and caspase-1 activation238.
Non-inflammasome forming NLRs
As highlighted above, much of the research on NLRs focused on their function in inflammasome assembly and processing of inflammatory cytokines. However, some NLRs regulate crucial inflammasome-independent processes. Several NLR proteins, including NLRX1, NLRC3, NLRC5, and NLRP12, can negatively regulate inflammation, especially by controlling type I interferon and NFκB signaling240,241,242,243. Notably, NLRX1 stands out from other NLR proteins by its localization within mitochondria, where it regulates mitochondrial antiviral immunity244,245. Moreover, NLRC3 acts as a key inhibitory sensor of PI3K–mTOR pathways, thereby mediating protection against colorectal cancer246. There is no general consensus on whether NLRP12 or NLRC5 are also capable of assembling an inflammasome. Although a few studies suggest that such a putative inflammasome-dependent NLRP12 activity modulates certain infections247 and obesity248, others suggest that NLRP12’s main roles are inflammasome-independent and involve regulation of infection and colon inflammation243,249,250,251. NLRC5 has been implicated to form an inflammasome by cooperating with NLRP3 in some studies252,253, while others suggest that NLRC5 modulates anti-pathogenic immunity in an inflammasome-independent manner 254,255.
Challenges and unknowns in inflammasome research
Despite the great advances in inflammasome research, many unknown challenging areas remain to be explored. First, although much knowledge has been gained about inflammasome activation upon bacterial pathogens, inflammasone sensing and reactivity to eukaryotic activation such as fungi, parasites and their products, awaits further investigation and discovery. Second, in addition to cytokine secretion, pyroptosis and apoptosis, the functional outputs of inflammasome activation on other cellular process, such as intestinal or skin barrier function, autophagy, mucus secretion, and more constitute attractive prospects for future research. Third, the context-specific potential for commensal microorganisms and of microbiome-associated metabolites to induce inflammasome activation needs to be further delineated in molecular detail. Given the existence of inter-facility variability in gut microbiome composition and function198, such regulation of inflammasome activation might be influenced by facility-dependent differences in microbiome configurations. Mechanisms mediated by commensal microbes or their products might also contribute to dampening or fine-tuning of inflammasome activation, which has yet to be determined in great details. Last, the function of many NLRs, including the NLRC, NLRP, NAIP subfamily members, remains unclarified to date. Future investigation on their inflammasome-forming ability, activation and functional consequences upon yet unknown triggers and the underlying regulating mechanisms, will greatly help to decode the complex inflammasome network and its integrative immune activities.
Human diseases associated with inflammasome dysregulation
As highlighted above, the innate immune system is programmed to carry out a prompt and vigorous immune response towards exogenous and endogenous noxious threats. As inflammasomes represent important sensors mediating innate immune responses, it is logical that their dysregulation is associated with excessive and self-perpetuating inflammation, at different clinical contexts, which can be detrimental to the host. It is now clear that regulated or dysregulated inflammasome activation participate in most types of inflammatory disorders256. Inflammasome participation in the pathogenesis of multiple clinical disorders is described in more detail elsewhere16,257,258. In short, mutations affecting inflammasome sensor-encoding genes cause several hereditary autoinflammatory disorders, such as familial cold autoinflammatory syndrome, Muckle–Wells-syndrome or familial Mediterranean fever259,260,261. Moreover, evidence from murine and human studies associated inappropriate inflammasome activation with enhanced risk of development or progression of several common acquired human diseases, such as neurodegenerative and metabolic disorders including, among others, multiple sclerosis, AD, atherosclerosis, and type 2 diabetes262,263,264,265,266. Another rapidly advancing area of inflammasome research is their role in initiation and progression of cancer. Low-grade inflammation sustained by inflammasome singling contributes to all stages of tumorigenesis. Conversely, inflammasome activation also participates in tumor control, highlighting the diverging signals and complex roles of inflammasomes in different parts of the cancer niche267. The central role of inflammasomes in such a large array of diseases renders them promising drug targets, thereby meriting future basic and clinical studies.
Recent advances in translating inflammasome research into therapeutics
To date, several inhibitors of inflammasomes have been identified and successfully implemented in disease models, with much of the research focusing on the NLRP3 inflammasome. Translation of basic inflammasome biology into therapeutic applications is a rapidly growing domain involving numerous diseases. Excellent recent reviews concisely cover this subject267,268. For example, sulforaphane is a naturally occurring phytochemical derived from cruciferous vegetables, such as broccoli. It inhibits caspase-1 proteolytic activation, IL-1β maturation and secretion downstream of several inflammasomes, including the NLRP1, NLRP3, and NAIP5–NLRC4 inflammasomes269, it and potentially also the AIM2 inflammasome269,270. Sulforaphane inhibits cell recruitment to the peritoneum and interleukin‐1β secretion in an in vivo model of gouty peritonitis model and reverses NLRP1‐mediated resistance to B. anthracis spore infection in mice269, as well as playing a preventive and protective role in high fat diet induced non-alcoholic steatohepatitis271, acute pancreatitis272 and diabetic retinopathy273. Besides, sulforaphane was suggested to be a promising drug candidate for AD therapy, as it inhibits cathepsin B- and caspase-1-dependent NLRP3 inflammasome activation induced by amyloid-β (Aβ)274. However, the importance of cathepsin B for crystal-induced NLRP3 responses has been debated by several groups showing normal NLRP3 activation in cathepsin B knockout macrophages275. Additionally, the roles of different cathepsins remain unclear due to compensatory upregulation of cathepsin activity and/or redundancy that has been suggested to account for the inability of genetic experiments to delineate a conclusive role for specific cathepsins in activating NLRP3276. To date, clinical studies in various inflammatory conditions utilizing sulforaphane or precursor-containing extracts have shown promising, yet inconsistent results. These have been recently reviewed elsewhere277. Further randomized controlled clinical studies are warranted to translate Sulforaphane use into clinical practice.
Moreover, the anti-type 2 diabetes drug glyburide, has been shown to inhibit NLRP3 inflammasome activation and consequent microbial ligand-, DAMP-, and crystal-induced IL-1β secretion278. Likewise, the small molecule MCC950 was demonstrated to block both canonical and noncanonical NLRP3 activation at nanomolar concentrations, to reduce IL-1β production in vivo and to attenuate the severity of experimental autoimmune encephalomyelitis (EAE) in mice, a disease model of multiple sclerosis279. Similarly, the small molecule CY-09 directly binds to the ATP-binding motif of NLRP3 NACHT domain and inhibits NLRP3 ATPase activity, which results in suppression of NLRP3 inflammasome assembly and activation. Administration of CY-09 showed considerable therapeutic efficacy in modulating mouse models of cryopyrin-associated autoinflammatory syndrome and type 2 diabetes280. Isoliquiritigenin (ILG), a flavonoid derived from the flowery plant Glycyrrhiza uralensis, potently inhibits LPS plus IAPP‐induced IL‐1β production in bone marrow-derived macrophages, hence improving HFD-induced obesity, lipid homeostasis, insulin resistance, and hepatic steatosis in mice281. Furthermore, ILG Inhibition of the NLRP3 inflammasome by triggering of Nrf2 activity was demonstrated to ameliorate LPS-induced acute lung injury282 and intracerebral hemorrhage in mice283.
