
Abstract
Tauopathies define a broad range of neurodegenerative diseases that encompass pathological aggregation of the microtubule-associated protein tau. Although tau aggregation is a central feature of these diseases, their underlying pathobiology is remarkably heterogeneous at the molecular level. In this review, we summarize critical differences that account for this heterogeneity and contrast the physiological and pathological functions of tau. We focus on the recent understanding of its prion-like behavior that accounts for its spread in the brain. Moreover, we acknowledge the limited appreciation about how upstream cellular changes influence tauopathy. Dysfunction of the highly conserved endosomal trafficking complex retromer is found in numerous tauopathies such as Alzheimer’s disease, Pick’s disease, and progressive supranuclear palsy, and we discuss how this has emerged as a major contributor to various aspects of neurodegenerative diseases. In particular, we highlight recent investigations that have elucidated the contribution of retromer dysfunction to distinct measures of tauopathy such as tau hyperphosphorylation, aggregation, and impaired cognition and behavior. Finally, we discuss the potential benefit of targeting retromer for modifying disease burden and identify important considerations with such an approach moving toward clinical translation.
Similar content being viewed by others

Facts
-
Tau aggregation in the brain is a common feature of numerous neurodegenerative diseases termed “tauopathies.”
-
Retromer is an evolutionarily conserved complex consisting of VPS26, VPS35, and VPS29.
-
Retromer abundance is inversely correlated with tau aggregation in the brain of people with tauopathies, including Alzheimer’s disease, Pick’s disease, and progressive supranuclear palsy.
-
Depletion of retromer subunit VPS35 enhances pathological tau hyperphosphorylation, aggregation, and cognitive/behavioral phenotypes in various preclinical models of tauopathy.
-
Pharmacological stabilization of retromer with small-molecule chaperones suppresses tau-mediated pathologies.
Open questions
-
In human tauopathies, does retromer depletion in the brain precede tau aggregation? Or vice versa?
-
Does reduced synthesis, increased turnover, or both account for retromer depletion in the brain of people with tauopathies?
-
What downstream function(s) of retromer account for its neuroprotective effects against tauopathies in vivo?
-
Will retromer chaperones enhance lifespan (or healthspan) in mouse models of tauopathy?
-
Will retromer chaperones be effective after the onset of disease hallmarks in mouse models of tauopathy?
Despite clear similarities tauopathies are heterogeneous
Pathological aggregation of the microtubule-associated protein tau is a common feature of several age-related neurodegenerative diseases, collectively termed “tauopathies” [1]. The tau aggregates found in these diseases are heterogeneous and differ considerably across several parameters such as location in the brain, pattern of spread, isoform composition, residing cell type, histological morphology, recognition by amyloid-binding dyes, posttranslational modifications, and structural conformation/strain [2]. It is therefore not surprising that with such marked heterogeneity, tauopathies are present as clinically distinct syndromes. Examples of tauopathies include Alzheimer’s disease (AD, the most common tauopathy), Pick’s disease, progressive supranuclear palsy, corticobasal degeneration, frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17), chronic traumatic encephalopathy, argyrophilic grain disease, globular glial tauopathy, and primary age-related tauopathy [1]. Although there is no relationship between MAPT variation and AD, more than 40 MAPT mutations are known to cause FTDP-17 [3,4,5,6,7]. In addition, MAPT mutations also cause corticobasal degeneration [8]. These mutations typically affect the microtubule-binding capacity of tau, increase its aggregation propensity, or alter its splicing pattern [3,4,5, 9, 10]. In addition, the H1 haplotype of MAPT is associated with increased risk of progressive supranuclear palsy and primary age-related tauopathy [11, 12].
Tau in physiology and pathophysiology
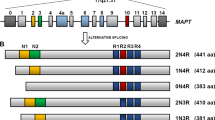
Topologically, tau is divided into two sections: its C-terminal microtubule-binding repeat domain (RD) and an N-terminal projection domain that extends away from microtubules to regulate microtubule spacing [13, 14]. In the adult human brain, tau is expressed as six isoforms that vary by alternative slicing of exons that encode the two N-terminal inserts (N1 and N2) in its projection domain and the second (R2), out of four repeats (4R) that make up the RD, to generate 3R- and 4R-containing isoforms [15,16,17,18]. Under physiological conditions, isoforms with and without R2 are equally abundant despite 4R isoforms having a greater microtubule-binding affinity/avidity [16, 19]. Tau regulates microtubule stability by binding to the interface of α–β tubulin heterodimers [20, 21]. Additionally, tau regulates axonal transport and the actin cytoskeleton [22,23,24,25]. In the central nervous system, tau is localized predominantly along axons, and to a lesser extent, within dendrites [14, 26]. In disease, tau is redistributed to other locations within the neuron where it forms hyperphosphorylated inclusions, such as neurofibrillary tangles in the soma, neuropil threads within neurites, and neuritic plaques in the vicinity of amyloid plaques [27,28,29,30]. Tau hyperphosphorylation is marked in the AD brain to the extent that paired-helical filaments of tau, that comprise larger aggregates, contain ~7 moles of phosphate per mole of protein, which is ~3.5 times more than control brains [31]. Tau is natively unfolded owing to its high composition of hydrophilic amino acids, and this lack of complexity in its structure makes it an ideal substrate for posttranslational modifications such as phosphorylation, methylation, acetylation, glycosylation, SUMOylation, O-linked N-acetylglucosamination, and cleavage [13, 32]. Because tau can be so heavily modified, with >40 amino acid residues identified by mass spectrometry as being phosphorylated, it is difficult to fully understand the contribution of individual site-specific phosphorylations [33]. However, the functional significance of several phosphorylation events is well-characterized. For example, phosphorylation of residues within or flanking the RD negatively affects the tau–microtubule interaction [34, 35]. Moreover, phosphorylated tau accumulates in the AD brain before the emergence of neurofibrillary tangles [36]. Tau hyperphosphorylation enhances its aggregation via self-assembly into paired-helical and straight filaments, which constitute neurofibrillary tangles [37,38,39]. While tau hyperphosphorylation promotes tau aggregation, tau aggregation may also enhance subsequent hyperphosphorylation. During embryonic development, tau is hyperphosphorylated but not aggregated, indicating that phosphorylation alone does not accurately predict aggregation [40]. Moreover, not all tau phosphorylation events are pathological. Indeed, phosphorylation at T205 by p38γ prevents amyloid-β-mediated toxicity in APP23 transgenic mice (human APP [isoform 751] with K670M and N671L [Swedish] mutations) [41]. Whether tau aggregation represents a loss- or a toxic gain-of-function phenotype is still heavily debated [42,43,44,45,46,47,48,49,50]. However, genetic deletion of endogenous tau in mouse and fruit fly models does not recapitulate key phenotypes observed in transgenic mouse models of tauopathy, which mirror hallmarks of human disease, arguing strongly against pathology being driven by a loss of function [51, 52].
Pathogenic tau displays prion-like spread in the brain
The spread of tau aggregation throughout the brain is tightly linked to disease severity [53]. In AD, neurofibrillary tangles composed of hyperphosphorylated and aggregated tau first appear in the locus coeruleus and the trans-entorhinal and entorhinal regions (Braak stages I and II) [53]. Following this, tau aggregation spreads to the hippocampal formation and parts of the neocortex (Braak stages III and IV) before eventually reaching more peripheral parts of the neocortex (Braak stages V and VI) [53]. Intriguingly, tau spread in the neocortex occurs in the opposite direction to cortical myelogenesis [54]. Tau spread throughout the brain correlates strongly with cognitive decline, whereas the accumulation of amyloid-β plaques, another proteinaceous hallmark of AD, does not [55]. Tau pathology does not spread stochastically but rather in a predictable, trans-neuronal manner [56,57,58,59,60]. This explains how tau aggregation can spread trans-synaptically to distant but anatomically connected parts of the brain [61].
The initial corrupting event(s) that converts natively folded tau into a pathological, aggregation-prone state in neurons remains poorly understood. Heparan sulfate polyanions and RNA are potent inducers of tau aggregation in vitro, although their in vivo contributions remain unclear [62, 63]. In contrast, injection of amyloid-β42 fibrils into the brain of pR5 transgenic mice (MAPT [isoform 2N4R] with P301L mutation) expressing P301L tau enhances tau aggregation in vivo [64]. Once this conversion occurs, small oligomers of misfolded tau can be trafficked to and internalized by connected neurons. Here, these misfolded species can gain access to natively folded—or naive—tau in the cytoplasm and promote their misfolding and aggregation by a process often called “seeding” (Fig. 1) [65]. Biochemical studies show that a tau trimer is the smallest assembly with the ability to seed aggregation and induce toxicity [66, 67]. Tau seeds can be shared between connected neurons through distinct routes, such as via extracellular vesicles, tunneling nanotubules, or unconventionally by direct penetration of the plasma membrane [68, 69]. In addition, microglia are involved in spreading tau throughout the brain [68, 70]. Following this, tau seeds can be internalized by neighboring neurons via macropinocytosis or endocytosis [71]. Recently, low-density lipoprotein receptor-related protein 1 was found to regulate tau internalization by neurons, whereas other studies indicate that heparan sulfate proteoglycans mediate this process [72, 73]. After internalization, tau seeds are physically separated from the cytoplasm in the endocytic compartment. Eventually, tau seeds escape this confine and can seed the aggregation of naive tau in the cytoplasm, propagating the spread of tau aggregation [74]. Presently, it is unclear how this escape mechanism is executed. Whilst it is conceivable that this may be a stochastic event, tau fibrils prepared in vitro can elicit endomembrane injury after cellular uptake [75, 76]. Although cells have evolved distinct mechanisms for repairing, removing and replacing damaged vesicles, these mechanisms presumably fail to completely abolish tau seeding, likely owing to the rapid kinetics of tau aggregation [74, 76, 77].

