
Abstract
The advent of organoid technology has enabled scientists and clinicians to utilize cells from primary tissues or pluripotent stem cells (PSCs) to grow self-organizing tissue systems, thus attaining cellular diversity, spatial organization, and functionality as found within digestive tracts. The development of human gastrointestinal (GI) and hepato-biliary-pancreatic organoids as an in-a-dish model present novel opportunities to study humanistic mechanisms of organogenesis, regeneration and pathogenesis. Herein, we review the recent portfolios of primary tissue-derived and PSC-derived organoids in the digestive systems. We also discuss the promise and challenges in disease modeling and drug development applications for digestive disorders.
Similar content being viewed by others


Facts
-
Digestive organoids can be generated from two types of stem cells: adult stem cells (AdSCs) or pluripotent stem cells (PSCs) which include embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs).
-
While AdSC-derived digestive organoids contain mainly epithelial component, PSC-derived digestive organoids harbor epithelial and non-epithelial components.
-
Patient-specific and gene-edited organoids have been utilized for in vitro digestive disease modeling and mechanistic studies.
-
Organoids have been utilized for disease phenotyping, drug screening, drug development, and disease modeling.
Open questions
-
What aspects of digestive diseases can and cannot be modeled in organoids?
-
What are critical quality control attributes for consistent and reproducible organoid generation?
-
Would phenotyping of organoid disease models lend itself to expedite the drug discovery process?
-
What are the key considerations involved in scaling up the organoid manufacturing process for downstream applications?
Introduction
Human organoids have received substantial attention as in vitro culture systems, where stem cells self-organize into three-dimensional structures resembling human organs [1]. Patient’s specific organoids offer a highly expandable and tractable resource that can also be genetically and pharmacologically manipulated. In the human body, access to the digestive tract is generally limited, and it is not trivial to test rapidly evolving knowledge associated with diagnosis and therapeutic intervention. Thus, digestive organoids can be used as an alternate model to study patients’ pathobiology and individualized responses to therapy, offering an innovative approach to drug development, toxicology, and precision medicine.
The disorders associated with digestive systems (also encompassing diseases of the liver and pancreas) affect millions of people of all ages and have enormous social and economic costs. Digestive organoids generated from diseased patients have already been proven to phenocopy aspects of molecular and cellular pathogenesis, and thus are an invaluable platform for subsequent mechanistic investigation and interrogation. Herein, we review digestive organoid technology, their potential therapeutic applications for diseases, and discuss challenges in clinical applications.
PSC-derived digestive organoids
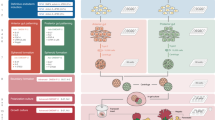
The definitive endoderm (DE) was first defined as the innermost tissue or germ layer found in metazoan embryos. The DE gives rise to a vast array of highly specialized epithelial cell types that line the respiratory and digestive systems and is fundamental for organs such as thyroid, thymus, lungs, liver, biliary system, and pancreas [2]. After gastrulation, a series of morphogenetic movements transforms the DE into a primitive gut tube that is surrounded by mesoderm. Endoderm patterning is controlled by a series of adjacent interactions with nearby mesoderm tissues. As development continues, broad gene expression patterns within the foregut, midgut, and hindgut become progressively refined into precise domains for the formation of specific organs. For example, WNT, BMP, and/or FGF are known to be signaling molecules that regulate the development of the foregut, midgut, and hindgut [3]. The foregut further forms the esophagus, trachea, stomach, lungs, thyroid, liver, bile duct, and pancreas, whereas the midgut forms the small intestine, and the hindgut forms the large intestine (Fig. 1a). Organ buds develop as outgrowths of endoderm epithelium intermingle with the surrounding mesenchyme. Although the precise role of region specific mesenchyme has been elusive, recent single cell level analyses indicates the presence of highly diverse and specialized mesenchyme during early endoderm organogenesis [4]. Together, the endoderm epithelium and mesenchyme are coordinated to establish early organ primordia during fetal development [2].