Other groups of compounds shown to effectively inhibit the NLRP3 inflammasome include several clinically approved non-steroidal anti-inflammatory drugs (NSAIDs) of the fenamate class, acting via inhibition of volume-regulated anion channels in macrophages, independent of their roles on inhibiting COX enzymes284. The therapeutic effects of fenamates were demonstrated in models of Aβ-induced memory loss and a transgenic mouse model of alzheimer's disease284. Recently, microRNA-based post-transcriptional control of NLRP3 became a focus of translational research285. MicroRNA-9 showed promising results in inhibiting NLRP3 inflammasome activation through the JAK1/STAT signaling pathway in human cell models of atherosclerosis-associated inflammation286. OLT1177, a β-sulfonyl nitrile compound suggested to act as an NLRP3 inflammasome inhibitor, ameliorated metabolic perturbations associated with inflammation and proved to be safe in humans287. Likewise, the ketone body beta-hydroxy-butirate (BHB) or a ketogenic diet attenuated caspase-1 activation and IL-1β secretion in mouse models of NLRP3-mediated diseases such as Muckle–Wells syndrome, familial cold autoinflammatory syndrome, and urate crystal-induced peritonitis, providing a potential mechanistic explanation for the well-known anti-inflammatory effects of caloric restriction and ketogenic dieting167. Their therapeutic utility in patients remains to be determined.
Despite that the clinical promise is presented by the increasing number of small molecule inflammasome inhibitor candidates discovered during the past decade, their medical benefits and long-term safety still largely await proof by randomised controlled human trials. Moreover, as comparative data is limited, there is currently little evidence supporting the choice of one compound over the other in a given medical condition. Arguably, selective inflammasome inhibitors may be preferable over non-selective ones (e.g., sulforaphane), by avoiding or minimizing activation of other inflammasomes, thereby maintaining crucial functions in host defense intact while minimizing adverse effects288. Whether such selective and nonselective inflammasome inhibitors differ in efficacy and safety merits future studies.
Perspectives and future directions
Recent advances in inflammasome research have greatly enhanced our understanding of how recognition of PAMPs and DMAPs by the innate immune system triggers inflammation. Given the intimate association between inflammasome dysregulation and various human diseases, it is both intriguing and challenging to unravel the complex mechanisms underlying inflammasome activation and regulation and their significance for human health and disease. First, as the structural mechanisms of canonical inflammasome assembly are progressively elucidated, future investigations will focus more on structural details regarding ligand-sensor binding, interaction between different inflammasome sensors and noncanonical inflammasome formation. Unraveling these will enable to identify how divergent signals activate inflammasomes, and how different types of inflammasomes interact with each other. Second, given the diversity of microbiome- and host-derived factors activating or inhibiting inflammasomes, particularly of the NLRP3 inflammasome, it is crucial to identify a unifying triggering pathways upstream of inflammasome activation, which will facilitate the development of potential therapeutic targets. Third, despite a growing body of research, our knowledge on molecular mechanisms orchestrating inflammasome activation remains incomplete. For example, the molecular events linking the K+ efflux and NLRP3–NEK7 inflammasome assembly are still elusive. Future studies are required to identify key molecules that can potently impede or promote inflammasome assembly or activation. Designing therapeutic drugs by targeting these key molecules may be a promising approach contributing to development of new disease treatments and prevention strategies. Finally, many studies focusing on inflammasome biology focused so far on small animal models. Translation to the humans is challenging, but critically necessary given the considerable interspecies divergence between murine and human inflammasomes. Therefore, it is imperative to expand future inflammasome investigations into the context of human health and disease.
References
Medzhitov, R. Origin and physiological roles of inflammation. Nature 454, 428–435 (2008).
Janeway, C. A. Jr & Medzhitov, R. Innate immune recognition. Annu Rev. Immunol. 20, 197–216 (2002).
Schroder, K. & Tschopp, J. The inflammasomes. Cell 140, 821–832 (2010).
Akira, S. & Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 4, 499–511 (2004).
Geijtenbeek, T. B. & Gringhuis, S. I. Signalling through C-type lectin receptors: shaping immune responses. Nat. Rev. Immunol. 9, 465–479 (2009).
Loo, Y. M. & Gale, M. Jr Immune signaling by RIG-I-like receptors. Immunity 34, 680–692 (2011).
Hornung, V., Hartmann, R., Ablasser, A. & Hopfner, K. P. OAS proteins and cGAS: unifying concepts in sensing and responding to cytosolic nucleic acids. Nat. Rev. Immunol. 14, 521–528 (2014).
Martinon, F., Burns, K. & Tschopp, J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10, 417–426 (2002).
Ting, J. P. et al. The NLR gene family: a standard nomenclature. Immunity 28, 285–287 (2008).
Cridland, J. A. et al. The mammalian PYHIN gene family: phylogeny, evolution and expression. BMC Evol. Biol. 12, 140 (2012).
Martinon, F., Mayor, A. & Tschopp, J. The inflammasomes: guardians of the body. Annu. Rev. Immunol. 27, 229–265 (2009).
Strowig, T., Henao-Mejia, J., Elinav, E. & Flavell, R. Inflammasomes in health and disease. Nature 481, 278–286 (2012).
Dinarello, C. A. A clinical perspective of IL-1beta as the gatekeeper of inflammation. Eur. J. Immunol. 41, 1203–1217 (2011).
Menu, P. & Vince, J. E. The NLRP3 inflammasome in health and disease: the good, the bad and the ugly. Clin. Exp. Immunol. 166, 1–15 (2011).
Walsh, J. G., Muruve, D. A. & Power, C. Inflammasomes in the CNS. Nat. Rev. Neurosci. 15, 84–97 (2014).
Man, S. M. Inflammasomes in the gastrointestinal tract: infection, cancer and gut microbiota homeostasis. Nat. Rev. Gastroenterol. Hepatol. 15, 721–737 (2018).
Zhao, Y. et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477, 596–600 (2011).
Levinsohn, J. L. et al. Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog. 8, e1002638 (2012).
Hayward, J. A., Mathur, A., Ngo, C. & Man, S. M. Cytosolic recognition of microbes and pathogens: inflammasomes in action. Microbiol. Mol. Biol. Rev. 82, e00015–e00018 (2018).
Rathinam, V. A. et al. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell 150, 606–619 (2012).
Munoz-Planillo, R. et al. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38, 1142–1153 (2013).
Hara, H. et al. The NLRP6 inflammasome recognizes lipoteichoic acid and regulates gram-positive pathogen infection. Cell 175, 1651–1664 (2018). e1614.
Khare, S. et al. An NLRP7-containing inflammasome mediates recognition of microbial lipopeptides in human macrophages. Immunity 36, 464–476 (2012).
Fernandes-Alnemri, T. et al. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat. Immunol. 11, 385–393 (2010).
Xu, H. et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 513, 237–241 (2014).
Kayagaki, N. et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341, 1246–1249 (2013).
Brewer, S. M., Brubaker, S. W. & Monack, D. M. Host inflammasome defense mechanisms and bacterial pathogen evasion strategies. Curr. Opin. Immunol. 60, 63–70 (2019).
Erttmann, S. F. & Gekara, N. O. Hydrogen peroxide release by bacteria suppresses inflammasome-dependent innate immunity. Nat. Commun. 10, 3493 (2019).
Negash, A. A., Olson, R. M., Griffin, S. & Gale, M. Jr Modulation of calcium signaling pathway by hepatitis C virus core protein stimulates NLRP3 inflammasome activation. PLoS Pathog. 15, e1007593 (2019).
Chen, I. Y., Moriyama, M., Chang, M. F. & Ichinohe, T. Severe acute respiratory syndrome coronavirus viroporin 3a activates the NLRP3 inflammasome. Front. Microbiol. 10, 50 (2019).
Ichinohe, T., Pang, I. K. & Iwasaki, A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat. Immunol. 11, 404–410 (2010).
Ito, M., Yanagi, Y. & Ichinohe, T. Encephalomyocarditis virus viroporin 2B activates NLRP3 inflammasome. PLoS Pathog. 8, e1002857 (2012).