Natively folded tau is hyperphosphorylated and undergoes sporadic misfolding that initiates the assembly of oligomeric species of tau. These can then seed the misfolding and aggregation of natively folded tau monomers in the cytoplasm. Meanwhile, phosphorylation of monomeric tau increases the amount of cytoplasmic tau available for incorporation, and in addition, phosphorylation of tau aggregates may also occur. Assemblies of oligomeric tau can continue to grow into repeating filamentous structures (e.g., paired-helical and straight filaments) that eventually coalesce into large intraneuronal inclusions (e.g., neurofibrillary tangles and neuropil threads). An example of neurofibrillary tangles and neuropil threads stained for phospho-tau (S202/T205, AT8) from an AD brain is shown.
Seeding as a mode of inheriting protein conformations has been extensively studied in the context of mammalian prion diseases [78]. In these diseases, pathological conformers of the prion protein PrPSc (Sc denotes Scrapie) induce the misfolding and aggregation of the naive form PrPC (C denotes cellular form) [78]. Despite the majority of prion diseases being sporadic, their initial corrupting event(s) are more well-established than in tauopathies and can occur through direct contact with PrPSc-contaminated materials (i.e., grafts, surgical equipment, and consumption of contaminated meat) and by rare mutations to the gene that encodes PrPC (PRNP) [78, 79]. Prions and aggregated tau can both exist in distinct conformational states, which are referred to as “strains” [59, 60, 78]. Indeed, tau strains are remarkably stable and can be propagated in and between transgenic mice and cell cultures, whilst maintaining their structural identity [59, 60]. However, despite these similarities, tauopathies are not considered a bone fide prion disease for the following reasons: (1) tau is not infectious and (2) naive endogenous tau is not a strict requirement for tau inclusions to propagate between neurons, but rather (3) is a requirement for toxicity [80, 81]. Instead, tau is referred to as being “prion-like.” Advances in cryoelectron microscopy have detailed distinct strains of tau fibrils purified from the brains of people with AD, Pick’s disease, and corticobasal degeneration [82,83,84]. Moreover, even aggregated tau from people with AD displays remarkable patient-to-patient heterogeneity across several parameters, including seeding capacity and posttranslational modifications [85]. Importantly, tau seeding correlates strongly with symptomatic severity in AD, a feature that covaries with phosphorylation of tau at T231, S235, and S262 [85].
Tau aggregation and neurotoxicity
Are neurofibrillary tangles themselves neurotoxic? In AD, loss of neurons in the temporal sulcus correlates with, but exceeds the deposition of neurofibrillary tangles, indicating that the majority of neurons die without neurofibrillary tangles [86]. Further, neurons in the human brain can survive for two decades with neurofibrillary tangles, arguing strongly against a mode of acute neurotoxicity [87]. Remarkably, in rTg4510-inducible transgenic mice, “switching off” P301L tau expression improves cognition and memory, even though neurofibrillary tangles still persist [88]. A caveat to this model is that some neurodegenerative phenotypes are nonspecific and due to insertional mutagenesis from the transgene [89]. In Drosophila, neuronal overexpression of human tau increases neurodegeneration and reduces lifespan in the absence of neurofibrillary tangles [90]. Cognitive function is unchanged in transgenic mice that express tau RD (ΔK280/I227P/I308P), a mutant incapable of aggregation, suggesting that aggregation, but not necessarily neurofibrillary tangles, may be required for neurotoxicity [91]. Intriguingly, injection of oligomeric, but not monomeric or fibrillar tau into the brain of wild-type mice leads to cognitive decline and synaptic dysfunction [92]. Cognitive decline is apparent immediately after injection of oligomeric tau into the hippocampus of wild-type mice [92]. Indeed, soluble tau aggregates but not large fibrils reduce cell viability following delivery to SH-SY5Y neuroblastoma cells [93]. Cell culture-based reporters detect seeding from PS19 mouse brain lysates before the emergence or NFTs and cognitive decline [94]. Further, dorsal root ganglion neurons containing aggregated tau P301S only die following coculture with phagocytes [95]. Together, these data support the idea that oligomeric species of tau, rather than neurofibrillary tangles per se mediate tau-induced neurotoxicity through crosstalk with immune cells.
Retromer dysfunction is associated with various neurodegenerative phenotypes
The consequences of tau hyperphosphorylation and aggregation on measures of neurodegeneration have been well-documented, whereas factors that modify these pathological changes remain poorly understood. Understanding how neurons and other cell types in “healthy” brains cope with insults from deleterious factors (e.g., aggregated tau) to curb pathological changes will be an important step forward for the field. In this regard, over the past 15 years, dysfunction of the retromer complex has emerged as an interesting trait of neurodegenerative diseases, especially AD, Parkinson’s disease, and more recently amyotrophic lateral sclerosis [96,97,98,99,100]. Importantly, retromer dysfunction recapitulates numerous disease-related phenotypes such as amyloid-β deposition, cell death, gliosis, synapse loss, impaired neurotransmission, and cognitive impairment [100,101,102,103,104]. More recently, retromer deficiency was found to exacerbate several measures of tauopathy, including hyperphosphorylation, aggregation, and cognitive decline [104,105,106]. Apart from enhancing tau aggregation, retromer dysfunction also enhances the accumulation of aggregated amyloid-β, α-synuclein, and huntingtin [96, 104, 106,107,108,109,110,111,112,113,114,115] (Table 1). Together, these findings suggest that retromer dysfunction is important for a broad range of proteopathies.
Retromer is a highly conserved endosomal receptor trafficking complex
A phenomenon observed across several tauopathies is a reduction of retromer complex components in the brain [96, 104]. Retromer is a highly conserved assembly of vacuolar protein sorting 35, −26, and −29 (VPS35, VPS26, and VPS29) that is recruited to the maturing endosome to orchestrate receptor sorting and transport to the trans-Golgi network or plasma membrane [116]. These trafficking itineraries are critical for the maintenance of receptor homeostasis and therefore downstream functions, otherwise these receptors are defaulted to the lysosome for destruction [117]. Selection of receptors at the endosomal membrane is a tightly regulated process with different receptors selected by distinct surfaces of the retromer complex [118,119,120]. Engagement with specific receptors can also be facilitated by subcomplexes of retromer with itinerary-specific adapters [118]. The trans-Golgi network trafficking function is achieved by a subcomplex of retromer with sorting nexin (SNX) 1 or -2 and SNX5 or -6 (SNX–BAR retromer), which mediates the generation of tubular endosomal carriers through the BAR (Bin-Amphiphysin-Rvs) domain of these SNX proteins [121,122,123]. Alternatively, a subcomplex of retromer with SNX3 (SNX3 retromer) achieves a similar effect, albeit through vesicular endosomal carriers [124]. On the other hand, plasma membrane trafficking is achieved by a subcomplex of retromer with SNX27 (SNX27 retromer) [125].
At the endosomal surface, retromer engages with the Wiskott–Aldrich syndrome protein and scar homolog (WASH) complex to facilitate local F-actin nucleation while also engaging with microtubules, albeit indirectly, through an interaction between SNX5/6 and the p150 subunit of the dynactin/dynein microtubule motor complex [126, 127]. Moreover, the SNX1-interactor DNAJ homolog subfamily C member 13 (RME8/DNAJC13) binds to the WASH complex subunit 2C (FAM21/WASHC2C) to support endosomal tubulation [128]. RME8 also segregates these endosomal domains away from degradative domains that are decorated with the hepatocyte growth factor-regulated tyrosine kinase substrate component of endosomal sorting complexes required for transport complex [128, 129]. Collectively, this enables outward protrusion of endosomal domains that are concentrated with cargo-engaged retromer complexes and long-range trafficking of these carriers following scission [126, 127]. Aided by tension from the protruding endosomal tubule, carrier scission is thought to be performed by Eps15 homology domain-containing protein 1, whereas capture and fusion of these carriers to target membranes appears to occur through distinct mechanisms [130, 131]. For example, endosomal carriers containing retromer cargo, the cation-independent mannose 6-phosphate receptor, are tethered to the trans-Golgi network by golgin 245 in an SNX1/2-dependent manner, or by GCC88 in an SNX3-dependent manner [132]. On the other hand, the plasma membrane-localized tethers for SNX27-retromer carriers are poorly defined. An inhibitory interaction between the retromer complex and TBC1 domain family member 5 (TBC1D5), a GTPase-activating protein for Rab7, controls the uncoating of retromer from endosomes [133]. Other additional but notable retromer cargoes include the amyloid-β precursor protein (APP), sortilin precursor (SORT1), sortilin-related receptor precursor (SORL1), divalent metal ion transporter 1-II, parathyroid hormone-related protein, glucose transporter 1 (GLUT1/SLC2A1), and the β2-adrenergic receptor (ADRB2) [130].
