
Abstract
Superoxide dismutase 1 (SOD1) modulates intestinal barrier integrity and intestinal homeostasis as an antioxidant enzyme. Intestinal homeostasis is maintained by the intestinal stem cells (ISCs). However, whether and how SOD1 regulates ISCs is unknown. In this study, we established intestinal organoids from tamoxifen–inducible intestinal epithelial cell–specific Sod1 knockout (Sod1f/f; Vil-creERT2) mice. We found that loss of Sod1 in organoids suppressed the proliferation and survival of cells and Lgr5 gene expression. SOD1 is known for nearly half a century for its canonical role as an antioxidant enzyme. We identified its enzyme-independent function in ISC: inhibition of SOD1 enzymatic activity had no impact on organoid growth, and enzymatically inactive Sod1 mutants could completely rescue the growth defects of Sod1 deficient organoids, suggesting that SOD1-mediated ISC growth is independent of its enzymatic activity. Moreover, Sod1 deficiency did not affect the ROS levels of the organoid, but induced the elevated WNT signaling and excessive Paneth cell differentiation, which mediates the occurrence of growth defects in Sod1 deficient organoids. In vivo, epithelial Sod1 loss induced a higher incidence of apoptosis in the stem cell regions and increased Paneth cell numbers, accompanied by enhanced expression of EGFR ligand Epiregulin (EREG) in the stromal tissue, which may compensate for Sod1 loss and maintain intestinal structure in vivo. Totally, our results show a novel enzyme-independent function of SOD1 in ISC growth under homeostasis.
Similar content being viewed by others

Introduction
Intestinal stem cell (ISC) populations are crucial for intestinal homeostasis by undergoing either self-renewing or differentiation divisions. Cells in different locations in the crypt can act as ISCs in the mouse intestine.
Crypt base columnar cells (CBCs) which are named because of their elongated/columnar appearance, reside at the bottom of the crypts, intermingled with the Paneth cells. CBCs are rapidly cycling and show characteristic expression of Lgr5 [1]) and Olfm4 [2]. The CBCs form rapidly proliferating transit-amplifying (TA) progenitors that differentiate downwards into Paneth cells at the crypt base, and upwards along the crypt-villus axis into enterocytes, goblet cells and enteroendocrine cells, which undergo apoptosis at the tips of the villi. Under normal conditions, continuous exposure to food and bacteria leads to enhanced apoptosis and removal of mucosal cells. The CBCs are responsible for replenishment of lost epithelial cell populations [1]. Another stem cell pool is present above the Paneth cells at position +4 relative to the base of the crypt. However, there is no consensus regarding their cycling dynamics and sensitivity to irradiation. Numerous reports have showed that +4 ISCs are slow-cycling, radioresistant, and are marked by the expression of Bmi1, mTert, and Hopx [3,4,5]. Under homeostatic conditions, quiescent Bmi1+ ISCs are slow-cycling. Upon intestinal injury or ablation of Lgr5, the quiescent Bmi1 or Hopx-expressing cells can enter a rapidly cycling state to replenish the Lgr5+ ISCs as the reserve stem cell pool [5, 6]. Potten et al., however, proposed that the position +4 harbors fast-cycling and radiosensitive ISCs based on lineage-tracing data. A dose of irradiation as low as 1 Gy is sufficient to eliminate all Potten’s +4 cells [7, 8] comprising both Lgr5-positive and Lgr5-negative cells [1, 9]. The radiosensitivity of +4 populations is a protective mechanism preventing the transmission of irradiation-induced damaged DNA to progenitors [10]. The radiosensitive +4 cells are distinct from active CBCs or quiescent +4 populations which have a low sensitivity to irradiation [3, 11, 12]. Once the radiosensitive +4 cells are eliminated, the neighboring proliferative progenitors, including CBCs, will efficiently replace them and assume their functions [9, 13].
Paneth cells are the only differentiated cells at the bottom of the crypt and protect the intestinal barrier by secreting antimicrobial granules containing defensin, cryptdins and lysozyme (LYZ) [14]. Besides the antimicrobial function, Paneth cells act as a critical component of the ISCs niche by secreting essential signals such as EGF, R-spondin1 (RSPO1), and WNT3a [15, 16]. Paneth cell-derived WNT signaling not only supports ISC growth [17, 18] but also functions as a differentiation signal for Paneth cells and drives Paneth cell maturation and differentiation. In the absence of WNT signals, Paneth cells exhibit improper localization and fail to undergo morphological maturation [19]. Upon activation of WNT signaling, β-catenin/TCF4 complex activates the transcription of Paneth cells markers and promotes ISCs differentiation towards the Paneth cell lineage [19, 20]. The ISCs growth and differentiation should be tightly regulated to keep homeostasis. Although multiple signaling pathways were involved in ISCs regulation [21], their exact mechanisms remain unclear.
The Superoxide dismutase (SOD) family of antioxidant enzymes consist of SOD1 (Cu/Zn superoxide dismutase, Cu/Zn-SOD), SOD2 (manganese superoxide dismutase, Mn-SOD), and SOD3 (extracellular superoxide dismutase, EC-SOD). SOD1 is distributed in the cytosol, nucleus, peroxisomes, and mitochondrial intermembrane space [22]. Its canonical function is as antioxidant enzymes sustaining redox balance by catalyzing the rapid conversion of the superoxide anion into molecular oxygen and hydrogen peroxide [23]. On the other hand, SOD1 also exerts an enzyme-independent function, such as the regulation of the genes expression as a transcriptional factor [24,25,26,27], and the regulation of ribosome biogenesis driving KRAS-driven lung tumorigenesis [28]. It is known that SOD1 is closely associated with epithelial barrier integrity. Sod1 deficiency or mutation causes redox imbalance which leads to disruption of intestinal tight junction in vivo [29, 30]. Sod1 overexpression significantly reduces intestinal damage induced by intestinal ischemia/reperfusion [31,32,33]. Given the critical role of ISCs in keeping the intestinal barrier integrity, we investigated the possible function of SOD1 in the proliferation and survival of ISC. We provided the first evidence that SOD1 promotes the ISC growth via suppressing WNT signaling and upregulating EREG expression, which is independent of SOD1 enzyme activity.
Results
SOD1 promotes organoid proliferation and survival
To elucidate SOD1 function in ISCs, we established 3D intestinal organoid cultures with freshly isolated crypts from tamoxifen–inducible intestinal epithelial cell (IEC)–specific Sod1 knockout mice (Sod1f/f; Vil-creERT2) (Fig. 1A, B). After treating the cultures with tamoxifen, SOD1 expression in organoids was time-dependently inhibited and deleted completely on day 3 (Fig. 1C), accompanying by a time-dependent increase of the percentage of dead organoid, which was determined based on the absence of a defined edge, no light bud, overall dark appearance, and very small size (Fig. 1D). To evaluate the function of SOD1 in the organoid growth, we detected the proliferation and survival of organoids after 5 days of tamoxifen treatment. Western blot showed that SOD1 expression was completely blocked on day 5 (Fig. 1E). Sod1ΔIEC organoids exhibited reduced Edu-positive cells (Fig. 1F) and decreased budding number per organoid (Fig. 1G, H), suggesting that Sod1 loss inhibits the proliferation of organoids. Compared to control group which had higher percentage of “healthy” organoids which exhibited clean and light appearance, tightly packed cells and integral epithelia, Sod1-deficient group had a higher percentage of “dead” organoids and “poor” organoids which displayed more loosely packed cells, fewer light buds, overall dark appearance, and smaller size (Fig. 1G, I). Moreover, Sod1 loss increased propidium iodide (PI)-positive cells in the epithelial layer of the organoids (Fig. S1A, B), and promoted fluorescent signals of active caspase-3, caspase-8 and caspase-9 in organoids (Fig. 1J), indicating that Sod1 deletion inhibits the survival of organoids and promotes its apoptosis. qPCR analysis showed that the loss of Sod1 significantly reduced the mRNA expression level of the ISC marker Lgr5 (Fig. 1K). To further confirm the role of SOD1 in organoid growth, Sod1f/f and Sod1f/f; Vil-creERT2 mice were intraperitoneally injected with tamoxifen, and crypts were isolated for organoid culture after 4 weeks post 5 consecutive tamoxifen injection. Sod1-deficient crypt was cultured under standard conditions and almost completely dead 2 days after seeding (Fig. 1L). Thus, SOD1 is indispensable for organoid proliferation and survival.