Zhu, S. et al. Nlrp9b inflammasome restricts rotavirus infection in intestinal epithelial cells. Nature 546, 667–670 (2017).
Briard, B. et al. Fungal ligands released by innate immune effectors promote inflammasome activation during Aspergillus fumigatus infection. Nat. Microbiol. 4, 316–327 (2019).
Ali, M. F., Dasari, H., Van Keulen, V. P. & Carmona, E. M. Canonical stimulation of the NLRP3 inflammasome by fungal antigens links innate and adaptive B-lymphocyte responses by modulating IL-1beta and IgM production. Front. Immunol. 8, 1504 (2017).
Kasper, L. et al. The fungal peptide toxin Candidalysin activates the NLRP3 inflammasome and causes cytolysis in mononuclear phagocytes. Nat. Commun. 9, 4260 (2018).
Rogiers, O. et al. Candidalysin crucially contributes to Nlrp3 inflammasome activation by Candida albicans hyphae. MBio. 10, e02221–18 (2019).
de Carvalho, R. V. H. et al. Leishmania lipophosphoglycan triggers caspase-11 and the non-canonical activation of the NLRP3 inflammasome. Cell Rep. 26, 429–437 (2019). e425.
Chaves, M. M. et al. Non-canonical NLRP3 inflammasome activation and IL-1beta signaling are necessary to L. amazonensis control mediated by P2X7 receptor and leukotriene B4. PLoS Pathog. 15, e1007887 (2019).
Celias, D. P. et al. Cathepsin L3 from Fasciola hepatica induces NLRP3 inflammasome alternative activation in murine dendritic cells. Front. Immunol. 10, 552 (2019).
Levy, M. et al. Microbiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell 163, 1428–1443 (2015).
Seo, S. U. et al. Distinct commensals induce interleukin-1beta via NLRP3 inflammasome in inflammatory monocytes to promote intestinal inflammation in response to injury. Immunity 42, 744–755 (2015).
Chen, K., Shanmugam, N. K., Pazos, M. A., Hurley, B. P. & Cherayil, B. J. Commensal bacteria-induced inflammasome activation in mouse and human macrophages is dependent on potassium efflux but does not require phagocytosis or bacterial viability. PLoS ONE 11, e0160937 (2016).
Malik, A. et al. SYK-CARD9 signaling axis promotes gut fungi-mediated inflammasome activation to restrict colitis and colon cancer. Immunity 49, 515–530 (2018). e515.
Tschopp, J. & Schroder, K. NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 10, 210–215 (2010).
Chen, J. & Chen, Z. J. PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature 564, 71–76 (2018).
Ruhl, S. & Broz, P. Caspase-11 activates a canonical NLRP3 inflammasome by promoting K(+) efflux. Eur. J. Immunol. 45, 2927–2936 (2015).
He, Y., Zeng, M. Y., Yang, D., Motro, B. & Nunez, G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 530, 354–357 (2016).
Shi, H. et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat. Immunol. 17, 250–258 (2016).
Tang, T. et al. CLICs-dependent chloride efflux is an essential and proximal upstream event for NLRP3 inflammasome activation. Nat. Commun. 8, 202 (2017).
Rossol, M. et al. Extracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors. Nat. Commun. 3, 1329 (2012).
Lee, G. S. et al. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 492, 123–127 (2012).
Katsnelson, M. A., Rucker, L. G., Russo, H. M. & Dubyak, G. R. K+ efflux agonists induce NLRP3 inflammasome activation independently of Ca2+ signaling. J. Immunol. 194, 3937–3952 (2015).
Baldwin, A. G. et al. Boron-based inhibitors of the NLRP3 inflammasome. Cell Chem. Biol. 24, 1321–1335 (2017). e1325.
Gong, T., Yang, Y., Jin, T., Jiang, W. & Zhou, R. Orchestration of NLRP3 inflammasome activation by ion fluxes. Trends Immunol. 39, 393–406 (2018).
Luo, H. et al. Mitochondrial stress-initiated aberrant activation of the NLRP3 inflammasome regulates the functional deterioration of hematopoietic stem cell aging. Cell Rep. 26, 945–954 (2019). e944.
Zhong, Z. et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 560, 198–203 (2018).
West, A. P. et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557 (2015).
Man, S. M. et al. The transcription factor IRF1 and guanylate-binding proteins target activation of the AIM2 inflammasome by Francisella infection. Nat. Immunol. 16, 467–475 (2015).
Dang, E. V., McDonald, J. G., Russell, D. W. & Cyster, J. G. Oxysterol restraint of cholesterol synthesis prevents AIM2 inflammasome activation. Cell 171, 1057–1071 (2017). e1011.
Nakahira, K. et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 12, 222–230 (2011).
Zhong, Z. et al. NF-kappaB restricts inflammasome activation via elimination of damaged mitochondria. Cell 164, 896–910 (2016).
Shimada, K. et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36, 401–414 (2012).
Allam, R. et al. Mitochondrial apoptosis is dispensable for NLRP3 inflammasome activation but non-apoptotic caspase-8 is required for inflammasome priming. EMBO Rep. 15, 982–990 (2014).
Vince, J. E. et al. The mitochondrial apoptotic effectors BAX/BAK activate caspase-3 and -7 to trigger NLRP3 inflammasome and caspase-8 driven IL-1beta activation. Cell Rep. 25, 2339–2353 (2018). e2334.
Zhou, R., Tardivel, A., Thorens, B., Choi, I. & Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 11, 136–140 (2010).
Dostert, C. et al. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320, 674–677 (2008).
van Bruggen, R. et al. Human NLRP3 inflammasome activation is Nox1-4 independent. Blood 115, 5398–5400 (2010).
Bauernfeind, F. et al. Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J. Immunol. 187, 613–617 (2011).
Dang, E. V. & Cyster, J. G. Loss of sterol metabolic homeostasis triggers inflammasomes—how and why. Curr. Opin. Immunol. 56, 1–9 (2019).
Prochnicki, T. & Latz, E. Inflammasomes on the crossroads of innate immune recognition and metabolic control. Cell Metab. 26, 71–93 (2017).
Sanchez-Lopez, E. et al. Choline uptake and metabolism modulate macrophage IL-1beta and IL-18 production. Cell Metab. 29, 1350–1362 (2019). e1357.
Cantuti-Castelvetri, L. et al. Defective cholesterol clearance limits remyelination in the aged central nervous system. Science 359, 684–688 (2018).
Hu, Z. et al. Crystal structure of NLRC4 reveals its autoinhibition mechanism. Science 341, 172–175 (2013).
Hu, Z. et al. Structural and biochemical basis for induced self-propagation of NLRC4. Science 350, 399–404 (2015).
Zhang, L. et al. Cryo-EM structure of the activated NAIP2-NLRC4 inflammasome reveals nucleated polymerization. Science 350, 404–409 (2015).
Tenthorey, J. L. et al. The structural basis of flagellin detection by NAIP5: a strategy to limit pathogen immune evasion. Science 358, 888–893 (2017).
Halff, E. F. et al. Formation and structure of a NAIP5-NLRC4 inflammasome induced by direct interactions with conserved N- and C-terminal regions of flagellin. J. Biol. Chem. 287, 38460–38472 (2012).
Yang, X. et al. Structural basis for specific flagellin recognition by the NLR protein NAIP5. Cell Res. 28, 35–47 (2018).
Schmid-Burgk, J. L. et al. A genome-wide CRISPR (clustered regularly interspaced short palindromic repeats) screen identifies NEK7 as an essential component of NLRP3 inflammasome activation. J. Biol. Chem. 291, 103–109 (2016).