a Overview of digestive organ formation. b Development of PSC-derived digestive organoids. a Endoderm cell lineages projected on to a schematic of the digestive organs. As development proceeds, broad gene expression patterns within the foregut, midgut, and hindgut become progressively refined into precise domains in which specific organs will form. The foregut gives rise to the esophagus, stomach, liver, and pancreas; whereas the midgut forms the small intestine and the hindgut forms the large intestine. b PSCs [embryonic stem cells (ESCs) or induced pluripotent stem cells (iPSCs)] can be derived into the endoderm in vitro with specific stepwise differentiation protocols. After the endoderm specification, progenitor cells are transferred into 3D systems and generate digestive organoids with developmental signals.
In mimicking the early patterning and morphogenesis, digestive organoids have been generated from PSCs including ESCs and iPSCs (Fig. 1b and Table 1) [5,6,7,8,9,10,11,12,13,14,15]. PSC-derived digestive organoids generally remain immature with a transcriptomic profile resembling fetal-like state [16]. Future efforts to direct continued organoid growth and tissue maturation will be critical before being translated into human studies [17, 18].
AdSC-derived digestive organoids
Generation of organoids derived from adult stem/progenitor cells relies on regenerative biology principles. Decades of studies identified the presence of adult stem/progenitor cells that possess self-renewal capacity and differentiation potential. In the regenerative conditions, recent advances in understanding the stem cell niche, and molecular and cellular factors have allowed for long-term in vitro cultures of adult stem/progenitor cells isolated from primary human tissues (Table 1) [19,20,21,22,23,24,25,26]. For example, intestinal organoids were among the first to be derived from primary gut tissues by introducing key niche signaling factors such as Wnt/R-spondin that control crypt proliferation in vivo [26]. Relative to PSC-based approach, conserved epigenetic signatures are one unique features in AdSC organoids that can be useful to model tissue homeostasis and its disruption during disease progression [27,28,29]. Generally, AdSC-derived organoids retain more mature signatures than similar to the in vivo tissue of origin than do PSC-dereived organoids. In the following section, we review both PSC- and AdSC-derived digestive organoid-based disease modeling for drug discovery and development.
Digestive organoid-based disease modeling for drug discovery and development
The use of disease-specific digestive organoids along with healthy organoids will facilitate our understanding of molecular mechanisms behind the disease, the identification of potential biomarkers, and the development of patient-specific platforms for drug testing or toxicology studies [30]. In the following subsections, we will discuss four major areas of diseases related to disorders in the digestive tracts: genetic diseases, infectious diseases, inflammatory diseases, and malignant diseases.
Genetic diseases
Cystic fibrosis (CF) is the most common monogenetic recessive disease in the Caucasian population and is caused by mutations in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene. The CFTR protein regulates trans-epithelial secretion of bicarbonate and chloride, and loss of function mutations in the gene result in aberrant fluid transport and abnormal mucus formation that affects the functionality of multiple organs, including the pancreas, lungs, and the intestines [31]. Over 2000 CFTR mutations have been identified, but the most dominant mutation (accounting for 70% of the worldwide CF population) is a deletion of phenylalanine at position 508 (CFTR F508del). In addition, heterogeneity in the response to modulators of CTFR carrying homozygous F508del-CFTR has been reported, therefore, optimal prediction of response to therapy on an individual level is desirable [32, 33].
Berkers et al. derived rectal organoids (ROs) from AdSCs in rectal biopsies that maintained patient-specific CFTR modulator responses in cultures for over 6 months. Furthermore, CF patient-derived ROs are stored as a living biobank for drug efficacy measurements based on forskolin-induced swelling (FIS) assays [34, 35]. The authors have also reported that in vitro drug efficacy measurements using FIS assays in ROs of individuals with CF correlated the best with the clinical efficacy of CFTR modulators (change in ppFEV1 and SCC). The data further suggest that thresholds can be established to prospectively identify clinical responders with acceptable positive and negative predictive values. In vitro assay using organoid cultures can prospectively identify efficacious treatments for patients and that biobanked stem cells and organoids will potentially be used to tailor individual treatments (Fig. 2).

Establishment of the individual patient tissue-derived digestive organoid biobank and in vitro drug testing using clinically relevant assay (e.g., FIS assay using rectal organoids for Cystic fibrosis) for personalized medicine approaches.