A Experimental scheme for generation of Sod1f/f; Vil-creERT2 mice. B Genotyping using PCR. C Western blot analysis of SOD1 protein in organoids from day 0 to day 5 after tamoxifen treatment. Sod1f/f and Sod1f/f; Vil-creERT2 organoids were treated with 2 μg ml−1 tamoxifen on day 0 (2 days after seeding) and analyzed for SOD1 expression from day 0 to day 5 (n = 4 mice per genotype). D Quantification of the percentage of dead organoids from day 0 to day 5 following tamoxifen treatment. TAM, tamoxifen. Data represent mean ± standard error of the mean (SEM), n = 6 wells per mouse; n = 4 mice per genotype. E Western blot analysis of SOD1 protein in organoids after 5 days of tamoxifen treatment (n = 4 mice per genotype). F Confocal images (nuclear staining, blue; and EdU staining, red) of Edu incorporation (1 h) in organoids after 5 days of tamoxifen or ethanol-only vehicle control treatment. Tamoxifen was dissolved in ethanol. EtOH, ethanol. G Brightfield images of organoids after 5 days of tamoxifen or ethanol-only vehicle controls treatment. Green arrowhead indicates “poor” organoids, and red arrowhead indicates dead organoids. H Percentage of organoids with 0, 1, 2, 3, or ≥4 crypts formed after 5 days of tamoxifen treatment. Data represent mean ± SEM; n = 6 wells per mouse; n = 4 mice per genotype; number of organoids counted: n = 504 (Sod1f/f), n = 557 (Sod1f/f;Vil-creERT2) organoids per group; one of three experiments. I Percentage of dead, poor, and healthy organoids analyzed after 5 days of tamoxifen or ethanol-only vehicle controls treatment. Data represent mean ± SEM; n = 6 wells per mouse; n = 4 mice per genotype; number of organoids counted: n = 511 (Sod1f/f + ethanol), n = 656 (Sod1f/f;Vil-creERT2 + ethanol), n = 498 (Sod1f/f + tamoxifen), n = 784 (Sod1f/f;Vil-creERT2 + tamoxifen) organoids per group; one of three experiments. J Fluorescence immunostaining to detect cleaved caspase-3 (yellow), cleaved caspase-8 (green) and cleaved caspase-9 (red) in the organoids after 5 days of tamoxifen treatment. Representative images of staining performed on organoids derived from 3 separate mice. K qPCR results showing relative mRNA expression of Sod1 and Lgr5 genes in organoids 3 and 5 days after tamoxifen treatment. Data represent mean ± SEM. n = 4 mice per genotype. L Organoids cultured for 2 days with Sod1-deficient crypts isolated from the Sod1f/f and Sod1f/f; Vil-creERT2 mice after 4 weeks post 5 consecutive tamoxifen injection (left); histogram showing quantification of the number of dead organoids on the indicated day after seeding (right). Data represent mean ± SEM. n = 3 mice per genotype. All results are representative of at least three independent experiments. All statistical significances were tested by two-way ANOVA. *P < 0.05, **P < 0.01, ***P < 0.001.
SOD1-mediated organoid growth is independent of its enzymatic activity
To determine the role of SOD1 enzymatic activity in SOD1-induced ISC growth, 0–20 μM of ATN-224, a specific inhibitor of SOD1 enzyme activity [34] was used. ATN-224 at 10 μM and 20 μM almost completely inhibited SOD1 activity in Sod1fl/fl; Vil-creERT2 organoids (Fig. 2A). However, ANT-224 treatment did not affect organoid proliferation determined by Edu incorporation (Fig. 2B) and budding number per organoid (Fig. 2C), or organoid survival measured by PI staining (Fig. S2A, B) and the percentage of dead organoids (Fig. 2D).

A Sod1f/f; Vil-creERT2 organoids were treated with 0 (PBS), 5, 10, and 20 μM ATN-224. SOD1 protein level and enzymatic activity were analyzed after 5 days of ATN-224 treatment (upper panel). Histogram showing quantification of the brightness of SOD1 activity band (lower panel). Data represent mean ± SEM; n = 4 mice per genotype. Statistical significances were tested by one-way ANOVA. ***P < 0.001. B Confocal images (nuclear staining, blue; and EdU staining, red) of Edu incorporation in organoids after 5 days of ATN-224 treatment. C Percentage of organoids with 0, 1, 2, 3, or ≥4 crypts formed after 5 days of ATN-224 treatment. Data represent mean ± SEM; n = 6 wells per group; n = 4 mice per genotype; organoids number counted: n = 375 (0 μM), n = 287 (5 μM), n = 363 (10 μM), n = 544 (20 μM) organoids per group; one of three experiments. D Brightfield images of organoids after 5 days of ATN-224 treatment. (upper panel). Percentage of dead organoids analyzed after 5 days of ATN-224 treatment (lower panel). Data represent mean ± SEM; n = 6 wells per group, n = 4 mice per genotype; organoids number counted: n = 1299 (0 μM), n = 1090 (5 μM), n = 1182 (10 μM), n = 1235 (20 μM) organoids per group; one of three experiments. E The endogenous SOD1 expression in Sod1fl/fl organoids and the doxycycline (Dox)-induced expression of wild-type SOD1, SOD1C58S, and SOD1C147S in Sod1fl/fl; Vil-creERT2 organoids were analyzed by Western blot after 5 days of tamoxifen and doxycycline induction. n = 3 mice per genotype. Vector, vector control; WT, wild-type Sod1; C58S, Sod1C58S; C147, Sod1C147S. F Percentage of organoids with 0, 1, 2, 3, or ≥4 crypts formed after 5 days of tamoxifen and doxycycline induction. Untransduced Sod1fl/fl organoids are taken along as a reference. Data represent mean ± SEM; n = 6 wells per group; n = 3 mice per genotype. Statistical significances were tested by two-way ANOVA. *P < 0.05, **P < 0.01, ***P < 0.001. Organoids number counted: n = 86 (Sod1fl/fl), n = 157 (Sod1fl/fl; Vil-creERT2), n = 410 (Sod1fl/fl; Vil-creERT2 + vector), n = 93 (Sod1fl/fl; Vil-creERT2 + wild-type Sod1), n = 77 (Sod1fl/fl; Vil-creERT2 + Sod1C58S), n = 81 (Sod1fl/fl; Vil-creERT2 + Sod1C147S) organoids per group; one of three experiments. G Quantification of the percentage of dead organoids after 5 days of tamoxifen and doxycycline induction. Data represent mean ± SEM. n = 6 wells per group; n = 3 mice per genotyping. Statistical significances were tested by one-way ANOVA. ***P < 0.001. Number of organoids counted: n = 1008 (Sod1fl/fl), n = 1244 (Sod1fl/fl; Vil-creERT2), n = 760 (Sod1fl/fl; Vil-creERT2 + vector), n = 1202 (Sod1fl/fl; Vil-creERT2 + wild-type Sod1), n = 896 (Sod1fl/fl; Vil-creERT2 + Sod1C58S), n = 978 (Sod1fl/fl; Vil-creERT2 + Sod1C147S) organoids per group; one of three experiments. H Brightfield images of organoids after 5 days of tamoxifen and doxycycline induction. Red arrowheads indicate the dead organoids. I Confocal images (nuclear staining, blue; and EdU staining, red) of Edu incorporation in organoids after 5 days of tamoxifen and doxycycline induction. All results are representative of at least three independent experiments.
Mutations in the catalytically active cysteine to serine (C58S or C147S) results in the loss of enzyme activity of SOD1 in yeast [35]. Both residues are conserved between mouse and yeast. To validate the functional significance of SOD1 enzymatic activity, we over-expressed Sod1C58S and Sod1C147S, which mimic the inactive state, and wild-type Sod1 in the Sod1ΔIEC organoids (Fig. 2E). Sod1fl/fl organoids, expressing endogenous levels of Sod1 was included as a reference. The results showed that inactive Sod1 (Sod1C58S, Sod1C147S) and wild-type Sod1 rescued the proliferation (Fig. 2F, I) and survival (Fig. 2G, H) of Sod1ΔIEC organoids. All these data indicate that SOD1-induced organoid growth is independent of its enzymatic activity.