Sharif, H. et al. Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature 570, 338–343 (2019).
Faustin, B. et al. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol. Cell 25, 713–724 (2007).
Jin, T., Curry, J., Smith, P., Jiang, J. & Xiao, T. S. Structure of the NLRP1 caspase recruitment domain suggests potential mechanisms for its association with procaspase-1. Proteins 81, 1266–1270 (2013).
Shen, C. et al. Molecular mechanism for NLRP6 inflammasome assembly and activation. Proc. Natl Acad. Sci. USA 116, 2052–2057 (2019).
Singer, H. et al. NLRP7 inter-domain interactions: the NACHT-associated domain is the physical mediator for oligomeric assembly. Mol. Hum. Reprod. 20, 990–1001 (2014).
Radian, A. D., Khare, S., Chu, L. H., Dorfleutner, A. & Stehlik, C. ATP binding by NLRP7 is required for inflammasome activation in response to bacterial lipopeptides. Mol. Immunol. 67, 294–302 (2015).
Jin, T. et al. Structures of the HIN domain:DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity 36, 561–571 (2012).
Jin, T., Perry, A., Smith, P., Jiang, J. & Xiao, T. S. Structure of the absent in melanoma 2 (AIM2) pyrin domain provides insights into the mechanisms of AIM2 autoinhibition and inflammasome assembly. J. Biol. Chem. 288, 13225–13235 (2013).
Lu, A. et al. Plasticity in PYD assembly revealed by cryo-EM structure of the PYD filament of AIM2. Cell Discov 1, 15013 (2015).
Morrone, S. R. et al. Assembly-driven activation of the AIM2 foreign-dsDNA sensor provides a polymerization template for downstream ASC. Nat. Commun. 6, 7827 (2015).
Morrone, S. R. et al. Cooperative assembly of IFI16 filaments on dsDNA provides insights into host defense strategy. Proc. Natl Acad. Sci. USA 111, E62–E71 (2014).
Lu, A. et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 156, 1193–1206 (2014).
Richards, N. et al. Interaction between pyrin and the apoptotic speck protein (ASC) modulates ASC-induced apoptosis. J. Biol. Chem. 276, 39320–39329 (2001).
Vajjhala, P. R. et al. Identification of multifaceted binding modes for pyrin and ASC pyrin domains gives insights into pyrin inflammasome assembly. J. Biol. Chem. 289, 23504–23519 (2014).
Li, Y. et al. Cryo-EM structures of ASC and NLRC4 CARD filaments reveal a unified mechanism of nucleation and activation of caspase-1. Proc. Natl Acad. Sci. USA 115, 10845–10852 (2018).
Kayagaki, N. et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671 (2015).
Shi, J. et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665 (2015).
Lamkanfi, M. & Dixit, V. M. Mechanisms and functions of inflammasomes. Cell 157, 1013–1022 (2014).
Howard, A. D. et al. IL-1-converting enzyme requires aspartic acid residues for processing of the IL-1 beta precursor at two distinct sites and does not cleave 31-kDa IL-1 alpha. J. Immunol. 147, 2964–2969 (1991).
Ghayur, T. et al. Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature 386, 619–623 (1997).
Van Gorp, H. & Lamkanfi, M. The emerging roles of inflammasome-dependent cytokines in cancer development. EMBO Rep. 20, e47575 (2019).
Kesavardhana, S. & Kanneganti, T. D. Mechanisms governing inflammasome activation, assembly and pyroptosis induction. Int Immunol. 29, 201–210 (2017).
Xia, X., Wang, X., Zheng, Y., Jiang, J. & Hu, J. What role does pyroptosis play in microbial infection? J. Cell Physiol. 234, 7885–7892 (2019).
Liu, X. et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158 (2016).
Shi, J., Gao, W. & Shao, F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem. Sci. 42, 245–254 (2017).
Miao, E. A. et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat. Immunol. 11, 1136–1142 (2010).
Jorgensen, I., Zhang, Y., Krantz, B. A. & Miao, E. A. Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J. Exp. Med. 213, 2113–2128 (2016).
Jorgensen, I., Lopez, J. P., Laufer, S. A. & Miao, E. A. IL-1beta, IL-18, and eicosanoids promote neutrophil recruitment to pore-induced intracellular traps following pyroptosis. Eur. J. Immunol. 46, 2761–2766 (2016).
Sagulenko, V. et al. AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ. 20, 1149–1160 (2013).
Rauch, I. et al. NAIP-NLRC4 inflammasomes coordinate intestinal epithelial cell expulsion with eicosanoid and IL-18 release via activation of caspase-1 and -8. Immunity 46, 649–659 (2017).
Lee, B. L. et al. ASC- and caspase-8-dependent apoptotic pathway diverges from the NLRC4 inflammasome in macrophages. Sci. Rep. 8, 3788 (2018).
Schneider, K. S. et al. The inflammasome drives GSDMD-independent secondary pyroptosis and IL-1 release in the absence of caspase-1 protease activity. Cell Rep. 21, 3846–3859 (2017).
Mascarenhas, D. P. A. et al. Inhibition of caspase-1 or gasdermin-D enable caspase-8 activation in the Naip5/NLRC4/ASC inflammasome. PLoS Pathog. 13, e1006502 (2017).
Van Opdenbosch, N. et al. Caspase-1 engagement and TLR-induced c-FLIP expression suppress ASC/caspase-8-dependent apoptosis by inflammasome sensors NLRP1b and NLRC4. Cell Rep. 21, 3427–3444 (2017).
Tsuchiya, K. et al. Caspase-1 initiates apoptosis in the absence of gasdermin D. Nat. Commun. 10, 2091 (2019).
Finger, J. N. et al. Autolytic proteolysis within the function to find domain (FIIND) is required for NLRP1 inflammasome activity. J. Biol. Chem. 287, 25030–25037 (2012).
Boyden, E. D. & Dietrich, W. F. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat. Genet. 38, 240–244 (2006).
Chavarria-Smith, J. & Vance, R. E. Direct proteolytic cleavage of NLRP1B is necessary and sufficient for inflammasome activation by anthrax lethal factor. PLoS Pathog. 9, e1003452 (2013).
Chavarria-Smith, J., Mitchell, P. S., Ho, A. M., Daugherty, M. D. & Vance, R. E. Functional and evolutionary analyses identify proteolysis as a general mechanism for NLRP1 inflammasome activation. PLoS Pathog. 12, e1006052 (2016).
Sandstrom, A. et al. Functional degradation: a mechanism of NLRP1 inflammasome activation by diverse pathogen enzymes. Science 364, eaau1330 (2019).
Chui, A. J. et al. N-terminal degradation activates the NLRP1B inflammasome. Science 364, 82–85 (2019).
Xu, H. et al. The N-end rule ubiquitin ligase UBR2 mediates NLRP1B inflammasome activation by anthrax lethal toxin. Embo J. 38, e101996 (2019).
Neiman-Zenevich, J., Stuart, S., Abdel-Nour, M., Girardin, S. E. & Mogridge, J. Listeria monocytogenes and Shigella flexneri activate the NLRP1B inflammasome. Infect. Immun. 85, e00338–17 (2017).
Ewald, S. E., Chavarria-Smith, J. & Boothroyd, J. C. NLRP1 is an inflammasome sensor for Toxoplasma gondii. Infect. Immun. 82, 460–468 (2014).
Liao, K. C. & Mogridge, J. Activation of the Nlrp1b inflammasome by reduction of cytosolic ATP. Infect. Immun. 81, 570–579 (2013).