The recent advances in genome editing, along with the development of precise and efficient CRISPR-Cas9 nucleases, have accelerated the gene correction efforts for genetic diseases, including CF. To restore CFTR function in CF patient-derived intestinal organoids generated from AdSCs, Schwank et al repaired the defective CFTR gene by using CRISPR-Cas9 and homology-directed repair [36]. CFTR-corrected CF organoids regained the swelling response to forskolin treatment with levels comparable to those of wild-type intestinal organoids. Cas9 RNP and Adeno-associated viruses have recently been used to efficiently gene edit human airway basal stem cells with a promising result of >30% allelic correction opening new treatment possibilities for CF [37]. In another scenario, Cas9-assisted adenine base editors repaired nonsense mutations in CFTR efficiently without causing detectable off-target effects during repair [38]. Therefore, CRISPR-mediated gene correction of organoids enables an accurate evaluation of causative mutations from identical genetic (isogenic) backgrounds free from other heritable and non-heritable factors.
Recently, hepato-biliary organoids have been used to model several monogenic liver diseases including polycystic liver disease [39], Alagille syndrome (ALGS) [40], Wilson’s disease [41] and α1-antitrypsin (A1AT) deficiency [40] to name a few. Guan et al. used iPSC-derived hepatic organoids (HOs) composed of hepatocytes and cholangiocyte-like cells organized as a layer of epithelia surrounding the lumen of bile duct-like structures to study the effect of mutations in Jagged1 which causes ALGS [42]. The ALGS patient-derived organoids fail to develop tubular structures and mainly consist of vesicles lined by hepatocytes, recapitulating the disease phenotype. Moreover, mRNA expression of NOTCH2 and its target genes HEY1 and HES1, which are involved in bile duct formation, are reduced, thus validating the mechanisms known from animal and humans.
Huch et al. used patient-derived HOs originating from adult bile duct stem cells in liver biopsies, to study the effect of SERPINA1 gene mutations responsible for A1AT deficiency [40]. In A1AT deficiency, the molecular pathogenesis of the liver disease relates to the aggregation of A1AT protein within the endoplasmic reticulum (ER) of hepatocytes. A1AT protein aggregates were readily observed within the cells of the patient-derived HOs similar to the findings from the original biopsy. The HOs from A1AT deficient patients mimicked the in vivo context and showed signs of ER stress, such as phosphorylation of eIF2a and increased apoptosis in the differentiated state. In a recent study, Gómez et al reported that oncostatin M (OSM), a well-known inducer of SERPINA1, significantly increased A1AT expression in patient-derived HOs generated from AdSCs [43]. These HOs recapitulate the disease phenotype in vitro and can be used not only as precision diagnostics but also for the identification of therapeutic targets for these rare diseases (Fig. 3).

Patient-derived digestive organoids can be used in phenotypic screening and subsequent in vitro preclinical studies (e.g., toxicology testing) for drug discovery and development. 3D phenotypic assay technologies, including organs/bodies-on-a-chip using microfluidic devices, will play a prominent role in hit validation and profiling. As the organoid-based strategies mature, preclinical animal experiments can be phased out.
Hirschsprung’s disease (HSCR) is a complex genetic illness, defined by the absence of enteric neurons at variable lengths of the bowel. When left untreated, it can lead to tonic colonic constriction and pseudo-obstruction leading to megacolon and potentially life-threatening intestinal perforation. Currently, the only treatment is surgical resection of the aganglionic segment of the bowel, with the aim of improving bowel function by anastomosing the ganglionic portion of the bowel with the normally innervated anal canal. However, while surgical resection is lifesaving, it is not curative [44]. Therefore, there is an unmet need for novel regenerative medicine strategies and drug discovery to treat HSCR and other enteric neuropathies.