Loss of Sod1 does not affect the ROS levels on the organoid
SOD1 plays a critical role in maintaining ROS homeostasis. ROS is known as a central regulator of ISC function [36]. Thus, we speculated that Sod1 loss would cause elevated ROS, and thereby mediate growth defects in the organoids. However, Sod1 loss did not alter the levels of superoxide and peroxide in the epithelial layer of organoids, as indicated by DHE staining for superoxide, DHR staining for peroxide and peroxynitrite, and CellROX staining for oxidative stress (Fig. 3A). To determine whether the growth defects in Sod1ΔIEC organoid were due to ROS dysregulation, we treated the organoids with antioxidants such as the general ROS inhibitor NAC and the mitochondrial ROS inhibitor MnTBAP after two days of tamoxifen administration. As shown in Fig. 3B, C, the death rate and budding number in Sod1ΔIEC organoids were not rescued after antioxidant treatment, even at higher concentrations. Organoids were then treated with tamoxifen and antioxidants simultaneously, which could not rescue the death rate of Sod1ΔIEC organoids too (Fig. S3), indicating that ROS levels were not involved in Sod1 loss-induced growth defects in the organoids.

A Sod1 deletion does not affect superoxide level, peroxide level, and oxidative stress in the epithelial layer of organoids. Sod1f/f and Sod1f/f; Vil-creERT2 organoids were stained with DHE, DHR, and CellRox after 5 days of tamoxifen treatment. Representative images of staining performed on organoids. n = 4 mice per genotyping. B Quantification of the percentage of dead organoids after the indicated induction and antioxidant treatment. Sod1f/f and Sod1f/f; Vil-creERT2 organoid cultures were induced by ethanol or tamoxifen for 2 days and then treated with the indicated antioxidants (NAC at 1 mM and 4 mM, MnTBAP at 50 μM and 100 μM) for another 2 days. Data represent mean ± SEM. n = 6 wells per group; n = 4 mice per genotyping; organoids number counted: n = 1094 (Sod1f/f + EtOH), n = 1070 (Sod1f/f + TAM), n = 1107 (Sod1f/f + TAM + NAC1mM), n = 1113 (Sod1f/f + TAM + NAC4mM), n = 1150 (Sod1f/f + TAM + MnTBAP50μM), n = 1202 (Sod1f/f + TAM + MnTBAP100μM), n = 1316 (Sod1f/f;Vil-creERT2 + EtOH), n = 841 (Sod1f/f;Vil-creERT2 + TAM), n = 916 (Sod1f/f;Vil-creERT2 + TAM + NAC1mM), n = 919 (Sod1f/f;Vil-creERT2 + TAM + NAC4mM), n = 898 (Sod1f/f;Vil-creERT2 + TAM + MnTBAP50μM), n = 936 (Sod1f/f;Vil-creERT2 + TAM + MnTBAP100μM) organoids per group; one of four experiments. C Percentage of organoids with 0, 1, 2, 3, or ≥4 crypts formed after the indicated induction and antioxidant treatment. Sod1f/f and Sod1f/f; Vil-creERT2 organoid cultures were induced by ethanol or tamoxifen for 2 days and then treated with the indicated antioxidants (NAC at 1 mM and 4 mM, MnTBAP at 50 μM and 100 μM) for another 2 days. Data represent mean ± SEM. n = 6 wells per group; n = 4 mice per genotyping; organoids number counted: n = 421 (Sod1f/f + EtOH), n = 391 (Sod1f/f + TAM), n = 398 (Sod1f/f + TAM + NAC1mM), n = 439 (Sod1f/f + TAM + NAC4mM), n = 491 (Sod1f/f + TAM + MnTBAP50μM), n = 472 (Sod1f/f + TAM + MnTBAP100μM), n = 568 (Sod1f/f;Vil-creERT2 + EtOH), n = 600 (Sod1f/f;Vil-creERT2 + TAM), n = 547 (Sod1f/f;Vil-creERT2 + TAM + NAC1mM), n = 521 (Sod1f/f;Vil-creERT2 + TAM + NAC4mM), n = 634 (Sod1f/f;Vil-creERT2 + TAM + MnTBAP50μM), n = 654 (Sod1f/f;Vil-creERT2 + TAM + MnTBAP100 μM) organoids per group; one of four experiments. All data are representative of four independent experiments. All statistical significances were tested by two-way ANOVA. *P < 0.05, **P < 0.01, ***P < 0.001.
SOD1 promotes organoid growth by suppressing both WNT signaling and ectopic Paneth cell differentiation
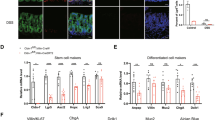
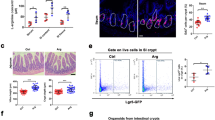
To elucidate the potential mechanism by which SOD1 promotes ISC growth, we performed RNA-seq using Sod1f/f and Sod1f/f;Vil-creERT2 organoids after 3 days of tamoxifen treatment. RNA-seq analysis showed that Paneth cell signature genes were 1.5−2.5 times higher in the Sod1-deficient organoids than in the controls (Fig. 4A). Consistently, the number of Paneth cells markedly increased in the Sod1ΔIEC organoids (Fig. 4B). qPCR confirmed that the mRNA levels of the Paneth cell marker Lyz and Wnt3 were significantly upregulated in Sod1ΔIEC organoids (Fig. 4C). To determine whether elevated WNT signaling contributes to the organoid growth defects after Sod1 knockout, we reduced WNT signaling in Sod1ΔIEC organoids by reducing the levels of the WNT agonist RSPO1 [37]; this improved organoids survival despite reduced overall growth (Fig. 4D–F). These results indicate that, in the absence of Sod1, high WNT signaling-induced excessive Paneth cell differentiation plays a pivotal role in the appearance of growth defects in the organoids.

A Relative expression of Paneth cell signature genes by RNA-seq analysis are shown as a histogram comparing Sod1ΔIEC and control organoids after 3 days of tamoxifen induction. Sod1f/f and Sod1f/f; Vil-creERT2 organoids were harvested after 3 days of tamoxifen induction for RNA-seq. n = 3 mice per genotype. B Fluorescence immunostaining to detect LYZ expression (red) in the organoids after 3 days of tamoxifen induction. Arrowheads indicate location of LYZ+ Paneth cells. Images are representative of stainings performed on organoids derived from 3 separate mice (left panel); Histogram (middle panel) shows the quantification of the number of LYZ+ Paneth cells per crypt, according to the scheme in the inset (right panel). The number of crypts measured per group was labeled on the histogram. One of three experiments. Data represent mean ± SEM; n = 6 wells per group; n = 3 mice per genotyping. Student t-test (two-tailed). ***P < 0.001. C qPCR analysis of Sod1, Lyz, and Wnt3a genes after 3 days of tamoxifen induction. Data represent mean±SEM; n = 3 mice per genotyping. Student t-test (two-tailed). **P < 0.01, ***P < 0.001. D Representative images of Sod1f/f and Sod1f/f; Vil-creERT2 organoids treated with tamoxifen and cultured in reducing RSPO1 concentrations for 5 days. E Percentage of dead organoids assayed after 5 days in culture using indicated concentrations of RSPO1. Data represent mean±SEM; n = 6 wells per group; n = 3 mice per genotyping. Statistical significances were tested by two-way ANOVA. ***P < 0.001. Organoids number counted: n = 246 (Sod1f/f + normal RSPO1), n = 385 (Sod1f/f; Vil-creERT2 + normal RSPO1), n = 244 (Sod1f/f + 1/2 RSPO1), n = 328 (Sod1f/f; Vil-creERT2 + 1/2 RSPO1), n = 256 (Sod1f/f + 1/4 RSPO1), n = 303 (Sod1f/f; Vil-creERT2 + 1/4 RSPO1), n = 265 (Sod1f/f + 1/8 RSPO1), n = 281 (Sod1f/f; Vil-creERT2 + 1/8 RSPO1), n = 229 (Sod1f/f + 1/16 RSPO1), n = 275 (Sod1f/f; Vil-creERT2 + 1/16 RSPO1) organoids per group; one of three experiments. F Quantification of percentage of organoids with 0, 1, 2, 3, or ≥4 crypts formed after 5 days in culture using indicated concentrations of RSPO1. Data represent mean±SEM; n = 6 wells per group; n = 3 mice per genotyping; organoids number counted: n = 189 (Sod1f/f + normal RSPO1), n = 203 (Sod1f/f; Vil-creERT2 + normal RSPO1), n = 197 (Sod1f/f + 1/2 RSPO1), n = 185 (Sod1f/f; Vil-creERT2 + 1/2 RSPO1), n = 186 (Sod1f/f + 1/4 RSPO1), n = 181 (Sod1f/f; Vil-creERT2 + 1/4 RSPO1), n = 196 (Sod1f/f + 1/8 RSPO1), n = 203 (Sod1f/f; Vil-creERT2 + 1/8 RSPO1), n = 166 (Sod1f/f + 1/16 RSPO1), n = 186 (Sod1f/f; Vil-creERT2 + 1/16 RSPO1) organoids per group; one of three experiments. All results are representative of at least three independent experiments.