Zhong, F. L. et al. Germline NLRP1 mutations cause skin inflammatory and cancer susceptibility syndromes via inflammasome activation. Cell 167, 187–202 (2016). e117.
Okondo, M. C. et al. Inhibition of Dpp8/9 activates the Nlrp1b inflammasome. Cell Chem. Biol. 25, 262–267.e5 (2018).
Zhong, F. L. et al. Human DPP9 represses NLRP1 inflammasome and protects against autoinflammatory diseases via both peptidase activity and FIIND domain binding. J. Biol. Chem. 293, 18864–18878 (2018).
Everett, B. M. et al. Anti-inflammatory therapy with canakinumab for the prevention and management of diabetes. J. Am. Coll. Cardiol. 71, 2392–2401 (2018).
Bauernfeind, F. G. et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 183, 787–791 (2009).
Gurung, P. et al. FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J. Immunol. 192, 1835–1846 (2014).
Juliana, C. et al. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J. Biol. Chem. 287, 36617–36622 (2012).
Kang, S. et al. Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nat. Commun. 6, 7515 (2015).
Lin, K. M. et al. IRAK-1 bypasses priming and directly links TLRs to rapid NLRP3 inflammasome activation. Proc. Natl Acad. Sci. USA 111, 775–780 (2014).
Wei, M. et al. NLRP3 activation was regulated by DNA methylation modification during Mycobacterium tuberculosis infection. Biomed. Res. Int. 2016, 4323281 (2016).
Liu, C. C. et al. Upregulation of NLRP3 via STAT3-dependent histone acetylation contributes to painful neuropathy induced by bortezomib. Exp. Neurol. 302, 104–111 (2018).
Bauernfeind, F. et al. NLRP3 inflammasome activity is negatively controlled by miR-223. J. Immunol. 189, 4175–4181 (2012).
Neudecker, V. et al. Myeloid-derived miR-223 regulates intestinal inflammation via repression of the NLRP3 inflammasome. J. Exp. Med. 214, 1737–1752 (2017).
Zhou, Y. et al. MicroRNA-7 targets Nod-like receptor protein 3 inflammasome to modulate neuroinflammation in the pathogenesis of Parkinson’s disease. Mol. Neurodegener. 11, 28 (2016).
Li, D. et al. MicroRNA-30e regulates neuroinflammation in MPTP model of Parkinson’s disease by targeting Nlrp3. Hum. Cell 31, 106–115 (2018).
Huang, W. Q., Wei, P., Lin, R. Q. & Huang, F. Protective effects of microrna-22 against endothelial cell injury by targeting NLRP3 through suppression of the inflammasome signaling pathway in a rat model of coronary heart disease. Cell Physiol. Biochem. 43, 1346–1358 (2017).
Feng, X., Luo, Q., Wang, H., Zhang, H. & Chen, F. MicroRNA-22 suppresses cell proliferation, migration and invasion in oral squamous cell carcinoma by targeting NLRP3. J. Cell Physiol. 233, 6705–6713 (2018).
Hu, J., Wu, H., Wang, D., Yang, Z. & Dong, J. LncRNA ANRIL promotes NLRP3 inflammasome activation in uric acid nephropathy through miR-122-5p/BRCC3 axis. Biochimie 157, 102–110 (2019).
Yu, S. Y., Dong, B., Tang, L. & Zhou, S. H. LncRNA MALAT1 sponges miR-133 to promote NLRP3 inflammasome expression in ischemia-reperfusion injured heart. Int. J. Cardiol. 254, 50 (2018).
Zhang, P., Cao, L., Zhou, R., Yang, X. & Wu, M. The lncRNA Neat1 promotes activation of inflammasomes in macrophages. Nat. Commun. 10, 1495 (2019).
Brocker, C. N., et al. Long non-coding RNA Gm15441 attenuates hepatic inflammasome activation in response to metabolic stress. bioRxiv https://doi.org/10.1101/675785 (2019).
Song, N. et al. NLRP3 phosphorylation is an essential priming event for inflammasome activation. Mol. Cell 68, 185–197 (2017). e186.
Humphries, F. et al. The E3 ubiquitin ligase Pellino2 mediates priming of the NLRP3 inflammasome. Nat. Commun. 9, 1560 (2018).
Zhang, Z. et al. Protein kinase D at the Golgi controls NLRP3 inflammasome activation. J. Exp. Med. 214, 2671–2693 (2017).
Guo, C. et al. Bile acids control inflammation and metabolic disorder through inhibition of NLRP3 inflammasome. Immunity 45, 802–816 (2016).
Stutz, A. et al. NLRP3 inflammasome assembly is regulated by phosphorylation of the pyrin domain. J. Exp. Med. 214, 1725–1736 (2017).
Spalinger, M. R. et al. NLRP3 tyrosine phosphorylation is controlled by protein tyrosine phosphatase PTPN22. J. Clin. Invest. 126, 1783–1800 (2016).
Py, B. F., Kim, M. S., Vakifahmetoglu-Norberg, H. & Yuan, J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol. Cell 49, 331–338 (2013).
Ren, G. et al. ABRO1 promotes NLRP3 inflammasome activation through regulation of NLRP3 deubiquitination. Embo J. 38, e100376 (2019).
Yan, Y. et al. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 160, 62–73 (2015).
Kawashima, A. et al. ARIH2 ubiquitinates NLRP3 and negatively regulates NLRP3 inflammasome activation in macrophages. J. Immunol. 199, 3614–3622 (2017).
Song, H. et al. The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nat. Commun. 7, 13727 (2016).
Mishra, B. B. et al. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1beta. Nat. Immunol. 14, 52–60 (2013).
Carpentier, S. J. et al. The signaling adaptor BCAP inhibits NLRP3 and NLRC4 inflammasome activation in macrophages through interactions with Flightless-1. Sci. Signal 12, eaau0615 (2019).
Mehto, S. et al. The Crohn’s disease risk factor IRGM limits NLRP3 inflammasome activation by impeding its assembly and by mediating its selective autophagy. Mol. Cell 73, 429–445 (2019). e427.
de Almeida, L. et al. The PYRIN domain-only protein POP1 inhibits inflammasome assembly and ameliorates inflammatory disease. Immunity 43, 264–276 (2015).
Ratsimandresy, R. A. et al. The PYRIN domain-only protein POP2 inhibits inflammasome priming and activation. Nat. Commun. 8, 15556 (2017).
Samir, P. et al. DDX3X acts as a live-or-die checkpoint in stressed cells by regulating NLRP3 inflammasome. Nature 573, 590–594 (2019).
Yang, C. S. et al. Small heterodimer partner interacts with NLRP3 and negatively regulates activation of the NLRP3 inflammasome. Nat. Commun. 6, 6115 (2015).
Martine, P. et al. HSP70 is a negative regulator of NLRP3 inflammasome activation. Cell Death Dis. 10, 256 (2019).
Eren, E., Berber, M. & Ozoren, N. NLRC3 protein inhibits inflammation by disrupting NALP3 inflammasome assembly via competition with the adaptor protein ASC for pro-caspase-1 binding. J. Biol. Chem. 292, 12691–12701 (2017).
Youm, Y. H. et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 21, 263–269 (2015).
Segovia, M. et al. Targeting TMEM176B enhances antitumor immunity and augments the efficacy of immune checkpoint blockers by unleashing inflammasome activation. Cancer Cell 35, 767–781 (2019). e766.
Tang, T., Gong, T., Jiang, W. & Zhou, R. GPCRs in NLRP3 inflammasome activation, regulation, and therapeutics. Trends Pharm. Sci. 39, 798–811 (2018).
Ip, W. K. E., Hoshi, N., Shouval, D. S., Snapper, S. & Medzhitov, R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science 356, 513–519 (2017).