Decades of developmental biology studies have revealed the vagal neural crests cells (VNCCs) as the main source of the developing enteric nervous system (ENS). Defects in the migration and differentiation of VNCCs into enteric neurons through the RET signaling pathway have been recognized as key in the pathogenesis of HSCR. PSC-derived human intestinal organoids (HIOs) that incorporate ENS-like tissue have been developed to further study human intestinal motility, HSCR, and other associated enteric neuropathies. VNCCs derived from iPSCs that have been treated with retinoic acid were subsequently incorporated into HIOs [45]. Immunofluorescence imaging of HIOs aggregated with VNCCs revealed that enteric neurons and glial cells populated the smooth muscle fibers in the HIOs, similar to the in vivo development of ENS ganglia. VNCC with PHOX2B mutations in HIOs displayed impaired differentiation and inhibited smooth muscle differentiation, which resembles the aganglionosis phenotype in patient’s bowel. Future studies will allow more detailed molecular mechanistic investigation of enteric neuropathic defects as well as the establishment of drug screening platform
Infectious diseases
Helicobacter pylori causes most cases of peptic ulcer disease by inducing hypersecretion of acid in the stomach, and the colonization of this bacteria has been associated with gastric cancer. Despite a pressing need to develop new experimental models of H. pylori-associated gastric pathology, there has been no gold standard experimental model of human gastric mucosa in which H. pylori can establish their replicative niche. Although animal models provide valuable insights, species differences in development and architecture of the adult stomach highlight the importance of studying pathogenesis of the human stomach [46]. Recently, a H. pylori infection model using PSC-derived human gastric organoids (GOs) that reproduces the in vivo pathological conditions has been reported [47]. These GOs formed primitive gastric gland- and pit-like domains, proliferative zones containing LGR5-expressing cells, surface and antral mucous cells, and a diversity of gastric endocrine cells. Gastric cancer cell 2D models that were classically used for H. pylori research, due to lack of diversity and polarity of these epithelial cells, cannot accurately recapitulate the combination of an adherent mucus layer and a polarized epithelial cell layer for H. pylori infection, thus, GOs reflect a more relevant in vivo-like physiology by displaying more key aspects of the human stomach. Many observable hallmarks of an in vivo H. pylori infection such as attachment, CagA-c-MET interaction, and host cell proliferation have been recapitulated in the GOs. Complementing PSC-derived GO model, GOs derived from AdSCs in gastric tissues have been utilized to study the epithelial response to H. Pylori infection. Microinjection of H. Pylori into the GOs induced inflammatory response mediated by NF-κB pathway and chemokine IL-8 [48]. Upon microinjection of H. Pylori, phenotypes on physiological epithelial cells could be recapitulated in both PSC- and AdSC-derived GOs. These results indicate that GOs are a new and valuable model of H. pylori-associated gastric pathology. However, AdSC-derived GOs are composed of a simple epithelium without mesenchymal component, which is known to establish a specialized niche in the stomach [49]. To address this, transwell-based co-culture system with the immortalized stomach mesenchymal cells helps in achieving the maintenance of mature cell lineages within GOs and the functional activity of the gastric epithelium [50]. Furthermore, to study H. Pylori-induced protection (metaplasia) of gastric epithelium from immune response via programmed death ligand 1 (PD-L1), cytotoxic T lymphocytes and dendritic cells were cocultured with GOs [51]. Continued efforts are necessary to further promote the maturation of GOs and the integration of disease-relevant cells will help establish patient-specific, personalized medicine approaches for peptic ulcer disease.
Therapies against hepatitis B virus (HBV) have improved in recent decades; however, due to the lack of infection models that capture the personalized genetic background of each patient, the development of individualized treatments has been limited. Therefore, researchers generated iPSC-derived liver organoids (iPSC-LOs) and evaluated the organoids’ ability to model HBV infection and virus–host interactions [52]. To establish functional iPSC-LOs, the authors used iPSC-derived endodermal, mesenchymal, and endothelial cells with a chemically defined medium in a three-dimensional microwell culture system. These cells could self-organize and differentiate into functional organoids, which exhibited stronger hepatic functions than iPSC-derived hepatocyte-like cells (iPSC-HLCs) [52]. Furthermore, the functional iPSC-LOs demonstrated more susceptibility to HBV infection than iPSC-HLCs, could maintain HBV propagation, and could produce infectious virus for 20 days post infection. The authors also found that HBV infection could cause hepatic dysfunction in iPSC-LOs, accompanied with downregulation of hepatic gene expression, induced release of early acute liver failure markers (ALT and LDH), and altered hepatic ultrastructure. Therefore, HBV infection in iPSC-LOs could recapitulate the virus life cycle and virus-induced hepatic dysfunction, suggesting that iPSC-LOs may provide a promising personalized infection model for the development of individualized antiviral treatments for hepatitis.