Retromer dysfunction is a common feature of several neurodegenerative diseases
The first link between retromer dysfunction and neurodegenerative disease was uncovered from a gene expression microarray that showed increased expression of VPS35 in the entorhinal cortex of postmortem human AD brains compared with controls [96]. However, closer inspection revealed depletion of VPS35 and VPS26 proteins [96]. Because the entorhinal cortex is particularly susceptible to the deposition of plaques and tangles in AD, it was posited that retromer dysfunction may contribute to AD pathogenesis [53, 96]. Indeed, numerous studies have demonstrated that retromer suppresses amyloid-β generation and plaque deposition through proper trafficking of its precursor APP and cleaving enzymes b-secretase 1 (BACE1) and γ-secretase [108, 109, 134]. Notably, pathogenic mutations in APP and PSEN1 (encodes a component of γ-secretase) cause familial early onset AD [135, 136]. In addition, genetic variants of retromer interactors KIAA1033, RAB7A, SNX1, and SNX3 are associated with increased risk of sporadic late-onset AD, whereas VPS26 is not [137, 138]. Albeit, these hits were not replicated in genome-wide association studies (GWAS) [139]. Additionally, SORL1 variation is genetically linked to AD in several GWAS and is a retromer cargo [140,141,142,143]. Further, a collection of 891 genes that are connected to the endolysosomal and autophagy network as a whole are diffusely associated with AD [144]. This is important given that the retromer complex functions at multiple stages throughout this network. In addition, a rare L625P variant of VPS35 was identified in a single patient with early onset sporadic AD from exome sequencing [145]. In cultured cells, overexpression of this mutant impairs its interaction with VPS29 and TBC1D5, while reducing the endosomal localization of retromer, indicating that this person with sporadic early onset AD had perturbed retromer function [145].
Other tauopathies, including Pick’s disease and progressive supranuclear palsy, also show depletion of all retromer components in the hippocampus and cortex of postmortem human brains [104]. Apart from tauopathies, retromer dysfunction is also implicated in other neurodegenerative diseases. The D620N mutation of VPS35 causes a highly penetrant autosomal dominant form Parkinson’s disease [97, 98]. In contrast, SNX27 expression is downregulated in Down’s syndrome, and mutations in RAB7, a protein involved in endosomal recruitment of retromer, cause the sensory neuropathy Charcot–Marie–Tooth 2B disease [146, 147]. In addition, mutations in KIAA0196, which encodes the WASH complex subunit 5 (WASHC5/strumpellin), cause hereditary spastic paraplegia [148]. Intriguingly, in the cortex of the Tg2576 transgenic mouse model of AD (human APP [isoform 695] with K670M and N671L [Swedish] mutations), retromer components become depleted in an age-dependent manner, as does, in this case Vps35 mRNA [149]. Further, primary neurons from hAPPJ20 transgenic mice (human APP with K670M, N671L [Swedish], and V717F [Indiana] mutations) show dysregulation of Vps35-positive endosomes, with a reduction of these endosomes in the soma and enrichment along axons hinting at perturbed retrograde trafficking [150]. Because amyloid plaques precede tau aggregation in AD, this may trigger retromer dysfunction to exacerbate or even initiate a tauopathy cascade [151]. There is a bidirectional relationship between retromer expression and tau, that is, reduced retromer enhances tau hyperphosphorylation and aggregation, and there is reduced expression of retromer components in tauopathies [96, 104, 106]. Everything considered, we are yet to know which is the cause, and which is the effect in human disease. Preclinical studies indicate it is likely that retromer lies upstream of tauopathy [104, 106].
Retromer dysfunction promotes tauopathy
Recent studies have investigated the relationship between the retromer component VPS35 and tauopathy. Cortex-specific delivery of shRNA against Vps35 in PS19 mice (human MAPT [isoform 1N4R] with P301S mutation), which are prone to developing tangle pathology in the absence of amyloid plaques, exacerbated cognitive/behavioral outcomes in Y-maze and Morris water maze tests [104]. Biochemical assessment of the cortex revealed gross enhancements of tau hyperphosphorylation at several sites (Table 2) [104]. Although tangle pathology/tau aggregation in these mice was not assessed, given that PS19 mice develop tangle pathology at 6 months, such pathology was likely present [104, 152].
Remarkably, a Vps35 D620N knock-in mouse model of Parkinson’s disease showed clear evidence of endogenous mouse tau pathology in the brain [153]. Both hetero- and homozygous mutant mice showed elevated tau hyperphosphorylation and conformational change in various regions of the brain such as the hippocampus and cortex (Table 2) [153]. The magnitude of several pathological changes was more pronounced in the heterozygous mutant mice [153]. These effects appeared specific to the Vps35 D620N mutation, as mice harboring a heterozygous Vps35-null allele were resistant to such changes, suggesting that a loss-of-function phenotype does not account for these effects, or alternatively, that these mice were haplosufficient [153]. Importantly, Vps35 D620N-expressing mice did not present with neurofibrillary tangles or neuritic pathology after histological assessment, nor did they show increased tau insolubility [153]. Rather, these mice displayed higher levels of total soluble tau, which corroborated elevations in pathological modifications such as phosphorylation and conformational change (Table 2) [153].
How does retromer dysfunction contribute to tauopathy? A recent mechanistic study attempted to answer this question. Taking advantage of seeding approaches, our laboratory used brain lysate from the rTg4510 transgenic mouse model (human MAPT [isoform 0N4R] with P301L mutation) to induce aggregation of tau RD (V337M/P301L) expressed in HEK293 cells [106]. The aggregates generated in this system conformed to several parameters observed in human tauopathies, i.e., the aggregates were hyperphosphorylated, detergent-insoluble, detectable with an amyloid-binding dye, recapitulated tangle-like morphology, and were capable of prion-like seeding/propagation [106]. In this model, depletion of VPS35 led to a block in the resolution of autophagy that was attributed to a reduction in autophagosome–lysosome fusion and lysosomal degradation [106]. This defect was not found in SNX27-null cells, ruling out defective endosome-to-plasma membrane trafficking as the cause. Consequently, VPS35 depletion led to marked accumulation of tau aggregates, whereas its overexpression had the opposite effect (Table 2) [106]. These observations were replicated in human embryonic stem cell (hESC)-derived cortical neurons seeded with tau aggregates following VPS35 depletion (Table 2) [106]. Pharmacological and genetic approaches revealed that the autophagy–lysosome axis, not the proteasome, was critical for suppressing tau aggregation [106]. Closer inspection revealed that tau aggregates colocalized with the autophagy cargo receptor sequestosome 1 (SQSTM1/p62) independently of ubiquitin, suggesting that they are turned over through a ubiquitin-independent form of selective autophagy [74, 106]. Accordingly, VPS35 depletion led to a reduction in the fraction of SQSTM1-decorated tau aggregates, indicating that their selection for autophagic degradation was impaired [106]. In addition, retromer dysfunction may also affect the early stages of autophagy including autophagosome formation. Indeed, the Parkinson’s disease-linked D620N VPS35 mutation reduces the targeting of ATG9A to autophagosomes and inhibits autophagy [112].
Retromer enhancement stages off tauopathy
Gene therapy directed at the cortex of 3 × Tg mice (human APP [isoform 695] with K670M and N671L [Swedish], human PSEN1 with M146V, and MAPT [isoform 0N4R] with P301L mutations), which are prone to developing both plaque and tangle pathology, was used to overexpress Vps35 [154]. This intervention boosted Vps26 levels, indicating enhanced retromer complex formation and reduced cognitive/behavioral deficits in the Morris water maze test [154]. Importantly, apart from reducing amyloid-β production and deposition in the cortex, Vps35 overexpression reduced insoluble tau and tau hyperphosphorylation at several sites (Table 3) [154]. However, it remains unclear whether Vps35-dependent reductions in amyloid-β deposition or tau phosphorylation were more potent in eliciting rescue of cognitive/behavioral phenotypes. Accordingly, rescue of similar cognitive/behavioral phenotypes in hAPPJ9 mice (human APP [isoforms 695, 751, and 770] with K670M and N671L [Swedish] and V717F [Indiana] mutations) can be achieved through depletion of endogenous tau, highlighting a more dominant and downstream role for tau in eliciting neurodegeneration [155].