Miao, H. et al. Macrophage CGI-58 deficiency activates ROS-inflammasome pathway to promote insulin resistance in mice. Cell Rep. 7, 223–235 (2014).
Kim, M. J. et al. SESN2/sestrin2 suppresses sepsis by inducing mitophagy and inhibiting NLRP3 activation in macrophages. Autophagy 12, 1272–1291 (2016).
Yue, S. et al. The myeloid heat shock transcription factor 1/beta-catenin axis regulates NLR family, pyrin domain-containing 3 inflammasome activation in mouse liver ischemia/reperfusion injury. Hepatology 64, 1683–1698 (2016).
Talty, A. et al. Inhibition of IRE1alpha RNase activity reduces NLRP3 inflammasome assembly and processing of pro-IL1beta. Cell Death Dis. 10, 622 (2019).
Kofoed, E. M. & Vance, R. E. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 477, 592–595 (2011).
Tenthorey, J. L., Kofoed, E. M., Daugherty, M. D., Malik, H. S. & Vance, R. E. Molecular basis for specific recognition of bacterial ligands by NAIP/NLRC4 inflammasomes. Mol. Cell 54, 17–29 (2014).
Goncalves, A. V. et al. Gasdermin-D and Caspase-7 are the key Caspase-1/8 substrates downstream of the NAIP5/NLRC4 inflammasome required for restriction of Legionella pneumophila. PLoS Pathog. 15, e1007886 (2019).
Qu, Y. et al. Phosphorylation of NLRC4 is critical for inflammasome activation. Nature 490, 539–542 (2012).
Matusiak, M. et al. Flagellin-induced NLRC4 phosphorylation primes the inflammasome for activation by NAIP5. Proc. Natl Acad. Sci. USA 112, 1541–1546 (2015).
Suzuki, S. et al. Shigella type III secretion protein MxiI is recognized by Naip2 to induce Nlrc4 inflammasome activation independently of Pkcdelta. PLoS Pathog. 10, e1003926 (2014).
Qu, Y. et al. NLRP3 recruitment by NLRC4 during Salmonella infection. J. Exp. Med. 213, 877–885 (2016).
Man, S. M. et al. Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proc. Natl Acad. Sci. USA 111, 7403–7408 (2014).
Karki, R. et al. IRF8 regulates transcription of Naips for NLRC4 inflammasome activation. Cell 173, 920–933 (2018). e913.
Grenier, J. M. et al. Functional screening of five PYPAF family members identifies PYPAF5 as a novel regulator of NF-kappaB and caspase-1. FEBS Lett. 530, 73–78 (2002).
Elinav, E. et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145, 745–757 (2011).
Wlodarska, M. et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell 156, 1045–1059 (2014).
Wang, P. et al. Nlrp6 regulates intestinal antiviral innate immunity. Science 350, 826–830 (2015).
Anand, P. K. et al. NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature 488, 389–393 (2012).
Normand, S. et al. Nod-like receptor pyrin domain-containing protein 6 (NLRP6) controls epithelial self-renewal and colorectal carcinogenesis upon injury. Proc. Natl Acad. Sci. USA 108, 9601–9606 (2011).
Chen, G. Y., Liu, M., Wang, F., Bertin, J. & Nunez, G. A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J. Immunol. 186, 7187–7194 (2011).
Birchenough, G. M. H., Nystrom, E. E. L., Johansson, M. E. V. & Hansson, G. C. A sentinel goblet cell guards the colonic crypt by triggering Nlrp6-dependent Muc2 secretion. in Science, Vol. 352, 1535–1542 (American Association for the Advancement of Science, 2016).
Sun, Y. et al. Stress-induced corticotropin-releasing hormone-mediated NLRP6 inflammasome inhibition and transmissible enteritis in mice. Gastroenterology 144, 1478–1487 (2013). 1487.e1471–1478.
Hu, B. et al. Microbiota-induced activation of epithelial IL-6 signaling links inflammasome-driven inflammation with transmissible cancer. Proc. Natl Acad. Sci. USA 110, 9862–9867 (2013).
Seregin, S. S. et al. NLRP6 protects Il10(-/-) mice from colitis by limiting colonization of Akkermansia muciniphila. Cell Rep. 19, 733–745 (2017).
Mamantopoulos, M. et al. Nlrp6- and ASC-dependent inflammasomes do not shape the commensal gut microbiota composition. Immunity 47, 339–348 (2017). e334.
Lemire, P. et al. The NLR protein NLRP6 does not impact gut microbiota composition. Cell Rep. 21, 3653–3661 (2017).
Volk, J. K.et al. The Nlrp6 inflammasome is not required for baseline colonic inner mucus layer formation or function. J. Exp. Med. 2602–2618 (2019). https://www.ncbi.nlm.nih.gov/pubmed/31420376.
Elinav, E., Henao-Mejia, J., Strowig, T. & Flavell, R. NLRP6 and dysbiosis: avoiding the luring attraction of over-simplification. Immunity 48, 603–604 (2018).
Galvez, E. J. C., Iljazovic, A., Gronow, A., Flavell, R. & Strowig, T. Shaping of intestinal microbiota in Nlrp6- and Rag2-deficient mice depends on community structure. Cell Rep. 21, 3914–3926 (2017).
Hornung, V. et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458, 514–518 (2009).
Lugrin, J. & Martinon, F. The AIM2 inflammasome: sensor of pathogens and cellular perturbations. Immunol. Rev. 281, 99–114 (2018).
Ludlow, L. E., Johnstone, R. W. & Clarke, C. J. The HIN-200 family: more than interferon-inducible genes? Exp. Cell Res 308, 1–17 (2005).
Man, S. M., Karki, R. & Kanneganti, T. D. AIM2 inflammasome in infection, cancer, and autoimmunity: role in DNA sensing, inflammation, and innate immunity. Eur. J. Immunol. 46, 269–280 (2016).
Rathinam, V. A. et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 11, 395–402 (2010).
Reinholz, M. et al. HPV16 activates the AIM2 inflammasome in keratinocytes. Arch. Dermatol. Res. 305, 723–732 (2013).
Ekchariyawat, P. et al. Inflammasome signaling pathways exert antiviral effect against Chikungunya virus in human dermal fibroblasts. Infect. Genet. Evol. 32, 401–408 (2015).
Karki, R. et al. Concerted activation of the AIM2 and NLRP3 inflammasomes orchestrates host protection against Aspergillus infection. Cell Host Microbe 17, 357–368 (2015).
Kalantari, P. et al. Dual engagement of the NLRP3 and AIM2 inflammasomes by plasmodium-derived hemozoin and DNA during malaria. Cell Rep. 6, 196–210 (2014).
Di Micco, A. et al. AIM2 inflammasome is activated by pharmacological disruption of nuclear envelope integrity. Proc. Natl Acad. Sci. USA 113, E4671–E4680 (2016).
Hu, B. et al. The DNA-sensing AIM2 inflammasome controls radiation-induced cell death and tissue injury. Science 354, 765–768 (2016).
Dorfleutner, A. et al. Cellular pyrin domain-only protein 2 is a candidate regulator of inflammasome activation. Infect. Immun. 75, 1484–1492 (2007).
Stehlik, C. et al. The PAAD/PYRIN-only protein POP1/ASC2 is a modulator of ASC-mediated nuclear-factor-kappa B and pro-caspase-1 regulation. Biochem. J. 373, 101–113 (2003).
Khare, S. et al. The PYRIN domain-only protein POP3 inhibits ALR inflammasomes and regulates responses to infection with DNA viruses. Nat. Immunol. 15, 343–353 (2014).
Roberts, T. L. et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 323, 1057–1060 (2009).