Epithelial Sod1 deficiency promotes apoptosis in the stem cell regions in vivo
To determine whether SOD1 is important for ISCs in vivo, we first tested SOD1 localization. Immunohistochemistry revealed that in control mice, TA cells showed both nuclear and cytoplasmic SOD1 expression, and the crypt base cells including Paneth cells and CBCs exhibited stronger nuclear SOD1 expression (Fig. 5A). The villous epithelial cells showed only cytoplasmic SOD1 expression (data not shown). After 24 h post 5 consecutive days of tamoxifen injections, the SOD1 immunostaining in all epithelial cells was lost (Fig. 5A). SOD1 deletion in the epithelium significantly promoted the activation of caspase-3 in the stem cell regions (Fig. 5B). Consistently, western blot analyses using separated crypt tissues showed that Sod1 loss induced higher expression of apoptotic proteins including cleaved caspase-3, cleaved caspase-7, cleaved caspase-8, and cleaved caspase-9 (Fig. 5C), suggesting that epithelial Sod1 deficiency in vivo promotes apoptosis of crypt stem cells. However, Sod1ΔIEC mice showed no significant changes in body weight, crypt-villus architecture, and cell proliferation in comparison to control mice (Fig. S4A, B and Fig. 5D). As depicted in Fig. 5E, F, Sod1 deficiency in vivo also led to a lower number of ISCs expressing OLFM4, a marker of LGR5+ ISCs, and an increased Paneth cell numbers per crypt.

A Representative IHC staining for SOD1 in small intestinal sections from Sod1f/f and Sod1f/f; Vil-creERT2 mice after 24 h post 5 consecutive tamoxifen injections. n = 4 mice per genotype. B Representative IHC staining for cleaved caspase-3 in small intestinal sections from Sod1f/f and Sod1f/f; Vil-creERT2 mice after 24 h post 5 consecutive tamoxifen injections (left panel); scoring of frequency of cleaved caspase-3-positive cells per crypt at specific positions relative to the crypt bottom, according to the scheme in the inset (middle panel). Statistical significances on the middle panel were tested by two-way ANOVA. Quantification of frequency of cleaved caspase-3-positive cells in three classes: CBC cells (located at position 0, 1′, 2′), +4 cells (located at position 4), and TA cells (located at position 5-15) (right panel). Statistical significance on the right panel was tested by Student t-test (two-tailed). ***P < 0.001. n = 4 mice per genotype; crypts number measured: n = 445 (CTR), n = 655 (Sod1∆IEC) crypts per group; one of three experiments. C Western blot for cleaved caspase-3, caspase-7, caspase-8, cleaved caspase-9 and Actin in the duodenal crypt samples of control and Sod1∆IEC mice after 24 h post 5 consecutive tamoxifen injections. n = 3 mice per genotype. D Representative IHC staining for Ki-67 in small intestinal sections from Sod1f/f and Sod1f/f; Vil-creERT2 mice after 24 h post 5 consecutive tamoxifen injections (left panel); scoring of frequency of Ki-67-positive cells per crypt at specific positions relative to the crypt bottom (middle panel). Statistical significance on the middle panel were tested by two-way ANOVA. Quantification of frequency of Ki-67-positive cells in three classes: CBC cells (located at position 0, 1′, 2′), +4 cells (located at position 4), and TA cells (located at position 5-22) (right panel). Statistical significance on the right panel was tested by Student t-test (two-tailed). n = 4 mice per genotype; crypts number measured: n = 117 (CTR), n = 135 (Sod1∆IEC) crypts per group; one of three experiments. E Representative IHC staining for OLFM4 after 4 weeks post 5 consecutive tamoxifen injection (left panel); scoring of frequency of OLFM4-positive cells per crypt at specific positions (right panel). Statistical significance was tested by Student t-test (two-tailed). ***P < 0.001. n = 4 mice per genotype; number of crypts counted per group was labeled on the histogram. One of three experiments. F Representative IHC staining for LYZ in the small intestines of Sod1f/f and Sod1f/f; Vil-creERT2 mice after 4 weeks post 5 consecutive tamoxifen injection. Arrowheads point to the location of LYZ+ Paneth cells per crypt (left panel); right panel is the quantification of LYZ+ Paneth cells per crypt. Statistical significance was tested by Student t-test (two-tailed). ***P < 0.001. n = 4 mice per genotype; number of crypts measured per group is labeled on the histogram. One of three experiments. All results are representative of at least three independent experiments.
EREG compensates for growth defects induced by Sod1 deficiency
The EGFR ligand EREG is highly expressed in mesenchymal cells, and supports ISC growth [38,39,40]. We separated the stromal compartment from the small intestine of Sod1f/f and Sod1ΔIEC mice after 24 h post 5 consecutive tamoxifen injections. Western blot analysis showed enhanced EREG expression in the stromal tissue of Sod1ΔIEC mice, and similar SOD1 expressions in the control and Sod1ΔIEC stromal compartment which was because tamoxifen induced deletion of Sod1 only in the epithelial cell lineages (Fig. 6A). Furthermore, treatment of Sod1ΔIEC organoids showed that EREG rescued the morphology and growth of Sod1ΔIEC organoids, and other EGFR ligands such as EGF and AREG did not (Fig. 6B, C, D, E). Thus, EREG could support the growth of Sod1ΔIEC organoids, and stromal EREG upregulation might compensate for Sod1 loss in vivo.

A Western blot analysis of EREG and SOD1 expression using the separated stromal compartment from Sod1f/f and Sod1f/f; Vil-creERT2 mice after 24 h post 5 consecutive tamoxifen injections. n = 4 mice per genotype. B Western blot analysis of SOD1 expression in Sod1f/f and Sod1f/f; Vil-creERT2 organoids after 5 days of tamoxifen with or without 0.5 μg ml-1 of recombinant EGF/AREG/EREG induction. n = 3 mice per genotype. C Representative images of organoids after 5 days of tamoxifen with or without EGF/AREG/EREG induction. n = 6 wells per group; n = 3 mice per genotype. Red arrowhead indicates dead organoids. D Percentage of organoids with 0, 1, 2, 3, or ≥4 crypts formed after 5 days of tamoxifen with or without EGF/AREG/EREG induction. Data represent mean ± SEM; n = 6 wells per group; n = 3 mice per genotyping. Statistical significances were tested by two-way ANOVA. *P < 0.05, ***P < 0.001. Number of organoids counted: n = 249 (Sod1f/f), n = 287 (Sod1f/f; Vil-creERT2), n = 307 (Sod1f/f; Vil-creERT2 + EGF), n = 309 (Sod1f/f; Vil-creERT2 + AREG), n = 272 (Sod1f/f; Vil-creERT2 + EREG) organoids per group; one of three experiments. E Percentage of dead organoids assayed after 5 days of tamoxifen with or without EGF/AREG/EREG induction. Data represent mean ± SEM; n = 6 wells per group; n = 3 mice per genotyping. Statistical significance was tested by one-way ANOVA. ***P < 0.001. Number of organoids counted: n = 393 (Sod1f/f), n = 475 (Sod1f/f; Vil-creERT2), n = 537 (Sod1f/f; Vil-creERT2 + EGF), n = 532 (Sod1f/f; Vil-creERT2 + AREG), n = 413 (Sod1f/f; Vil-creERT2 + EREG) organoids per group; one of three experiments. All results are representative of at least three independent experiments.