Overexpression of Vps35 in the mouse neuroblastoma N2a cell line reduced tau phosphorylation in a manner that was abolished by the aspartyl protease inhibitor pepstatin A (Table 3) [104]. This suggests that lysosomal function is required for Vps35-mediated suppression of tau phosphorylation. Consistent with this, the lysosomal inhibitor chloroquine failed to further enhance tau aggregation in HEK293 cells lacking VPS35 [106]. The important difference between these studies was that Carosi et al. employed seeded aggregation of human tau RD (V337M/P301L), whereas Vagnozzi et al. analyzed soluble endogenous murine tau [104, 106]. However, the former study demonstrated that aggregated, not soluble tau RD (V337M/P301L) is highly dependent on turnover through the autophagy–lysosome axis [106]. Moreover, lysates from Vps35 overexpressing cells showed elevated cathepsin D/E (CTSD/E) activity in vitro, which are lysosomal proteases known to be inhibited by pepstatin A [104]. Conversely, VPS35-null cells show impaired intralysosomal proteolytic maturation of CTSD [106, 132].
Retromer chaperones stage off tauopathy
Pharmacological stabilization of retromer with the chemically related small-molecule chaperones R55 and R33 has emerged as a promising avenue for suppressing amyloid-β pathology [101, 156]. Induced pluripotent stem cell (IPSC)-derived neuronal cultures from people with sporadic AD found that the retromer chaperone R55 reduces tau phosphorylation at T231, whereas shRNA-mediated depletion of VPS35 had the opposite effect (Table 3) [105]. Despite retromer dysfunction promoting pathological tau phosphorylation, it also enhances amyloid-β generation and deposition that can in turn promote tau phosphorylation and aggregation [64, 105]. Using a combination of genetic (i.e., APP gene duplication and CRISPR/Cas9-mediated APP knockout) and pharmacological (i.e., γ-secretase inhibitor compound E) approaches to modulate amyloid-β production in IPSC-derived neurons, it was revealed that the suppressive effect of retromer on tau phosphorylation occurred in an amyloid-β-independent manner (Table 3) [105]. The related retromer chaperone R33 also reduced tau phosphorylation at numerous sites in N2a cells (Table 3) [104]. When R33 was administered to 3 × Tg mice, it was similarly effective in reducing tau phosphorylation and amyloid-β generation and deposition [101]. In addition, pharmacological stabilization of retromer led to improvements across several cognitive/behavioral tests such as Y-maze, Morris water maze, and fear conditioning tests [101]. The R33 treatment and Vps35 overexpression in the 3 × Tg mouse model showed the opposite effects on tau phosphorylation to PS19 mice with cortex-specific depletion of Vps35, suggesting that tau phosphorylation is sensitive to retromer levels (Table 3) [101, 104, 154]. Further, R33 did not alter the levels of phosphatases and kinases that modify tau phosphorylation, including serine/threonine-protein phosphatase 2A catalytic subunit-α, glycogen synthase kinase-3β, and cyclin-dependent-like kinase 5 (Cdk5), as well as Cdk5 activators p35/25 in the cortex of 3 × Tg mice [101]. It remains unclear whether the activities of other tau kinases or phosphatases are altered by this intervention. Retromer promotes growth factor signaling and amino acid sensing for the mechanistic target of rapamycin kinase complex 1 (mTORC1) pathway [157, 158]. Both mTORC1 and its downstream effector S6K1 can phosphorylate tau [159, 160]. The generation of amino acids through lysosomal protein catabolism is critical for mTORC1 activation [161]. This may explain why VPS35-dependent reductions in tau phosphorylation are sensitive to lysosomal inhibition.
A proposed model for retromer dysfunction in tauopathies
While the mechanisms of retromer-mediated control of tauopathy are slowly emerging, most studies addressing this question are complementary. The consensus is that enhancement or reduction of retromer function suppresses or exacerbates various measures of tauopathy, respectively. From these data, we propose a model whereby tau hyperphosphorylation and aggregation are suppressed through the autophagy–lysosome axis (Fig. 2a). In tauopathies, where retromer function is known to be compromised, we posit that perturbed selection, trafficking, and degradation of newly formed tau aggregates (or their precursors) enhance the hyperphosphorylation and aggregation of tau (Fig. 2b).

a In a healthy neuron, (i) the retromer complex is functional and regulates endosomal trafficking of receptors to the trans-Golgi network and plasma membrane. In addition, (ii) the majority of tau is unphosphorylated and stabilizes microtubules along the axon. (iii) Sporadically formed tau aggregates are efficiently labeled with autophagy cargo receptors (e.g., SQSTM1) and targeted to the phagophore that fuses with lysosomes to facilitate their degradation and clearance, limiting further seeding and aggregation. b In a pathological neuron, (i) there is a reduction of retromer complex components, which results in the degradation of retromer cargo receptors. ILV intraluminal vesicle. Meanwhile, (ii) retromer dysfunction enhances tau phosphorylation, reducing its affinity for microtubules resulting in microtubule destabilization, and increases its local cytoplasmic concentration. (iii) Sporadically formed tau aggregates can seed the aggregation of tau, which is enhanced by defective selection of aggregates by autophagy cargo receptors (e.g., SQSTM1) and perturbs their targeting to the phagophore. Moreover, retromer deficiency results in impaired autophagosome–lysosome fusion and destruction. This increases the likelihood of lysosomal injury to release tau aggregates and enable subsequent seeding. Eventually, tau aggregates coalesce into large inclusions in the soma (e.g., neurofibrillary tangles). In addition, (iv) smaller misfolded/aggregate species of tau can be passed on to anatomically connected neurons, where they can propagate tau seeding and aggregation.
Moving forward with retromer
The retromer complex is a promising therapeutic target for ameliorating numerous aspects of pathology associated with neurological diseases such as amyloid β-deposition, microglial dysfunction, α-synuclein aggregation, and more recently tau hyperphosphorylation and aggregation [101, 104,105,106, 111, 154, 162, 163]. Future efforts should be directed toward a deeper mechanistic insight of how retromer dysfunction contributes to tau phosphorylation, altered autophagy/lysosomal function, and clearance of tau aggregates. This will improve understanding of the etiology of tauopathies. Although retromer chaperones are a promising therapeutic avenue, only a few studies have used these therapies in vivo [101, 154]. A recent study detailed the synthesis of a novel retromer chaperone called 2a, which was derived from R55 and has enhanced affinity for the VPS35–VPS29 interface over its precursor [99]. In the G1H transgenic mouse model of amyotrophic lateral sclerosis (human SOD1 with G93A mutation), 2a was found to stabilize the retromer complex and increased motor neuron survival and locomotor function [99]. With that said, several important questions about the safety/tolerance and efficacy of retromer chaperones remain unanswered. Given that retromer chaperones modify numerous disease-related phenotypes, it will be important to understand if single or combined functions of retromer chaperones account for its benefits. In addition, side-by-side comparisons between current retromer chaperones must be carried out to establish superiority in vivo. Moreover, it is imperative that future studies assess lifespan/survival with retromer chaperones in preclinical models. This is important as VPS35 has an apparent oncogenic function in hepatocellular carcinoma [157]. Further, understanding the contribution of retromer in cell and tissue metabolism is still in its infancy, and may have translational implications given that a high proportion of people with dementia have metabolic comorbidities including type 2 diabetes [164]. Significantly, VPS26A has been identified as a susceptibility locus for type 2 diabetes in people of South Asian ancestry [165]. Toward clinical translation, it remains unclear whether retromer chaperones can demonstrate benefits after the onset of disease-related features in preclinical models, otherwise their utility may be limited to prevention, akin to drugs such as rapamycin [166, 167]. With that said, cognitive/behavioral benefits must be the primary endpoint of future studies, rather than distinct biochemical or histological readouts to avoid yet another failed clinical trial.
Moving forward, critical differences between preclinical models of age-related dementias must be considered and carefully understood. For example, the 3 × Tg transgenic mouse line that showed positive outcomes for retromer chaperone therapy presents with cognitive impairments before the appearance of plaques and tangles [101]. This directly conflicts with clinical observations in people with AD, where such hallmarks emerge years or even decades before the clinical onset of dementia [168,169,170]. With that said, it has been reported that soluble oligomers of amyloid-β are more neurotoxic than plaques, and mild cognitive impairments appear with the onset of amyloid plaques [171, 172]. The other transgenic mouse line used to interrogate the utility of a retromer chaperone therapy was the PS19 model, which shows tangle pathology around the same time that cognitive decline is first apparent [104, 173]. However, seeding activity from PS19 mouse brains occurs prior to the emergence of tangles and cognitive decline in this model [94, 173]. This indicates that smaller, likely oligomeric, species of tau that are not detected by conventional histological approaches may be primary neurodegenerative effectors [94]. Indeed, tau oligomers are observed in the early stages of AD, and small tau aggregates (<200 nm2) are overrepresented in VPS35-deficient hESC-derived cortical neurons [106, 174]. Also, it is important to consider that preclinical models of tauopathies (i.e., animal and cell models) mimic features of such diseases, but are not complete models of the disease itself. The factors that make each tauopathy distinct are not accounted for in these systems. For example, most mouse models of “Alzheimer’s disease” employ mutants of tau that cause FTDP-17 [173]. Moreover, most transgenic mice overexpress a single isoform of tau, which limits their relevance to AD (all isoforms present in aggregates) or Pick’s disease (3R-containing isoforms present in aggregates), for example. These factors must be considered when attributing disease relevance to experimental findings, even in the case of retromer. To better understand the utility of retromer chaperones, the fact that tau aggregates can exist as distinct strains must also be considered. Modeling this heterogeneity in vivo, likely through injection of structurally distinct tau “seeds” into the brains of mice, will be an important step forward.