Yin, Q. et al. Molecular mechanism for p202-mediated specific inhibition of AIM2 inflammasome activation. Cell Rep. 4, 327–339 (2013).
Kaminski, J. J. et al. Synthetic oligodeoxynucleotides containing suppressive TTAGGG motifs inhibit AIM2 inflammasome activation. J. Immunol. 191, 3876–3883 (2013).
Heilig, R. & Broz, P. Function and mechanism of the pyrin inflammasome. Eur. J. Immunol. 48, 230–238 (2018).
Chae, J. J. et al. Targeted disruption of pyrin, the FMF protein, causes heightened sensitivity to endotoxin and a defect in macrophage apoptosis. Mol. Cell 11, 591–604 (2003).
Gao, W., Yang, J., Liu, W., Wang, Y. & Shao, F. Site-specific phosphorylation and microtubule dynamics control Pyrin inflammasome activation. Proc. Natl Acad. Sci. USA 113, E4857–E4866 (2016).
Chung, L. K. et al. The yersinia virulence factor yopm hijacks host kinases to inhibit type iii effector-triggered activation of the pyrin inflammasome. Cell Host Microbe 20, 296–306 (2016).
Mansfield, E. et al. The familial Mediterranean fever protein, pyrin, associates with microtubules and colocalizes with actin filaments. Blood 98, 851–859 (2001).
Van Gorp, H. et al. Familial mediterranean fever mutations lift the obligatory requirement for microtubules in Pyrin inflammasome activation. Proc. Natl Acad. Sci. USA 113, 14384–14389 (2016).
Sharma, D. et al. Pyrin inflammasome regulates tight junction integrity to restrict colitis and tumorigenesis. Gastroenterology 154, 948–964 (2018). e948.
Alimov, I. et al. Bile acid analogues are activators of pyrin inflammasome. J. Biol. Chem. 294, 3359–3366 (2019).
Kayagaki, N. et al. Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121 (2011).
Russo, A. J., Behl, B., Banerjee, I. & Rathinam, V. A. K. Emerging insights into noncanonical inflammasome recognition of microbes. J. Mol. Biol. 430, 207–216 (2018).
Shi, J. et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514, 187–192 (2014).
Hagar, J. A., Powell, D. A., Aachoui, Y., Ernst, R. K. & Miao, E. A. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science 341, 1250–1253 (2013).
Zanoni, I. et al. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science 352, 1232–1236 (2016).
Lupfer, C. R. et al. Reactive oxygen species regulate caspase-11 expression and activation of the non-canonical NLRP3 inflammasome during enteric pathogen infection. PLoS Pathog. 10, e1004410 (2014).
Chung, H. et al. NLRP3 regulates a non-canonical platform for caspase-8 activation during epithelial cell apoptosis. Cell Death Differ. 23, 1331–1346 (2016).
Chen, M. et al. Internalized Cryptococcus neoformans activates the canonical caspase-1 and the noncanonical caspase-8 inflammasomes. J. Immunol. 195, 4962–4972 (2015).
Zhang, C. J. et al. TLR-stimulated IRAKM activates caspase-8 inflammasome in microglia and promotes neuroinflammation. J. Clin. Invest. 128, 5399–5412 (2018).
Gringhuis, S. I. et al. Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1beta via a noncanonical caspase-8 inflammasome. Nat. Immunol. 13, 246–254 (2012).
Kerur, N. et al. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection. Cell Host Microbe 9, 363–375 (2011).
Doitsh, G. et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 505, 509–514 (2014).
Murdoch, S. et al. Mutations in NALP7 cause recurrent hydatidiform moles and reproductive wastage in humans. Nat. Genet. 38, 300–302 (2006).
Radian, A. D., de Almeida, L., Dorfleutner, A. & Stehlik, C. NLRP7 and related inflammasome activating pattern recognition receptors and their function in host defense and disease. Microbes Infect. 15, 630–639 (2013).
Wang, C. M. et al. Identification of 13 novel NLRP7 mutations in 20 families with recurrent hydatidiform mole; missense mutations cluster in the leucine-rich region. J. Med. Genet. 46, 569–575 (2009).
Schneider, M. et al. The innate immune sensor NLRC3 attenuates Toll-like receptor signaling via modification of the signaling adaptor TRAF6 and transcription factor NF-kappaB. Nat. Immunol. 13, 823–831 (2012).
Xia, X. et al. NLRX1 negatively regulates TLR-induced NF-kappaB signaling by targeting TRAF6 and IKK. Immunity 34, 843–853 (2011).
Benko, S., Magalhaes, J. G., Philpott, D. J. & Girardin, S. E. NLRC5 limits the activation of inflammatory pathways. J. Immunol. 185, 1681–1691 (2010).
Allen, I. C. et al. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-kappaB signaling. Immunity 36, 742–754 (2012).
Moore, C. B. et al. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature 451, 573–577 (2008).
Feng, H. et al. NLRX1 promotes immediate IRF1-directed antiviral responses by limiting dsRNA-activated translational inhibition mediated by PKR. Nat. Immunol. 18, 1299–1309 (2017).
Karki, R. et al. NLRC3 is an inhibitory sensor of PI3K-mTOR pathways in cancer. Nature 540, 583–587 (2016).
Vladimer, G. I. et al. The NLRP12 inflammasome recognizes Yersinia pestis. Immunity 37, 96–107 (2012).
Truax, A. D. et al. The inhibitory innate immune sensor NLRP12 maintains a threshold against obesity by regulating gut microbiota homeostasis. Cell Host Microbe 24, 364–378 (2018). e366.
Zaki, M. H., Man, S. M., Vogel, P., Lamkanfi, M. & Kanneganti, T. D. Salmonella exploits NLRP12-dependent innate immune signaling to suppress host defenses during infection. Proc. Natl Acad. Sci. USA 111, 385–390 (2014).
Chen, S. T. et al. NLRP12 regulates Anti-viral RIG-I activation via interaction with TRIM25. Cell Host Microbe 25, 602–616 (2019). e607.
Chen, L. et al. NLRP12 attenuates colon inflammation by maintaining colonic microbial diversity and promoting protective commensal bacterial growth. Nat. Immunol. 18, 541–551 (2017).
Davis, B. K. et al. Cutting edge: NLRC5-dependent activation of the inflammasome. J. Immunol. 186, 1333–1337 (2011).
Triantafilou, K., Kar, S., van Kuppeveld, F. J. & Triantafilou, M. Rhinovirus-induced calcium flux triggers NLRP3 and NLRC5 activation in bronchial cells. Am. J. Respir. Cell Mol. Biol. 49, 923–934 (2013).
Tong, Y. et al. Enhanced TLR-induced NF-kappaB signaling and type I interferon responses in NLRC5 deficient mice. Cell Res. 22, 822–835 (2012).
Yao, Y. et al. NLRC5 regulates MHC class I antigen presentation in host defense against intracellular pathogens. Cell Res. 22, 836–847 (2012).
Di Virgilio, F. The therapeutic potential of modifying inflammasomes and NOD-like receptors. Pharm. Rev. 65, 872–905 (2013).
Lamkanfi, M. & Dixit, V. M. Inflammasomes and their roles in health and disease. Annu Rev. Cell Dev. Biol. 28, 137–161 (2012).
Voet, S., Srinivasan, S., Lamkanfi, M. & van Loo, G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol. Med. 11, e10248 (2019).
Hoffman, H. M., Mueller, J. L., Broide, D. H., Wanderer, A. A. & Kolodner, R. D. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat. Genet. 29, 301–305 (2001).