Inflammatory diseases
Steatohepatitis is a common liver disease that lacks an approved drug therapy and is marked by increased liver fat leading to inflammation and fibrosis. Steatohepatitis-related research has been mostly performed in various animal models, and has been unsuccessful in predicting clinical efficacy, highlighting a critical need for a predictive human model to study disease mechanisms and to discover potential new anti-inflammatory and anti-fibrotic therapies [53, 54]. Recently, Ouchi et al. developed human liver organoids (HLO) composed of iPSC-derived multiple key hepatic cell types (hepatocyte-, stellate-, and Kupffer-like cells) (Fig. 4 and Table 2) [55]. Under oleic acid treatment, the authors succeeded in recapitulating key features of steatohepatitis, including lipid accumulation, inflammation, and fibrotic phenotypes. Using atomic force microscopy, the authors demonstrated that organoid stiffening correlated with fibrosis severity as is seen in clinic elastography of patients with fibrosis.

Foregut-derived spheroids from PSCs do not merely generate epithelial cell types but also co-differentiate mesenchymal cell components, which may possess a capability to become supportive lineages such as hepatic stellate cells and Kupffer cells, thus can be used for inflammatory disease modeling, while AdSC-derived digestive organoids include no mesenchymal cell component, thus co-culture system with mesenchymal cells is required.
Wolman disease is caused by defective lysosomal acid lipase activity, and hepatocytes from patients have massive lipid accumulation accompanied by lethal steatohepatitis and fibrosis. Consistent with the clinical phenotype, HLO from Wolman disease patient-specific iPSCs phenocopied severe steatohepatitis and fibrosis. It was shown that this phenotype was rescued by FGF19, which is a downstream target of obeticholic acid, a known FXR-agonist, and is currently in phase III clinical trial (ClinicalTrials.gov Identifier: NCT02548351) as a potential drug for non-alcoholic steatohepatitis [56]. These results suggested that the in-a-dish model system can be utilized for steatohepatitis modeling and drug testing, allowing for the elucidation of pathological mechanisms and for the discovery of effective treatments against steatohepatitis that will ultimately benefit patients.
The inflammatory bowel diseases (IBD), Crohn’s disease (CD), and ulcerative colitis (UC), are chronic relapsing-remitting disorders with ill-defined etiology [57, 58]. IBD is thought to result from a dysregulated, inappropriate immune response against commensal microbes in a genetically susceptible host with additional environmental factors (dietary factors, smoking, drugs, etc.) also playing roles [59]. Despite the use of currently approved biological agents such as anti-tumor necrosis factor (TNF), and alpha4beta7 monoclonal antibodies, non-responders are still encountered in IBD patients, which impedes mucosal healing and often necessitates surgical intervention [60]. Therefore, it is of paramount importance that drug resistance mechanisms are elucidated and effective therapies are developed to support mucosal healing and to avoid potentially invasive surgeries in IBD patients.
Recently, by using single cell RNA-sequencing data of colon biopsies from UC patients, Smillie et al. revealed that inflammatory monocytes and inflammation-associated fibroblasts are enriched in samples from anti-TNF non-responders and are associated with anti-TNF resistance via expression of OSM and oncostatin M receptor that phenocopy TNF [61]. The authors also reported the notable expansion of CD8 + IL-17+ T cell in UC that are also supported by the mapping of GWAS-implicated disease risk genes. Nanki et al. found somatic mutations in multiple genes related to IL-17 signaling in the inflamed epithelium using whole-exome sequencing (WES) data of colon organoids from UC patients. These mutations confer resistance to the IL-17 induced pro-apoptotic response through unbiased CRISPR-based knockout screening in colon organoids [62]. These two papers share findings related to disruption of IL-17 signaling in UC. Thus, in vitro WES analyses of organoids from GI disease patients will provide valuable insights for linking disease risk genes with specific cell types and disease-associated pathways that are concordant with in vivo disease progression.