Discussion
The mouse intestinal organoid is a useful ex vivo model for the study of ISCs, because it retains critical in vivo characteristics and normal numbers of ISCs and differentiated cell lineages [41]. ISCs, Paneth cells, and TA cells are localized at the bud site of organoids (crypt region). The crypt region undergoes continuous budding events surrounding a central lumen lined by a villus-like epithelium (villus region) [41]. The enterocyte, goblet cells and enteroendocrine cells are located at the villus region, and apoptotic cells are shed into the lumen in a manner similar to cell loss at the villus tips in vivo. Organoid cultures can be generated from dissociated crypts or single Lgr5+ cells in vitro, and passaged for more than 8 months without genetic alterations [41]. In this work, we used the organoid model to study the function of SOD1 in ISCs. Our study first demonstrates that SOD1 is critical for organoid growth and Lgr5 gene expression. SOD1 is known for its canonical role as an antioxidant enzyme. Thus, we studied the role of SOD1 enzymatic activity in SOD1-induced ISC growth, and showed that the SOD1 inhibitor ATN-224, which almost completely blocks the enzymatic activity of SOD1, did not affect the proliferation and survival of organoid. Moreover, catalytically inactive-Sod1 could reverse the growth defects induced by Sod1 deficiency. These observations revealed that the well-known enzyme SOD1 has a novel function in ISCs which is independent of its enzymatic activity.
ROS signaling plays an important role in ISC function, as reviewed [36]. ROS levels in ISCs are low in comparison to those in differentiated cells in Drosophila [42]. Intracellular ROS levels must be tightly regulated; too much or too little ROS could inhibit cell growth [43]. Deletion of autophagy related 5 (Atg5)-mediated ROS production could reduce ISC count [44], whereas repression of Nrf2, a regulator of anti-oxidative responses, could promote proliferative ROS in Drosophila ISC lineages [42]. Although SOD1 is known as a ROS-scavenging enzyme, there have been conflicting reports on the impact of Sod1 loss on intracellular ROS levels. Sod1 loss in mice caused oxidative stress, which mediates multiple organ pathologies [45,46,47,48]. However, Wang et al. recently found that SOD1-induced reduction of tumor burden in KRAS-driven NSCLC mice model did not alter the ROS levels [28]. In this study, we also showed that Sod1 deletion did not significantly affect ROS levels in the organoids, and treatment with general or mitochondrial antioxidants could not rescue the growth defects in Sod1-deficient organoids. These data indicate that ROS signaling is not involved in SOD1-induced organoid growth.
Paneth cell-derived WNT signaling is essential for ISC growth. Inhibition of WNT signaling or genetic removal of Paneth cells in vivo results in the loss of ISCs [15, 17, 18]. On the other hand, activation of WNT signaling or excessive Paneth cell differentiation also promotes apoptosis of the crypt region cells [20] or exhausts the ISC pool upon damage [40]. Here, we found notable increases in WNT signaling and Paneth cell number after Sod1 deletion in organoids. This increase occurred before reduction of Lgr5 gene expression (Fig. 1K and Fig. 4C). Furthermore, decreasing WNT signaling could block the growth defects in Sod1-deficient organoids. All these data indicate that redundant WNT signaling plays an important role in Sod1 loss-induced organoid growth defect.
Epithelial Sod1 deficiency in vivo inhibited the survival of the crypt stem cells but not the proliferation and crypt-villus architecture, while Sod1 deficiency in organoids induced a significant inhibition of survival and proliferation. The Sod1-deficient crypt-derived organoids were almost completely dead 2 days after seeding (Fig. 1L). To explain this discrepancy, we studied the difference between intestinal organoids and the small intestine in vivo. Stromal/mesenchymal cells are absent in organoids [49], but present in vivo. These stromal cells provide essential niche factors for ISCs in vivo [50], and stromal EGFR signaling promotes ISC proliferation [40]. EGFR has seven known ligands: EGF, AREG, EREG, TGFA, HB-EGF, BTC, and EPGN [51]. Unlike the other ligands, EREG has more potent bioactivity [52, 53]. In this study, we also showed enhanced stromal EREG expression in Sod1ΔIEC mice. Furthermore, supplementation of EREG, but not AREG and EGF, in vitro could rescue the growth of Sod1 deficient organoids. These data indicate that elevated stromal EREG may compensate for Sod1 loss in vivo, thereby preserving intestinal crypt-villus architecture.
In summary, our results showed that SOD1 promotes ISC growth independent of its enzyme activity, by suppressing both WNT signaling and excessive Paneth cell differentiation, while maintaining intestinal structure in vivo that may include EREG support. Paneth cells are frequently found in human colorectal adenomas and adenocarcinomas [54, 55]. Thus, our finding that SOD1 suppresses ectopic Paneth cell differentiation could suggest a self-sustaining mechanism that prevents intestinal tumor formation.
Material and methods
Mice
All mice were maintained in SPF housing, with a maximum of five mice per cage, under a 12 h light–dark cycle at a temperature range of 21 ± 2 °C. All mice were of C57/B6 and 129 mixed background. Sod1flox/flox mice were obtained from Dr. Holly Van Remmen [56]. Sod1flox/flox mice were bred with Vil-creERT2 mice (JAX stock ♯ 020282) to generate Sod1flox/flox; Vil-creERT2 mice. All mouse experiments were conducted in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. The study procedures were approved by the Institutional Animal Care and Use Committee of Renji Hospital, Shanghai Jiao Tong University School of Medicine.
To genotype the animals, 2 mm of the ear or tail tissue was placed directly into 75 μl alkaline lysis buffer and heated in 95 °C 1 h. After heating, samples were cooled to 4 °C, treated with 75 μl neutralization buffer and analyzed by PCR. Experimental mice (8–16 weeks old) were injected intraperitoneally with 50 mg kg−1 tamoxifen (Sigma-Aldrich, Missouri, USA, T5648) in sunflower seed oil (Sigma-Aldrich, 88921) once a day for 5 consecutive injections. Both males and females were used. All mouse experiments were exclusively performed on littermate animals, with 3 to 5 mice used for each genotype. No randomization was done to determine how samples/ animals were allocated. All analyses of mouse phenotype were performed blinded.
Organoid culture
Crypt isolation and culture were performed as previously described [57]. Briefly, duodenal crypts were obtained from Sod1flox/flox; Vil-creERT2 and Sod1flox/flox mice, seeded in growth factor reduced matrigel (BD, California, USA, 356231), and cultured in ENR (EGF, noggin, RSPO1) medium. Working ENR medium contained advanced DMEM/F12 (Thermo Scientific, Kansas, USA, 12634010), 1× Glutamax (Thermo Scientific, 35050061), 1× penicillin/streptomycin (Thermo Scientific, 15070063), 1× N2 Supplement (Thermo Scientific, 17502048), 1× B27 Supplement (Thermo Scientific, 12587010), 10 mol L−1 HEPES (Thermo Scientific, 15630-106), 1 mol L−1 N-acetyl l-cysteine (NAC) (Sigma-Aldrich, A9165), 0.05 μg ml−1 mouse recombinant EGF (Peprotech, New Jersey, USA, 315-09), 0.1 μg ml−1 mouse recombinant noggin (Peprotech, 250–38), and 5% RSPO1 (v/v). RSPO1 was produced using Cultrex HA-R-Spondin1-Fc 293 T (Trevigen, Maryland, USA). The organoids were passaged every 7 day by mechanically disrupting with a 20-Ga needle (Sigma-Aldrich, Z118052-100EA).
To stop SOD1 expression in the organoids, 2 μg ml−1 tamoxifen (Sigma-Aldrich, T5648) dissolved in ethanol was added to the culture medium on day 0 (2 days post seeding). The ethanol-only treatment is used as the vehicle control. The media was changed the next day, and subsequently every 2 days. The following inhibitors were used: ATN-224 (kindly provided by Dr. X.F.Steven Zheng, diluted in PBS), NAC (Sigma-Aldrich, A9165), and MnTBAP (Sigma-Aldrich, 475870). EGF (Peprotech, 315-09), Amphiregulin (AREG) (R&D Systems, Minnesota, USA, 989-AR-100), and EREG (R&D Systems, 1068-EP-050) were added to the culture media at a final concentration of 0.5 μg ml−1 on day 0 and were supplemented by media changes.