References
Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–59.
Irwin DJ. Tauopathies as clinicopathological entities. Parkinsonism Relat Disord. 2016;22 Suppl 1:S29–33.
Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, et al. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–5.
Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA. 1998;95:7737–41.
Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, et al. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43:815–25.
Poorkaj P, Grossman M, Steinbart E, Payami H, Sadovnick A, Nochlin D, et al. Frequency of tau gene mutations in familial and sporadic cases of non-Alzheimer dementia. Arch Neurol. 2001;58:383–7.
Hong M, Zhukareva V, Vogelsberg-Ragaglia V, Wszolek Z, Reed L, Miller BI, et al. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science. 1998;282:1914–7.
Kouri N, Carlomagno Y, Baker M, Liesinger AM, Caselli RJ, Wszolek ZK, et al. Novel mutation in MAPT exon 13 (p.N410H) causes corticobasal degeneration. Acta Neuropathol. 2014;127:271–82.
Perez M, Lim F, Arrasate M, Avila J. The FTDP-17-linked mutation R406W abolishes the interaction of phosphorylated tau with microtubules. J Neurochem. 2000;74:2583–9.
Shammas SL, Garcia GA, Kumar S, Kjaergaard M, Horrocks MH, Shivji N, et al. A mechanistic model of tau amyloid aggregation based on direct observation of oligomers. Nat Commun. 2015;6:7025.
Hoglinger GU, Melhem NM, Dickson DW, Sleiman PM, Wang LS, Klei L, et al. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet. 2011;43:699–705.
Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I, et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. 2014;128:755–66.
Lee G, Cowan N, Kirschner M. The primary structure and heterogeneity of tau protein from mouse brain. Science. 1988;239:285–8.
Chen J, Kanai Y, Cowan NJ, Hirokawa N. Projection domains of MAP2 and tau determine spacings between microtubules in dendrites and axons. Nature. 1992;360:674–7.
Goedert M, Spillantini MG, Potier MC, Ulrich J, Crowther RA. Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J. 1989;8:393–9.
Goedert M, Jakes R. Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 1990;9:4225–30.
Himmler A, Drechsel D, Kirschner MW, Martin DW Jr. Tau consists of a set of proteins with repeated C-terminal microtubule-binding domains and variable N-terminal domains. Mol Cell Biol. 1989;9:1381–8.
Goedert M, Wischik CM, Crowther RA, Walker JE, Klug A. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau. Proc Natl Acad Sci USA. 1988;85:4051–5.
Boutajangout A, Boom A, Leroy K, Brion JP. Expression of tau mRNA and soluble tau isoforms in affected and non-affected brain areas in Alzheimer’s disease. FEBS Lett. 2004;576:183–9.
Drubin DG, Kirschner MW. Tau protein function in living cells. J Cell Biol. 1986;103:2739–46.
Kadavath H, Hofele RV, Biernat J, Kumar S, Tepper K, Urlaub H, et al. Tau stabilizes microtubules by binding at the interface between tubulin heterodimers. Proc Natl Acad Sci USA. 2015;112:7501–6.
Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow EM. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol. 2002;156:1051–63.
Dixit R, Ross JL, Goldman YE, Holzbaur EL. Differential regulation of dynein and kinesin motor proteins by tau. Science. 2008;319:1086–9.
Zhang B, Maiti A, Shively S, Lakhani F, McDonald-Jones G, Bruce J, et al. Microtubule-binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proc Natl Acad Sci USA. 2005;102:227–31.
Cabrales Fontela Y, Kadavath H, Biernat J, Riedel D, Mandelkow E, Zweckstetter M. Multivalent cross-linking of actin filaments and microtubules through the microtubule-associated protein tau. Nat Commun. 2017;8:1981.
Li X, Kumar Y, Zempel H, Mandelkow EM, Biernat J, Mandelkow E. Novel diffusion barrier for axonal retention of tau in neurons and its failure in neurodegeneration. EMBO J. 2011;30:4825–37.
Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem. 1986;261:6084–9.
Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA. 1986;83:4913–7.
Braak H, Braak E, Grundke-Iqbal I, Iqbal K. Occurrence of neuropil threads in the senile human brain and in Alzheimer’s disease: a third location of paired helical filaments outside of neurofibrillary tangles and neuritic plaques. Neurosci Lett. 1986;65:351–5.
Li T, Braunstein KE, Zhang J, Lau A, Sibener L, Deeble C, et al. The neuritic plaque facilitates pathological conversion of tau in an Alzheimer’s disease mouse model. Nat Commun. 2016;7:12082.
Ksiezak-Reding H, Liu WK, Yen SH. Phosphate analysis and dephosphorylation of modified tau associated with paired helical filaments. Brain Res. 1992;597:209–19.
Wang Y, Mandelkow E. Tau in physiology and pathology. Nat Rev Neurosci. 2016;17:5–21.
Hanger DP, Betts JC, Loviny TL, Blackstock WP, Anderton BH. New phosphorylation sites identified in hyperphosphorylated tau (paired helical filament-tau) from Alzheimer’s disease brain using nanoelectrospray mass spectrometry. J Neurochem. 1998;71:2465–76.
Schneider A, Biernat J, von Bergen M, Mandelkow E, Mandelkow EM. Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments. Biochemistry. 1999;38:3549–58.
Sengupta A, Kabat J, Novak M, Wu Q, Grundke-Iqbal I, Iqbal K. Phosphorylation of tau at both Thr 231 and Ser 262 is required for maximal inhibition of its binding to microtubules. Arch Biochem Biophys. 1998;357:299–309.
Bancher C, Brunner C, Lassmann H, Budka H, Jellinger K, Wiche G, et al. Accumulation of abnormally phosphorylated tau precedes the formation of neurofibrillary tangles in Alzheimer’s disease. Brain Res. 1989;477:90–9.
Alonso A, Zaidi T, Novak M, Grundke-Iqbal I, Iqbal K. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc Natl Acad Sci USA. 2001;98:6923–8.
Alonso AD, Zaidi T, Novak M, Barra HS, Grundke-Iqbal I, Iqbal K. Interaction of tau isoforms with Alzheimer’s disease abnormally hyperphosphorylated tau and in vitro phosphorylation into the disease-like protein. J Biol Chem. 2001;276:37967–73.
Ihara Y, Nukina N, Miura R, Ogawara M. Phosphorylated tau protein is integrated into paired helical filaments in Alzheimer’s disease. J Biochem. 1986;99:1807–10.
Yu Y, Run X, Liang Z, Li Y, Liu F, Liu Y, et al. Developmental regulation of tau phosphorylation, tau kinases, and tau phosphatases. J Neurochem. 2009;108:1480–94.
Ittner A, Chua SW, Bertz J, Volkerling A, van der Hoven J, Gladbach A, et al. Site-specific phosphorylation of tau inhibits amyloid-beta toxicity in Alzheimer’s mice. Science. 2016;354:904–8.
Cowan CM, Mudher A. Are tau aggregates toxic or protective in tauopathies? Front Neurol. 2013;4:114.
Trojanowski JQ, Lee VM. Pathological tau: a loss of normal function or a gain in toxicity? Nat Neurosci. 2005;8:1136–7.
Goedert M, Eisenberg DS, Crowther RA. Propagation of tau aggregates and neurodegeneration. Annu Rev Neurosci. 2017;40:189–210.
Goedert M. The ordered assembly of tau is the gain-of-toxic function that causes human tauopathies. Alzheimers Dement. 2016;12:1040–50.
DeVos SL, Corjuc BT, Commins C, Dujardin S, Bannon RN, Corjuc D, et al. Tau reduction in the presence of amyloid-β prevents tau pathology and neuronal death in vivo. Brain. 2018;141:2194–212.
DeVos SL, Goncharoff DK, Chen G, Kebodeaux CS, Yamada K, Stewart FR, et al. Antisense reduction of tau in adult mice protects against seizures. J Neurosci. 2013;33:12887–97.