Kitamura, A., Sasaki, Y., Abe, T., Kano, H. & Yasutomo, K. An inherited mutation in NLRC4 causes autoinflammation in human and mice. J. Exp. Med. 211, 2385–2396 (2014).
Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. The International FMF Consortium. Cell 90, 797–807 (1997).
Gris, D. et al. NLRP3 plays a critical role in the development of experimental autoimmune encephalomyelitis by mediating Th1 and Th17 responses. J. Immunol. 185, 974–981 (2010).
Inoue, M., Williams, K. L., Gunn, M. D. & Shinohara, M. L. NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis. Proc. Natl Acad. Sci. USA 109, 10480–10485 (2012).
Halle, A. et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 9, 857–865 (2008).
Duewell, P. et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361 (2010).
Jourdan, T. et al. Activation of the Nlrp3 inflammasome in infiltrating macrophages by endocannabinoids mediates beta cell loss in type 2 diabetes. Nat. Med. 19, 1132–1140 (2013).
Karki, R. & Kanneganti, T. D. Diverging inflammasome signals in tumorigenesis and potential targeting. Nat. Rev. Cancer 19, 197–214 (2019).
Swanson, K. V., Deng, M. & Ting, J. P. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 19, 477–489 (2019).
Greaney, A. J., Maier, N. K., Leppla, S. H. & Moayeri, M. Sulforaphane inhibits multiple inflammasomes through an Nrf2-independent mechanism. J. Leukoc. Biol. 99, 189–199 (2016).
Lee, J. et al. Sulforaphane attenuates activation of NLRP3 and NLRC4 inflammasomes but not AIM2 inflammasome. Cell Immunol. 306-307, 53–60 (2016).
Yang, G., Lee, H. E. & Lee, J. Y. A pharmacological inhibitor of NLRP3 inflammasome prevents non-alcoholic fatty liver disease in a mouse model induced by high fat diet. Sci. Rep. 6, 24399 (2016).
Dong, Z. et al. Sulforaphane protects pancreatic acinar cell injury by modulating Nrf2-mediated oxidative stress and NLRP3 inflammatory pathway. Oxid. Med. Cell Longev. 2016, 7864150 (2016).
Li, S., Yang, H. & Chen, X. Protective effects of sulforaphane on diabetic retinopathy: activation of the Nrf2 pathway and inhibition of NLRP3 inflammasome formation. Exp. Anim. 68, 221–231 (2019).
An, Y. W., Jhang, K. A., Woo, S. Y., Kang, J. L. & Chong, Y. H. Sulforaphane exerts its anti-inflammatory effect against amyloid-beta peptide via STAT-1 dephosphorylation and activation of Nrf2/HO-1 cascade in human THP-1 macrophages. Neurobiol. Aging 38, 1–10 (2016).
Orlowski, G. M. et al. Multiple cathepsins promote Pro-IL-1beta synthesis and NLRP3-mediated IL-1beta activation. J. Immunol. 195, 1685–1697 (2015).
Rashidi, M. et al. The pyroptotic cell death effector gasdermin D is activated by gout-associated uric acid crystals but is dispensable for cell death and IL-1beta release. J. Immunol. 203, 736–748 (2019).
Mazarakis, N., Snibson, K., Licciardi, P. V. & Karagiannis, T. C. The potential use of l-sulforaphane for the treatment of chronic inflammatory diseases: a review of the clinical evidence. Clin. Nutr. 664–675 (2019). https://www.ncbi.nlm.nih.gov/pubmed/30954362.
Lamkanfi, M. et al. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J. Cell Biol. 187, 61–70 (2009).
Coll, R. C. et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 21, 248–255 (2015).
Jiang, H. et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J. Exp. Med. 214, 3219–3238 (2017).
Honda, H. et al. Isoliquiritigenin is a potent inhibitor of NLRP3 inflammasome activation and diet-induced adipose tissue inflammation. J. Leukoc. Biol. 96, 1087–1100 (2014).
Liu, Q., Lv, H., Wen, Z., Ci, X. & Peng, L. Isoliquiritigenin activates nuclear factor erythroid-2 related factor 2 to suppress the NOD-like receptor protein 3 inflammasome and inhibits the NF-kappaB pathway in macrophages and in acute lung injury. Front. Immunol. 8, 1518 (2017).
Zeng, J. et al. Isoliquiritigenin alleviates early brain injury after experimental intracerebral hemorrhage via suppressing ROS- and/or NF-kappaB-mediated NLRP3 inflammasome activation by promoting Nrf2 antioxidant pathway. J. Neuroinflamm. 14, 119 (2017).
Daniels, M. J. et al. Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer’s disease in rodent models. Nat. Commun. 7, 12504 (2016).
Tezcan, G. et al. MicroRNA post-transcriptional regulation of the NLRP3 inflammasome in immunopathologies. Front. Pharm. 10, 451 (2019).
Wang, Y. et al. MicroRNA-9 inhibits NLRP3 inflammasome activation in human atherosclerosis inflammation cell models through the JAK1/STAT. Cell Physiol. Biochem. 41, 1555–1571 (2017).
Marchetti, C. et al. OLT1177, a beta-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc. Natl Acad. Sci. USA 115, E1530–e1539 (2018).
Netea, M. G. & Joosten, L. A. Inflammasome inhibition: putting out the fire. Cell Metab. 21, 513–514 (2015).
Acknowledgements
We thank the members of the Elinav lab for discussions and apologize to authors whose work was not included due to space constraints. D.Z. is the recipient of the European Crohn’s and Colitis Organization (ECCO) Fellowship, and is supported by the Ke Lin Program of the First Affiliated Hospital, Sun Yat‐sen University. T.L. is funded as postdoctoral fellow by the German Research Foundation (DFG, 420943353). E.E. is supported by Yael and Rami Ungar; the Leona M. and Harry B. Helmsley Charitable Trust; Adelis Foundation; Pearl Welinsky Merlo Scientific Progress Research Fund; Lawrence and Sandra Post Family Foundation; Daniel Morris Trust; Park Avenue Charitable Fund; The Hanna and Dr. Ludwik Wallach Cancer Research Fund; Howard and Nancy Marks Charitable Fund; Aliza Moussaieff; Estate of Malka Moskowitz; Estate of Myron H. Ackerman; Estate of Bernard Bishin for the WIS-Clalit Program; Donald and Susan Schwarz; and by grants funded by the European Research Council; Israel Science Foundation; Israel Ministry of Science and Technology; Israel Ministry of Health; the Helmholtz Foundation; Else Kroener Fresenius Foundation; Garvan Institute; European Crohn’s and Colitis Organization; Deutsch-Israelische Projektkooperation; and Welcome Trust. E.E. is the incumbent of the Sir Marc and Lady Tania Feldmann Professorial Chair; a senior fellow, Canadian Institute of Advanced Research (CIFAR); and an international scholar, The Bill & Melinda Gates Foundation and Howard Hughes Medical Institute (HHMI).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zheng, D., Liwinski, T. & Elinav, E. Inflammasome activation and regulation: toward a better understanding of complex mechanisms. Cell Discov 6, 36 (2020). https://doi.org/10.1038/s41421-020-0167-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41421-020-0167-x
This article is cited by
-
NLRP inflammasomes in health and disease
Molecular Biomedicine (2024)
-
The interaction of inflammasomes and gut microbiota: novel therapeutic insights
Cell Communication and Signaling (2024)
-
The role of the NLRP3 inflammasome and pyroptosis in cardiovascular diseases
Nature Reviews Cardiology (2024)
-
Mechanistic insights from inflammasome structures
Nature Reviews Immunology (2024)
-
Inflammasomes in neurological disorders — mechanisms and therapeutic potential
Nature Reviews Neurology (2024)