Quantification of crypt number per organoid
Several random non-overlapping pictures were acquired from each well by using a Zeiss Axio Observer Z1 inverted microscope, and counted manually. Percentage of organoids with 0, 1, 2, 3, or ≥4 buddings were scored from 6 independent cultures for each mouse. There are 3-4 different mice per genotype. Organoids touching the edge of the images were excluded. The number of analyzed organoids is total number per group from one experiment.
PI staining of organoids
PI staining was performed as described previously [58]. Briefly, the organoids were incubated with 500 nM propidium iodide (Sigma-Aldrich, P4170) in PBS solution at 37 °C for 20 min. The organoids were then washed with PBS and covered with new medium. Images were captured by Zeiss confocal microscope.
Immunofluorescence
Organoids plated in 4-well chamber slides were fixed in 4% paraformaldehyde (PFA) for 30 min, permeabilized in 0.5% Triton for 20 min and blocked in 2% BSA. Mouse anti-lysozyme (Dako, Copenhagen, Denmark, A009902-2, 1:10000) primary antibody, cleaved caspase-3 (Cell Signaling Technology, Massachusetts, USA, 9664, 1:500) primary antibody, cleaved caspase-8 (Cell Signaling Technology, 8592, 1:500) primary antibody, cleaved caspase-9 (Cell Signaling Technology, 9507, 1:500) primary antibody and donkey anti-rabbit (Thermo Scientific, 1:2000) secondary antibody were used for immunostaining. Organoids were mounted with antifade mounting medium with DAPI (SouthernBiotech, Alabama, USA, 0100-20) for visualization. For EdU incorporation experiments, the intact organoid cultures were subjected to 1 h pre-incubation with EdU in ENR medium before fixing. Cells were stained using a Click-it EDU Imaging kit (Thermo Scientific, C10338), according to the manufacturer’s instructions. Images were acquired using a Zeiss confocal microscope.
Detection of intracellular ROS, superoxide levels, and oxidative stress
Dihydroethidium (DHE) (Thermo Scientific, D23107), Dihydrorhodamine (DHR) (Thermo Scientific, D23806), and CellROX (Thermo Scientific, C10422) staining were performed using kits according to the manufacturer’s instructions. Briefly, ROS was monitored by staining the organoids with 10 μM DHR for 15 min at 37 °C. Organoids were then washed with PBS and covered with new ENR medium. To monitor intracellular superoxide levels, organoids were incubated with 1 μM DHE for 10 min at 37 °C. Organoids were then washed with PBS and covered with new ENR medium. To measure intracellular oxidative stress, the organoids were treated with 5 μM CellROX for 30 min at 37 °C. Organoids were then washed with PBS and covered with new ENR medium. Images were obtained using a Zeiss confocal microscope.
Crypt and organoid protein extraction and western blotting
Duodenal crypts were insolated according to O’Rourke et al. [59]. Organoids were washed once with cold PBS and collected in cell recovery solution (Corning, New York, USA, 354253) to remove the matrigel. Duodenal crypts or organoid pellets were homogenized in cold RIPA lysis buffer (Beyotime Biotechnology, China, P0013C) containing proteinase and phosphatase inhibitors (Beyotime Biotechnology) for 10–20 min, sonicated at 4 °C using a Bioruptor® Plus device, mixed with 6×SDS loading buffer and heated at 100 °C for 5 min. Protein concentrations were measured using a Pierce™ BCA Protein Assay Kit (Thermo Scientific, 23225). Western blot analysis was performed using standard techniques [60]. The primary antibodies used were SOD1 (Santa Cruz Biotechnology, Texas, USA, sc-11407, 1:2000), epiregulin (Santa Cruz Biotechnology, sc-376284, 1:500), cleaved caspase-3 (Cell Signaling Technology, 9664, 1:1000), caspase-7 (Cell Signaling Technology, 9492, 1:1000), caspase-8 (Cell Signaling Technology, 4790, 1:1000), cleaved caspase-9 (Cell Signaling Technology, 9507, 1:1000) and β-Actin (Cell Signaling Technology, 4970, 1:1000).
RNA extraction and qPCR
RNA from the organoids was extracted using the RNeasy Kit (Qiagen, Limburg, Netherlands, 74104) with on-column DNase treatment. cDNA was synthesized using a RETROscript® Reverse Transcription Kit (Thermo Scientific, AM1710). qPCR was performed using PowerUp™ SyGreen Mix (Thermo Scientific, A25741). Normalization was done using the housekeeping gene Hmbs. Sod1-F: 5′-GAGGGTAGCAGATGAGTCTGAG-3′, Sod1-R: 5′-GAGTCTTGTTGCTAAGTAGAG-3′;Lgr5-F:5′-CCTACTCGAAGACTTACCCAGT-3′, Lgr5-R: 5′-GCATTGGGGTGAATGATAGCA-3′;Lyz-F:5′-ACTCCTCCTTGCTTTCTGTC-3′, Lyz-R: 5′-GTCGGTGCTTCGGTCTC-3′; Wnt3-F: 5′-TGGAACTGTACCACCATAGATGAC-3′, Wnt3-R: 5′-ACACCAGCCGAGGCGATG-3′; Hmbs-F:5′-GATGGGCAACTGTACCTGACTG-3′, Hmbs-R: 5′-CTGGGCTCCTCTTGGAATG-3′.
Lentiviral transduction in organoids
Mouse wild-type Sod1, Sod1C58S, and Sod1C147S (kindly provided by Dr. X.F.Steven Zheng) plasmids were cloned into the tet-inducible lentiviral vector pINDUCER20 (Addgene, Massachusetts, USA, 44012). For virus production, the Sod1 plasmids were co-transfected with psPAX2 (Addgene, 12260), and pMD2.G (Addgene, 12259) into HEK293T cells using FuGENE® 6 Transfection Reagent (Promega, Wisconsin, USA, E2691). Viral supernatants were collected 24 and 48 h after transfection by passage through a 0.45 mm filter, and concentrated with lentivirus concentration solution (OriGene Technologies, Maryland, USA, TR30025) according to the manufacturer’s instructions.
Organoids were dissociated into single cells and transfected with concentrated lentiviruses as described previously [61]. Briefly, concentrated viral supernatants were added to cells in 48-well plates, incubated at 37 °C for 3 h, collected, and reseeded in Matrigel-containing medium. Infected organoids were selected with G418 (Thermo Scientific, 10131035) at 72 h after viral transduction for 2 weeks.
To induce exogenous vector-, wild-type Sod1, Sod1C58S, and Sod1C147S expression in Sod1-deficient organoids, 1 μg ml−1 doxycycline (Sigma-Aldrich, D9891) was added to the culture media on day 0 and supplemented with media changes at 2-day intervals.
Immunohistochemistry
Dissected intestines were fixed overnight in 4% paraformaldehyde and subsequently embedded in paraffin. Immunohistochemistry (IHC) was carried out according to standard procedures. The following primary antibodies were used for immunostaining: SOD1 (Abcam, Massachusetts, USA, ab16831, 1:500), cleaved caspase-3 (Cell Signaling Technology, 9664, 1:500), Ki-67 (Cell Signaling Technology, 12202, 1:500), OLFM4 (Cell Signaling Technology, 39141, 1:500), lysozyme (Dako, A009902-2, 1:10000) and goat anti-rabbit IgG secondary antibody (Vector Laboratories, DI-1594-1.5, 1:5000). VECTASTAIN Elite ABC HRP Kit (Vector Laboratories, California, USA, PK-6100) and ImmPACT DAB Peroxidase (HRP) Substrate (Vector Laboratories, SK-4105) were used to detect primary antibodies for Immunohistochemistry.
SOD1 activity assay
SOD1 activity assay was carried out as previously described [62]. Briefly, organoids were harvested in cold PBS, lysed by sonication in phosphate buffer (0.05 M KH2PO4 and K2HPO4, pH 7.8) supplemented with 0.1% Triton X-100, and protease and phosphatase inhibitor cocktails. Organoid proteins (150–200 μg per sample) were loaded in 12% native gel. The native PAGE gels were stained with 2.43 mM nitro blue tetrazolium chloride (Sigma-Aldrich, N6876), 0.14 M riboflavin-5′-phosphate (Sigma-Aldrich, F1392), and 28 mM TEMED (Bio-Rad Laboratories, California, USA, 1610800) for 20 min at room temperature in darkness. To visualize SOD1 activity, gels were rinsed with water twice and placed on a light box for 15–120 min.