DeVos SL, Miller RL, Schoch KM, Holmes BB, Kebodeaux CS, Wegener AJ, et al. Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci Transl Med. 2017;9:eaag0481. https://doi.org/10.1126/scitranslmed.aag0481.
Kepp KP. Alzheimer’s disease due to loss of function: a new synthesis of the available data. Prog Neurobiol. 2016;143:36–60.
Bolkan BJ, Kretzschmar D. Loss of tau results in defects in photoreceptor development and progressive neuronal degeneration in Drosophila. Dev Neurobiol. 2014;74:1210–25.
Holth JK, Bomben VC, Reed JG, Inoue T, Younkin L, Younkin SG, et al. Tau loss attenuates neuronal network hyperexcitability in mouse and Drosophila genetic models of epilepsy. J Neurosci. 2013;33:1651–9.
Burnouf S, Gronke S, Augustin H, Dols J, Gorsky MK, Werner J, et al. Deletion of endogenous Tau proteins is not detrimental in Drosophila. Sci Rep. 2016;6:23102.
Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–59.
Braak H, Braak E. Development of Alzheimer-related neurofibrillary changes in the neocortex inversely recapitulates cortical myelogenesis. Acta Neuropathol. 1996;92:197–201.
Brier MR, Gordon B, Friedrichsen K, McCarthy J, Stern A, Christensen J, et al. Tau and Abeta imaging, CSF measures, and cognition in Alzheimer’s disease. Sci Transl Med. 2016;8:338–66.
Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11:909–13.
Clavaguera F, Hench J, Lavenir I, Schweighauser G, Frank S, Goedert M, et al. Peripheral administration of tau aggregates triggers intracerebral tauopathy in transgenic mice. Acta Neuropathol. 2014;127:299–301.
Peeraer E, Bottelbergs A, Van Kolen K, Stancu IC, Vasconcelos B, Mahieu M, et al. Intracerebral injection of preformed synthetic tau fibrils initiates widespread tauopathy and neuronal loss in the brains of tau transgenic mice. Neurobiol Dis. 2015;73:83–95.
Kaufman SK, Sanders DW, Thomas TL, Ruchinskas AJ, Vaquer-Alicea J, Sharma AM, et al. Tau prion strains dictate patterns of cell pathology, progression rate, and regional vulnerability in vivo. Neuron. 2016;92:796–812.
Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A, et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron. 2014;82:1271–88.
Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, et al. Trans-synaptic spread of tau pathology in vivo. PLoS ONE. 2012;7:e31302.
Goedert M, Jakes R, Spillantini MG, Hasegawa M, Smith MJ, Crowther RA. Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature. 1996;383:550–3.
Dinkel PD, Holden MR, Matin N, Margittai M. RNA binds to tau fibrils and sustains template-assisted growth. Biochemistry. 2015;54:4731–40.
Gotz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science 2001;293:1491–5.
Guo JL, Lee VM. Seeding of normal tau by pathological tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem. 2011;286:15317–31.
Mirbaha H, Holmes BB, Sanders DW, Bieschke J, Diamond MI. Tau trimers are the minimal propagation unit spontaneously internalized to seed intracellular aggregation. J Biol Chem. 2015;290:14893–903.
Tian H, Davidowitz E, Lopez P, Emadi S, Moe J, Sierks M. Trimeric tau is toxic to human neuronal cells at low nanomolar concentrations. Int J Cell Biol. 2013;2013:260787.
Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci. 2015;18:1584–93.
Katsinelos T, Zeitler M, Dimou E, Karakatsani A, Muller HM, Nachman E, et al. Unconventional secretion mediates the trans-cellular spreading of tau. Cell Rep. 2018;23:2039–55.
Maphis N, Xu G, Kokiko-Cochran ON, Jiang S, Cardona A, Ransohoff RM, et al. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain. 2015;138:1738–55.
Evans LD, Wassmer T, Fraser G, Smith J, Perkinton M, Billinton A, et al. Extracellular monomeric and aggregated tau efficiently enter human neurons through overlapping but distinct pathways. Cell Rep. 2018;22:3612–24.
Rauch JN, Luna G, Guzman E, Audouard M, Challis C, Sibih YE, et al. LRP1 is a master regulator of tau uptake and spread. Nature. 2020;580:381–5.
Holmes BB, DeVos SL, Kfoury N, Li M, Jacks R, Yanamandra K, et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci USA. 2013;110:E3138–47.
Falcon B, Noad J, McMahon H, Randow F, Goedert M. Galectin-8-mediated selective autophagy protects against seeded tau aggregation. J Biol Chem. 2018;293:2438–51.
Papadopoulos C, Kirchner P, Bug M, Grum D, Koerver L, Schulze N, et al. VCP/p97 cooperates with YOD1, UBXD1 and PLAA to drive clearance of ruptured lysosomes by autophagy. EMBO J. 2017;36:135–50.
Jia J, Claude-Taupin A, Gu Y, Choi SW, Peters R, Bissa B, et al. Galectin-3 coordinates a cellular system for lysosomal repair and removal. Dev Cell. 2020;52:69–87.e8.
Elbaum-Garfinkle S, Cobb G, Compton JT, Li XH, Rhoades E. Tau mutants bind tubulin heterodimers with enhanced affinity. Proc Natl Acad Sci USA. 2014;111:6311–6.
Aguzzi A, Heikenwalder M, Polymenidou M. Insights into prion strains and neurotoxicity. Nat Rev Mol Cell Biol. 2007;8:552–61.
Bagyinszky E, Giau VV, Youn YC, An SSA, Kim S. Characterization of mutations in PRNP (prion) gene and their possible roles in neurodegenerative diseases. Neuropsychiatr Dis Treat. 2018;14:2067–85.
Wegmann S, Maury EA, Kirk MJ, Saqran L, Roe A, DeVos SL, et al. Removing endogenous tau does not prevent tau propagation yet reduces its neurotoxicity. EMBO J. 2015;34:3028–41.
Polanco JC, Gotz J. No full admission for tau to the exclusive prion club yet. EMBO J. 2015;34:2990–2.
Fitzpatrick AWP, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, et al. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature. 2017;547:185–90.
Falcon B, Zhang W, Murzin AG, Murshudov G, Garringer HJ, Vidal R, et al. Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature. 2018;561:137–40.
Zhang W, Tarutani A, Newell KL, Murzin AG, Matsubara T, Falcon B, et al. Novel tau filament fold in corticobasal degeneration. Nature. 2020;580:283–7.
Dujardin S, Commins C, Lathuiliere A, Beerepoot P, Fernandes AR, Kamath TV, et al. Tau molecular diversity contributes to clinical heterogeneity in Alzheimer’s disease. Nat Med. 2020;26:1256–63.
Gomez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, et al. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol. 1997;41:17–24.
Morsch R, Simon W, Coleman PD. Neurons may live for decades with neurofibrillary tangles. J Neuropathol Exp Neurol. 1999;58:188–97.
Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–81.
Gamache J, Benzow K, Forster C, Kemper L, Hlynialuk C, Furrow E, et al. Factors other than hTau overexpression that contribute to tauopathy-like phenotype in rTg4510 mice. Nat Commun. 2019;10:2479.
Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, et al. Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science. 2001;293:711–4.
Van der Jeugd A, Hochgrafe K, Ahmed T, Decker JM, Sydow A, Hofmann A, et al. Cognitive defects are reversible in inducible mice expressing pro-aggregant full-length human tau. Acta Neuropathol. 2012;123:787–805.
Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Clos AL, Jackson GR, Kayed R. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol Neurodegener. 2011;6:39.
Ghag G, Bhatt N, Cantu DV, Guerrero-Munoz MJ, Ellsworth A, Sengupta U, et al. Soluble tau aggregates, not large fibrils, are the toxic species that display seeding and cross-seeding behavior. Protein Sci. 2018;27:1901–9.
Holmes BB, Furman JL, Mahan TE, Yamasaki TR, Mirbaha H, Eades WC, et al. Proteopathic tau seeding predicts tauopathy in vivo. Proc Natl Acad Sci USA. 2014;111:E4376–85.
Brelstaff J, Tolkovsky AM, Ghetti B, Goedert M, Spillantini MG. Living neurons with tau filaments aberrantly expose phosphatidylserine and are phagocytosed by microglia. Cell Rep. 2018;24:1939–48.e4.
Small SA, Kent K, Pierce A, Leung C, Kang MS, Okada H, et al. Model-guided microarray implicates the retromer complex in Alzheimer’s disease. Ann Neurol. 2005;58:909–19.
Zimprich A, Benet-Pages A, Struhal W, Graf E, Eck SH, Offman MN, et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet. 2011;89:168–75.
Vilarino-Guell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ, et al. VPS35 mutations in Parkinson disease. Am J Hum Genet. 2011;89:162–7.
Muzio L, Sirtori R, Gornati D, Eleuteri S, Fossaghi A, Brancaccio D, et al. Retromer stabilization results in neuroprotection in a model of amyotrophic lateral sclerosis. Nat Commun. 2020;11:3848.