RNA sequencing
Whole crypts were isolated from 9-weeks-old mice for organoid cultures, with three mice per genotype (Sod1f/f and Sod1f/f;Vil-creERT2). Total RNA was isolated from organoids after 3 days of tamoxifen treatment using Rneasy mini plus kit (Qiagen). The concentration and quality of total RNA samples was first assessed using Agilent 2100 Bioanalyzer. A RIN (RNA Integrity Number) of five or higher was required to pass the quality control. Then four hundred nanograms of RNA per sample were used to prepared dual-indexed strand-specific cDNA library using TruSeq RNA Access Library Prep Kit (Illumina). Samples were processed on an Illumina HiSeq 4000 for 2 × 75 bp paired-end sequencing. Analysis of RNA-Seq data was carried out using the following protocol: the sequencing data were first assessed using FastQC (Babraham Bioinformatics, Cambridge, UK) for quality control. Then all sequenced libraries were mapped to the mouse genome (UCSC mm10) using STAR RNA-seq aligner [63]. The reads distribution across the genome was assessed using bamutils (from ngsutils) [64]. Uniquely mapped sequencing reads were assigned to mm10 refGene genes using featureCounts (from subread) [65]. Quality control of sequencing and mapping results was summarized using MultiQC [66]. Genes with read count per million (CPM) > 0.5 in more than 3 of the samples were kept. The data was normalized using TMM (trimmed mean of M values) method. Differential expression analysis was performed using edgeR [67, 68]. False discovery rate (FDR) was computed from p-values using the Benjamini-Hochberg procedure. All expression data were deposited in Gene Expression Omnibus (GEO accession number GSE211035).
Statistical analysis
All data were presented as mean ± SEM and were analyzed using the GraphPad Prism 9 software. Statistical analyses were performed with two-way ANOVA, one-way ANOVA or Student’s t test. *P < 0.05 was considered to be statistically significant.
Data availability
The authors declare that all the other data supporting the findings of this study are available within the article and its Supplementary Information files and from the corresponding author on request.
References
Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007;449:1003–7.
van der Flier LG, Haegebarth A, Stange DE, van de Wetering M, Clevers H. OLFM4 is a robust marker for stem cells in human intestine and marks a subset of colorectal cancer cells. Gastroenterology 2009;137:15–7.
Montgomery RK, Carlone DL, Richmond CA, Farilla L, Kranendonk ME, Henderson DE, et al. Mouse telomerase reverse transcriptase (mTert) expression marks slowly cycling intestinal stem cells. Proc Natl Acad Sci. 2011;108:179–84.
Sangiorgi E, Capecchi MR. Bmi1 is expressed in vivo in intestinal stem cells. Nat Genet. 2008;40:915–20.
Takeda N, Jain R, LeBoeuf MR, Wang Q, Lu MM, Epstein JA. Interconversion between intestinal stem cell populations in distinct niches. Science 2011;334:1420–4.
Metcalfe C, Kljavin NM, Ybarra R, de Sauvage FJ. Lgr5+ stem cells are indispensable for radiation-induced intestinal regeneration. Cell Stem Cell. 2014;14:149–59.
Potten CS. Extreme sensitivity of some intestinal crypt cells to X and gamma irradiation. Nature 1977;269:518–21.
Marshman E, Booth C, Potten CS. The intestinal epithelial stem cell. Bioessays 2002;24:91–8.
Zhu Y, Huang YF, Kek C, Bulavin DV. Apoptosis differently affects lineage tracing of Lgr5 and Bmi1 intestinal stem cell populations. Cell Stem Cell. 2013;12:298–303.
Potten CS, Booth C, Pritchard DM. The intestinal epithelial stem cell: the mucosal governor. Int J Exp Pathol. 1997;78:219–43.
Hua G, Thin TH, Feldman R, Haimovitz-Friedman A, Clevers H, Fuks Z, et al. Crypt base columnar stem cells in small intestines of mice are radioresistant. Gastroenterology 2012;143:1266–76.
Yan KS, Chia LA, Li X, Ootani A, Su J, Lee JY, et al. The intestinal stem cell markers Bmi1 and Lgr5 identify two functionally distinct populations. Proc Natl Acad Sci. 2012;109:466–71.
Potten CS, Gandara R, Mahida YR, Loeffler M, Wright NA. The stem cells of small intestinal crypts: where are they? Cell Prolif. 2009;42:731–50.
Porter EM, Bevins CL, Ghosh D, Ganz T. The multifaceted Paneth cell. Cell Mol Life Sci. 2002;59:156–70.
Sato T, van Es JH, Snippert HJ, Stange DE, Vries RG, van den Born M, et al. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 2011;469:415–8.
Zhao J, de Vera J, Narushima S, Beck EX, Palencia S, Shinkawa P, et al. R-spondin1, a novel intestinotrophic mitogen, ameliorates experimental colitis in mice. Gastroenterology 2007;132:1331–43.
Korinek V, Barker N, Moerer P, van Donselaar E, Huls G, Peters PJ, et al. Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4. Nat Genet. 1998;19:379–83.
Pinto D, Gregorieff A, Begthel H, Clevers H. Canonical Wnt signals are essential for homeostasis of the intestinal epithelium. Genes Dev. 2003;17:1709–13.
van Es JH, Jay P, Gregorieff A, van Gijn ME, Jonkheer S, Hatzis P, et al. Wnt signalling induces maturation of Paneth cells in intestinal crypts. Nat Cell Biol. 2005;7:381–6.
Andreu P, Colnot S, Godard C, Gad S, Chafey P, Niwa-Kawakita M, et al. Crypt-restricted proliferation and commitment to the Paneth cell lineage following Apc loss in the mouse intestine. Development 2005;132:1443–51.
Beumer J, Clevers H. Regulation and plasticity of intestinal stem cells during homeostasis and regeneration. Development 2016;143:3639–49.
Okado-Matsumoto A, Fridovich I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-SOD in mitochondria. J Biol Chem. 2001;276:38388–93.
Sheng Y, Abreu IA, Cabelli DE, Maroney MJ, Miller AF, Teixeira M, et al. Superoxide dismutases and superoxide reductases. Chem Rev. 2014;114:3854–918.
Hu S, Xie Z, Onishi A, Yu X, Jiang L, Lin J, et al. Profiling the human protein-DNA interactome reveals ERK2 as a transcriptional repressor of interferon signaling. Cell 2009;139:610–22.
Li X, Qiu S, Shi J, Wang S, Wang M, Xu Y, et al. A new function of copper zinc superoxide dismutase: as a regulatory DNA-binding protein in gene expression in response to intracellular hydrogen peroxide. Nucleic Acids Res. 2019;47:5074–85.
Tsang CK, Liu Y, Thomas J, Zhang Y, Zheng XF. Superoxide dismutase 1 acts as a nuclear transcription factor to regulate oxidative stress resistance. Nat Commun. 2014;5:3446.
Xu J, Su X, Burley SK, Zheng XFS. Nuclear SOD1 in Growth Control, Oxidative Stress Response, Amyotrophic Lateral Sclerosis, and Cancer. Antioxidants (Basel). 2022;11:427.
Wang X, Zhang H, Sapio R, Yang J, Wong J, Zhang X, et al. SOD1 regulates ribosome biogenesis in KRAS mutant non-small cell lung cancer. Nat Commun. 2021;12:2259.
Wu S, Yi J, Zhang YG, Zhou J, Sun J. Leaky intestine and impaired microbiome in an amyotrophic lateral sclerosis mouse model. Physiol Rep. 2015;3:e12356.
Sagi H, Shibuya S, Kato T, Nakanishi Y, Tsuboi A, Moriya S, et al. SOD1 deficiency alters gastrointestinal microbiota and metabolites in mice. Exp Gerontol. 2020;130:110795.
Lee H, Ko EH, Lai M, Wei N, Balroop J, Kashem Z, et al. Delineating the relationships among the formation of reactive oxygen species, cell membrane instability and innate autoimmunity in intestinal reperfusion injury. Mol Immunol. 2014;58:151–9.
Kurose I, Wolf RE, Grisham MB, Granger DN. Hypercholesterolemia enhances oxidant production in mesenteric venules exposed to Ischemia/Reperfusion. Arterioscler Thromb Vasc Biol. 1998;18:1583–8.
Russell J, Epstein CJ, Grisham MB, Alexander JS, Yeh KY, Granger DN. Regulation of E-selectin expression in postischemic intestinal microvasculature. Am J Physiol Gastrointest Liver Physiol. 2000;278:G878–85.
Juarez JC, Betancourt O Jr, Pirie-Shepherd SR, Guan X, Price ML, Shaw DE, et al. Copper binding by tetrathiomolybdate attenuates angiogenesis and tumor cell proliferation through the inhibition of superoxide dismutase 1. Clin Cancer Res. 2006;12:4974–82.
Furukawa Y, Torres AS, O’Halloran TV. Oxygen-induced maturation of SOD1: a key role for disulfide formation by the copper chaperone CCS. EMBO J. 2004;23:2872–81.
Morris O, Jasper H. Reactive Oxygen Species in intestinal stem cell metabolism, fate and function. Free Radic Biol Med. 2021;166:140–6.
Farin HF, Van Es JH, Clevers H. Redundant sources of Wnt regulate intestinal stem cells and promote formation of Paneth cells. Gastroenterology 2012;143:1518–29. e7
Biteau B, Jasper H. EGF signaling regulates the proliferation of intestinal stem cells in Drosophila. Development 2011;138:1045–55.
Lee D, Pearsall RS, Das S, Dey SK, Godfrey VL, Threadgill DW. Epiregulin is not essential for development of intestinal tumors but is required for protection from intestinal damage. Mol Cell Biol. 2004;24:8907–16.
Gregorieff A, Liu Y, Inanlou MR, Khomchuk Y, Wrana JL. Yap-dependent reprogramming of Lgr5(+) stem cells drives intestinal regeneration and cancer. Nature 2015;526:715–8.
Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009;459:262–5.
Hochmuth CE, Biteau B, Bohmann D, Jasper H. Redox regulation by Keap1 and Nrf2 controls intestinal stem cell proliferation in Drosophila. Cell Stem Cell. 2011;8:188–99.
Cheung EC, Lee P, Ceteci F, Nixon C, Blyth K, Sansom OJ, et al. Opposing effects of TIGAR- and RAC1-derived ROS on Wnt-driven proliferation in the mouse intestine. Genes Dev. 2016;30:52–63.
Asano J, Sato T, Ichinose S, Kajita M, Onai N, Shimizu S, et al. Intrinsic Autophagy Is Required for the Maintenance of Intestinal Stem Cells and for Irradiation-Induced Intestinal Regeneration. Cell Rep. 2017;20:1050–60.
Elchuri S, Oberley TD, Qi W, Eisenstein RS, Jackson Roberts L, Van, et al. CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene 2005;24:367–80.
Muller FL, Song W, Liu Y, Chaudhuri A, Pieke-Dahl S, Strong R, et al. Absence of CuZn superoxide dismutase leads to elevated oxidative stress and acceleration of age-dependent skeletal muscle atrophy. Free Radic Biol Med. 2006;40:1993–2004.
Fischer LR, Igoudjil A, Magrane J, Li Y, Hansen JM, Manfredi G, et al. SOD1 targeted to the mitochondrial intermembrane space prevents motor neuropathy in the Sod1 knockout mouse. Brain 2011;134:196–209.
Fischer LR, Glass JD. Oxidative stress induced by loss of Cu,Zn-superoxide dismutase (SOD1) or superoxide-generating herbicides causes axonal degeneration in mouse DRG cultures. Acta Neuropathol. 2010;119:249–59.
Sato T, Clevers H. Primary mouse small intestinal epithelial cell cultures. Methods Mol Biol. 2013;945:319–28.
Li L, Xie T. Stem cell niche: structure and function. Annu Rev Cell Dev Biol. 2005;21:605–31.
Singh B, Carpenter G, Coffey RJ. EGF receptor ligands: recent advances. F1000Res. 2016;5:F1000 Faculty Rev-2270.
Riese DJ 2nd, Komurasaki T, Plowman GD, Stern DF. Activation of ErbB4 by the bifunctional epidermal growth factor family hormone epiregulin is regulated by ErbB2. J Biol Chem. 1998;273:11288–94.
Shelly M, Pinkas-Kramarski R, Guarino BC, Waterman H, Wang LM, Lyass L, et al. Epiregulin is a potent pan-ErbB ligand that preferentially activates heterodimeric receptor complexes. J Biol Chem. 1998;273:10496–505.
Joo M, Shahsafaei A, Odze RD. Paneth cell differentiation in colonic epithelial neoplasms: evidence for the role of the Apc/beta-catenin/Tcf pathway. Hum Pathol. 2009;40:872–80.
Pai RK, Rybicki LA, Goldblum JR, Shen B, Xiao SY, Liu X. Paneth cells in colonic adenomas: association with male sex and adenoma burden. Am J Surg Pathol. 2013;37:98–103.
Zhang Y, Davis C, Sakellariou GK, Shi Y, Kayani AC, Pulliam D, et al. CuZnSOD gene deletion targeted to skeletal muscle leads to loss of contractile force but does not cause muscle atrophy in adult mice. FASEB J. 2013;27:3536–48.
Chen L, Vasoya RP, Toke NH, Parthasarathy A, Luo S, Chiles E, et al. HNF4 Regulates Fatty Acid Oxidation and Is Required for Renewal of Intestinal Stem Cells in Mice. Gastroenterology 2020;158:985–99. e9
Sakamori R, Yu S, Zhang X, Hoffman A, Sun J, Das S, et al. CDC42 inhibition suppresses progression of incipient intestinal tumors. Cancer Res. 2014;74:5480–92.
O’Rourke KP, Ackerman S, Dow LE, Lowe SW. Isolation, Culture, and Maintenance of Mouse Intestinal Stem Cells. Bio Protoc. 2016;6:e1733.
Yan H, Wang YC, Li D, Wang Y, Liu W, Wu YL, et al. Arsenic trioxide and proteasome inhibitor bortezomib synergistically induce apoptosis in leukemic cells: the role of protein kinase Cdelta. Leukemia 2007;21:1488–95.
Van Lidth de Jeude JF, Vermeulen JL, Montenegro-Miranda PS, Van den Brink GR, Heijmans J. A protocol for lentiviral transduction and downstream analysis of intestinal organoids. J Vis Exp. 2015;20:52531.
Weydert CJ, Cullen JJ. Measurement of superoxide dismutase, catalase and glutathione peroxidase in cultured cells and tissue. Nat Protoc. 2010;5:51–66.
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21.
Breese MR, Liu Y. NGSUtils: a software suite for analyzing and manipulating next-generation sequencing datasets. Bioinformatics 2013;29:494–6.
Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014;30:923–30.
Ewels P, Magnusson M, Lundin S, Kaller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016;32:3047–8.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010;26:139–40.
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–78.
Acknowledgements
The authors would like to thank X. F. Steven Zheng and Michael P.Verzi for excellent technical support.
Funding
This project was supported in part by grants from National Key R&D Program of China (2020YFA0509200), National Natural Science Foundation of China (81421001, 81530072, 81830081, 31970718), Shanghai Municipal Health Commission, Collaborative Innovation Cluster Project (2019CXJQ02), National Natural Science Foundation of China (81772519), Excellent Youth Program of Shanghai Municipal Commission of Health and Family Planning (2018YQ29), Natural Science Foundation of Qinghai (2018-ZJ-942Q).
Author information
Authors and Affiliations
Contributions
YCW, HMC and JYF conceived and designed the experiments. YCW, XXL, CBZ and SYL performed the experiments and analyzed data. CKT, JX and MMZ aided in the early design and revision of this project. YCW, and HMC, JYF wrote and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Our studies did not include human participants, human data, or human tissue. All mouse experiments were conducted in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. The study procedures were approved by the Institutional Animal Care and Use Committee of Renji Hospital, Shanghai Jiao Tong University School of Medicine.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Professor Paolo Pinton
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, YC., Leng, XX., Zhou, CB. et al. Non-enzymatic role of SOD1 in intestinal stem cell growth. Cell Death Dis 13, 882 (2022). https://doi.org/10.1038/s41419-022-05267-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-022-05267-w