Zhang QY, Tan MS, Yu JT, Tan L. The role of retromer in Alzheimer’s disease. Mol Neurobiol. 2016;53:4201–9.
Li JG, Chiu J, Ramanjulu M, Blass BE, Pratico D. A pharmacological chaperone improves memory by reducing Abeta and tau neuropathology in a mouse model with plaques and tangles. Mol Neurodegener. 2020;15:1.
Farmer T, O’Neill KL, Naslavsky N, Luo X, Caplan S. Retromer facilitates the localization of Bcl-xL to the mitochondrial outer membrane. Mol Biol Cell. 2019;30:1138–46.
Temkin P, Morishita W, Goswami D, Arendt K, Chen L, Malenka R. The retromer supports AMPA receptor trafficking during LTP. Neuron. 2017;94:74–82.e5.
Vagnozzi AN, Li JG, Chiu J, Razmpour R, Warfield R, Ramirez SH, et al. VPS35 regulates tau phosphorylation and neuropathology in tauopathy. Mol Psychiatry. 2019. https://doi.org/10.1038/s41380-019-0453-x.
Young JE, Fong LK, Frankowski H, Petsko GA, Small SA, Goldstein LSB. Stabilizing the retromer complex in a human stem cell model of Alzheimer’s disease reduces TAU phosphorylation independently of amyloid precursor protein. Stem Cell Rep. 2018;10:1046–58.
Carosi JM, Hein LK, van den Hurk M, Adams R, Milky B, Singh S, et al. Retromer regulates the lysosomal clearance of MAPT/tau. Autophagy. 2020:1–21. https://doi.org/10.1080/15548627.2020.1821545. [Online ahead of print].
Ansell-Schultz A, Reyes JF, Samuelsson M, Hallbeck M. Reduced retromer function results in the accumulation of amyloid-beta oligomers. Mol Cell Neurosci. 2018;93:18–26.
Muhammad A, Flores I, Zhang H, Yu R, Staniszewski A, Planel E, et al. Retromer deficiency observed in Alzheimer’s disease causes hippocampal dysfunction, neurodegeneration, and Abeta accumulation. Proc Natl Acad Sci USA. 2008;105:7327–232.
Sullivan CP, Jay AG, Stack EC, Pakaluk M, Wadlinger E, Fine RE, et al. Retromer disruption promotes amyloidogenic APP processing. Neurobiol Dis. 2011;43:338–45.
Choy RW, Cheng Z, Schekman R. Amyloid precursor protein (APP) traffics from the cell surface via endosomes for amyloid β (Aβ) production in the trans-Golgi network. Proc Natl Acad Sci USA. 2012;109:E2077–82.
Follett J, Bugarcic A, Yang Z, Ariotti N, Norwood SJ, Collins BM, et al. Parkinson disease-linked Vps35 R524W mutation impairs the endosomal association of retromer and induces alpha-synuclein aggregation. J Biol Chem. 2016;291:18283–98.
Zavodszky E, Seaman MN, Moreau K, Jimenez-Sanchez M, Breusegem SY, Harbour ME, et al. Mutation in VPS35 associated with Parkinson’s disease impairs WASH complex association and inhibits autophagy. Nat Commun. 2014;5:1–16.
Miura E, Hasegawa T, Konno M, Suzuki M, Sugeno N, Fujikake N, et al. VPS35 dysfunction impairs lysosomal degradation of alpha-synuclein and exacerbates neurotoxicity in a Drosophila model of Parkinson’s disease. Neurobiol Dis. 2014;71:1–13.
Tang FL, Liu W, Hu JX, Erion JR, Ye J, Mei L, et al. VPS35 deficiency or mutation causes dopaminergic neuronal loss by impairing mitochondrial fusion and function. Cell Rep. 2015;12:1631–43.
Tang FL, Erion JR, Tian Y, Liu W, Yin DM, Ye J, et al. VPS35 in dopamine neurons is required for endosome-to-Golgi retrieval of Lamp2a, a receptor of chaperone-mediated autophagy that is critical for alpha-synuclein degradation and prevention of pathogenesis of Parkinson’s disease. J Neurosci. 2015;35:10613–28.
Bonifacino JS, Rojas R. Retrograde transport from endosomes to the trans-Golgi network. Nat Rev Mol Cell Biol. 2006;7:568–79.
Yang Z, Follett J, Kerr MC, Clairfeuille T, Chandra M, Collins BM, et al. Sorting nexin 27 (SNX27) regulates the trafficking and activity of the glutamine transporter ASCT2. J Biol Chem. 2018;293:6802–11.
Lucas M, Gershlick DC, Vidaurrazaga A, Rojas AL, Bonifacino JS, Hierro A. Structural mechanism for cargo recognition by the retromer complex. Cell. 2016;167:1623–35.
Yong X, Zhao L, Deng W, Sun H, Zhou X, Mao L, et al. Mechanism of cargo recognition by retromer-linked SNX-BAR proteins. PLoS Biol. 2020;18:e3000631.
Purushothaman LK, Ungermann C. Cargo induces retromer-mediated membrane remodeling on membranes. Mol Biol Cell. 2018;29:2709–19.
Carlton J, Bujny M, Peter BJ, Oorschot VM, Rutherford A, Mellor H, et al. Sorting nexin-1 mediates tubular endosome-to-TGN transport through coincidence sensing of high-curvature membranes and 3-phosphoinositides. Curr Biol. 2004;14:1791–800.
Rojas R, Kametaka S, Haft CR, Bonifacino JS. Interchangeable but essential functions of SNX1 and SNX2 in the association of retromer with endosomes and the trafficking of mannose 6-phosphate receptors. Mol Cell Biol. 2007;27:1112–24.
Wassmer T, Attar N, Bujny MV, Oakley J, Traer CJ, Cullen PJ. A loss-of-function screen reveals SNX5 and SNX6 as potential components of the mammalian retromer. J Cell Sci. 2007;120:45–54.
Harterink M, Port F, Lorenowicz MJ, McGough IJ, Silhankova M, Betist MC, et al. A SNX3-dependent retromer pathway mediates retrograde transport of the Wnt sorting receptor Wntless and is required for Wnt secretion. Nat Cell Biol. 2011;13:914–23.
Steinberg F, Gallon M, Winfield M, Thomas EC, Bell AJ, Heesom KJ, et al. A global analysis of SNX27-retromer assembly and cargo specificity reveals a function in glucose and metal ion transport. Nat Cell Biol. 2013;15:461–71.
Wassmer T, Attar N, Harterink M, van Weering JR, Traer CJ, Oakley J, et al. The retromer coat complex coordinates endosomal sorting and dynein-mediated transport, with carrier recognition by the trans-Golgi network. Dev Cell. 2009;17:110–22.
Gomez TS, Billadeau DD. A FAM21-containing WASH complex regulates retromer-dependent sorting. Dev Cell. 2009;17:699–711.
Freeman CL, Hesketh G, Seaman MN. RME-8 coordinates the activity of the WASH complex with the function of the retromer SNX dimer to control endosomal tubulation. J Cell Sci. 2014;127:2053–70.
Norris A, Tammineni P, Wang S, Gerdes J, Murr A, Kwan KY, et al. SNX-1 and RME-8 oppose the assembly of HGRS-1/ESCRT-0 degradative microdomains on endosomes. Proc Natl Acad Sci USA. 2017;114:E307–16.
Cullen PJ, Korswagen HC. Sorting nexins provide diversity for retromer-dependent trafficking events. Nat Cell Biol. 2012;14:29–37.
Gokool S, Tattersall D, Seaman MN. EHD1 interacts with retromer to stabilize SNX1 tubules and facilitate endosome-to-Golgi retrieval. Traffic. 2007;8:1873–86.
Cui Y, Carosi JM, Yang Z, Ariotti N, Kerr MC, Parton RG, et al. Retromer has a selective function in cargo sorting via endosome transport carriers. J Cell Biol. 2019;218:615–31.
Seaman MNJ, Mukadam AS, Breusegem SY. Inhibition of TBC1D5 activates Rab7a and can enhance the function of the retromer cargo-selective complex. J Cell Sci. 2018;131:jcs217398. https://doi.org/10.1242/jcs.217398.
Vieira SI, Rebelo S, Esselmann H, Wiltfang J, Lah J, Lane R, et al. Retrieval of the Alzheimer’s amyloid precursor protein from the endosome to the TGN is S655 phosphorylation state-dependent and retromer-mediated. Mol Neurodegener. 2010;5:1–21.
Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, et al. Mutation of the beta-amyloid precursor protein in familial Alzheimer’s disease increases beta-protein production. Nature. 1992;360:672–4.
Alzheimer’s Disease Collaborative Group. The structure of the presenilin 1 (S182) gene and identification of six novel mutations in early onset AD families. Nat Genet. 1995;11:219–22.
Vardarajan BN, Bruesegem SY, Harbour ME, Inzelberg R, Friedland R, St George-Hyslop P, et al. Identification of Alzheimer disease-associated variants in genes that regulate retromer function. Neurobiol Aging. 2012;33:2231.e15– e30.
Riemenschneider M, Schoepfer-Wendels A, Friedrich P, Konta L, Laws SM, Mueller JC, et al. No association of vacuolar protein sorting 26 polymorphisms with Alzheimer’s disease. Neurobiol Aging. 2007;28:883–4.
Bertram L, Tanzi RE. Genome-wide association studies in Alzheimer’s disease. Hum Mol Genet. 2009;18:R137–45.
Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, et al. Author correction: genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat Genet. 2019;51:1423–4.
Escott-Price V, Bellenguez C, Wang LS, Choi SH, Harold D, Jones L, et al. Gene-wide analysis detects two new susceptibility genes for Alzheimer’s disease. PLoS ONE. 2014;9:e94661.
Rogaeva E, Meng Y, Lee JH, Gu Y, Kawarai T, Zou F, et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet. 2007;39:168–77.
Fjorback AW, Seaman M, Gustafsen C, Mehmedbasic A, Gokool S, Wu C, et al. Retromer binds the FANSHY sorting motif in SorLA to regulate amyloid precursor protein sorting and processing. J Neurosci. 2012;32:1467–80.
Gao S, Casey AE, Sargeant TJ, Makinen VP. Genetic variation within endolysosomal system is associated with late-onset Alzheimer’s disease. Brain. 2018;141:2711–20.
Rovelet-Lecrux A, Charbonnier C, Wallon D, Nicolas G, Seaman MN, Pottier C, et al. De novo deleterious genetic variations target a biological network centered on Abeta peptide in early-onset Alzheimer disease. Mol Psychiatry. 2015;20:1046–56.
Wang X, Zhao Y, Zhang X, Badie H, Zhou Y, Mu Y, et al. Loss of sorting nexin 27 contributes to excitatory synaptic dysfunction by modulating glutamate receptor recycling in Down’s syndrome. Nat Med. 2013;19:473–80.
Cherry S, Jin EJ, Ozel MN, Lu Z, Agi E, Wang D, et al. Charcot-Marie-Tooth 2B mutations in rab7 cause dosage-dependent neurodegeneration due to partial loss of function. Elife. 2013;2:e01064.
Valdmanis PN, Meijer IA, Reynolds A, Lei A, MacLeod P, Schlesinger D, et al. Mutations in the KIAA0196 gene at the SPG8 locus cause hereditary spastic paraplegia. Am J Hum Genet. 2007;80:152–61.
Chu J, Pratico D. The retromer complex system in a transgenic mouse model of AD: influence of age. Neurobiol Aging. 2017;52:32–8.
Tammineni P, Jeong YY, Feng T, Aikal D, Cai Q. Impaired axonal retrograde trafficking of the retromer complex augments lysosomal deficits in Alzheimer’s disease neurons. Hum Mol Genet. 2017;26:4352–66.
Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256:184–5.
Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–51.
Chen X, Kordich JK, Williams ET, Levine N, Cole-Strauss A, Marshall L, et al. Parkinson’s disease-linked D620N VPS35 knockin mice manifest tau neuropathology and dopaminergic neurodegeneration. Proc Natl Acad Sci USA. 2019;116:5765–74.
Li JG, Chiu J, Pratico D. Full recovery of the Alzheimer’s disease phenotype by gain of function of vacuolar protein sorting 35. Mol Psychiatry. 2020;25:2630–40.
Roberson ED, Scearce-Levie S, Palop JJ, Yan F, Cheng IH, Wu T, et al. Reducing endogenous tau ameliorates amyloid ß-induced deficits in an Alzheimer’s disease mouse model. Science. 2007;316:750–4.
Mecozzi VJ, Berman DE, Simoes S, Vetanovetz C, Awal MR, Patel VM, et al. Pharmacological chaperones stabilize retromer to limit APP processing. Nat Chem Biol. 2014;10:443–9.
Zhang G, Tang X, Liang L, Zhang W, Li D, Li X, et al. DNA and RNA sequencing identified a novel oncogene VPS35 in liver hepatocellular carcinoma. Oncogene. 2020;39:3229–44.
Kvainickas A, Nagele H, Qi W, Dokladal L, Jimenez-Orgaz A, Stehl L, et al. Retromer and TBC1D5 maintain late endosomal RAB7 domains to enable amino acid-induced mTORC1 signaling. J Cell Biol. 2019;218:3019–38.
Tang Z, Bereczki E, Zhang H, Wang S, Li C, Ji X, et al. Mammalian target of rapamycin (mTor) mediates tau protein dyshomeostasis: implication for Alzheimer disease. J Biol Chem. 2013;288:15556–70.
Pei JJ, An WL, Zhou XW, Nishimura T, Norberg J, Benedikz E, et al. P70 S6 kinase mediates tau phosphorylation and synthesis. FEBS Lett. 2006;580:107–14.
Wyant GA, Abu-Remaileh M, Wolfson RL, Chen WW, Freinkman E, Danai LV, et al. mTORC1 activator SLC38A9 is required to efflux essential amino acids from lysosomes and use protein as a nutrient. Cell. 2017;171:642–54.e12.
Lucin KM, O’Brien CE, Bieri G, Czirr E, Mosher KI, Abbey RJ, et al. Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer’s disease. Neuron. 2013;79:873–86.
Yin J, Liu X, He Q, Zhou L, Yuan Z, Zhao S. Vps35-dependent recycling of Trem2 regulates microglial function. Traffic. 2016;17:1286–96.
Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM. Diabetes mellitus and the risk of dementia: the Rotterdam Study. Neurology. 1999;53:1937–42.
Kooner JS, Saleheen D, Sim X, Sehmi J, Zhang W, Frossard P, et al. Genome-wide association study in individuals of South Asian ancestry identifies six new type 2 diabetes susceptibility loci. Nat Genet. 2011;43:984–9.
Carosi JM, Sargeant TJ. Rapamycin and Alzheimer disease: a double-edged sword? Autophagy. 2019;15:1460–2.
Majumder S, Richardson A, Strong R, Oddo S. Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS ONE. 2011;6:e25416.
Farrell ME, Kennedy KM, Rodrigue KM, Wig G, Bischof GN, Rieck JR, et al. Association of longitudinal cognitive decline with amyloid burden in Middle-aged and Older Adults: Evidence for a Dose-Response Relationship. JAMA Neurol. 2017;74:830–8.
Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM. Intraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron 2005;45:675–88.
Jacobsen JS, Wu CC, Redwine JM, Comery TA, Arias R, Bowlby M, et al. Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. 2006;103:5161–6.
Gandy S, Simon AJ, Steele JW, Lublin AL, Lah JJ, Walker LC, et al. Days to criterion as an indicator of toxicity associated with human Alzheimer amyloid-beta oligomers. Ann Neurol. 2010;68:220–30.
Amariglio RE, Becker JA, Carmasin J, Wadsworth LP, Lorius N, Sullivan C, et al. Subjective cognitive complaints and amyloid burden in cognitively normal older individuals. Neuropsychologia. 2012;50:2880–6.
Takeuchi H, Iba M, Inoue H, Higuchi M, Takao K, Tsukita K, et al. P301S mutant human tau transgenic mice manifest early symptoms of human tauopathies with dementia and altered sensorimotor gating. PLoS ONE. 2011;6:e21050.
Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Sarmiento J, Troncoso J, Jackson GR, et al. Identification of oligomers at early stages of tau aggregation in Alzheimer’s disease. FASEB J. 2012;26:1946–59.
Acknowledgements
We thank all lab members for insightful discussions and advice. We thank Sofia Hassiotis for the image of tau staining in Fig. 1. Illustrations were generated under license with Biorender.com.
Funding
This investigation was supported by Lysosomal Health in Ageing at the South Australian Health & Medical Research Institute. JMC was supported by a Research Training Stipend and a Commonwealth Scholarship from the Australian Government, and Research Degree Excellence Grant from the University of South Australia, DD was supported by a National Health & Medical Research Council project grant (1124490), and SK is a National Health & Medical Research Council Senior Principal Research Fellow (1103006).
Author information
Authors and Affiliations
Contributions
JMC conceived, wrote, drafted, and approved the final paper, and created figures. DD, SK, and TJS discussed content, critiqued the draft paper and figures for intellectual content, and approved the final paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
Work with human brain was approved by the WCHN Human Research Ethics Committee (HREC/15/WCHN/198, SSA30/2016).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by G. Melino
Rights and permissions
About this article

Cite this article
Carosi, J.M., Denton, D., Kumar, S. et al. Retromer dysfunction at the nexus of tauopathies. Cell Death Differ 28, 884–899 (2021). https://doi.org/10.1038/s41418-020-00727-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-020-00727-2
This article is cited by
-
A comprehensive analysis of copy number variation in a Turkish dementia cohort
Human Genomics (2021)