Malignant diseases
Gastrointestinal (GI) cancers account for one-third of the total global cancer incidences and mortality. Therefore, it is essential to translate knowledge from basic research into health benefits by advancing therapeutics for GI cancers. Since transformed cell lines cannot recapitulate the complexity of the original human tumors and developmental, genetic, and physiological differences limit animal models, organoid technology has emerged as a powerful alternative method for culturing GI tumors. To create cancer model using human organoids, patient tumor-derived organoids have been utilized. These organoids are biobanked and used for drug screening and personalized medicine approach [63]. Organoids derived from oncogenic driver gene-edited AdSCs using CRISPR-Cas9 technology facilitate our understanding of multistep carcinogenesis (e.g., aberrant growth, metastatic potential, niche factor dependence) [64,65,66]. Recently, PSC-derived organoids have begun utilized for studying gene functions in driving tumorigenesis or inherited cancer-causing mutations. PSCs undergo directed differentiation to a cell type of interest that represents a likely cell-of-origin for a given cancer type and can be used to generate cancer organoids. Cancer-associated genetic alterations, if not already present in the genome of the source material, are introduced at the PSC stage, following PSC-directed differentiation [67]. Organoid models have been used to model tumor initiation, metastatic progression, and therapy response of the common GI cancers, including primary liver cancer (PLC) [68], colorectal cancer (CRC) [63, 69], and pancreatic ductal adenocarcinoma (PDAC) [70, 71]. The majority of PLC cases are classified into either hepatocellular carcinoma (HCC) or cholangiocarcinoma (CC), and there is also a combined hepatocellular-cholangiocarcinoma (CHC) subtype [68]. The development of effective treatments for PLC has been hindered by a shortage of reproducible human models in which to assess the efficacy of candidate therapeutic agents. Therefore, PLC organoids which were composed of HCC, CC, and CHC tumors were developed from patient biopsies [68]. Organoids generated from tumor-needle biopsies of various etiologies/tumor stages of patients with HCC, preserved the morphology and genetic heterogeneity of the source tumors [72]. The PLC organoids recapitulated many features of these subtypes ranging from histological architecture to genetic and transcriptomic features and displayed metastatic characteristics upon transplantation.
CRC, also known as bowel cancer and colon cancer, is the development of cancer from the colon or rectum. CRC pathogenesis has been classically portrayed as the stepwise progression of cancerous lesions from potentially malignant precursors, predominantly tubular adenomas. Familial adenomatous polyposis (FAP) is an inherited disorder characterized by CRC. Crespo et al. developed FAP patient-specific iPSC harboring germline mutations in the APC gene and generated colonic organoids (FAP COs) that exhibit enhanced WNT activity and increased epithelial cell proliferation [73]. Also, they revealed that a read-through drug, geneticin, can restore APC protein expression levels, can significantly decrease WNT overactivation, and can inhibit epithelial cell hyperproliferation specifically in FAP COs without affecting the normal phenotype of WT COs. Oncogenic driver gene-mutated COs from patient-derived iPSCs or using CRISPR-Cas9 technology can be utilized for drug discovery/development platforms.
PDAC has the worst survival rate of common malignancies due to late diagnosis and lack of curative therapeutic options. PDAC is characterized by a desmoplastic reaction resulting in dense, fibrotic stroma. Epithelial organoids from primary cancers, while useful for determining cancer-intrinsic sensitivities, miss these important stromal cues. Ohlund et al. established co-cultures of PDAC organoids and pancreatic stellate cells (PaSCs) and recapitulated the desmoplastic reaction of PDAC with PaSCs converting from a resting quiescent state to activated, stroma-producing fibroblasts [74]. They identified culture conditions in which the PDAC organoids and fibroblasts were mutually supportive to each another such that the co-cultures, but not monocultures, could proliferate. Also, using this co-culture system, they identified two mutually exclusive subpopulations of fibroblasts with reversible features (inflammatory fibroblasts and myofibroblasts) present in the PDAC microenvironment that might act in tumor-supportive and tumor-restrictive roles. These results suggest novel therapeutic approaches against PDAC by inhibiting tumor-supportive and activating tumor-restraining fibroblasts. Co-cultures of PDAC organoids and these cancer-associated fibroblasts can be utilized as an assay system for drug discovery. GI cancer organoid, assisted with cells consisting the tumor microenvironment, provide a new means to emulate some aspects of therapeutic responses against in vivo cancer.
Hopes, hypes, and future challenges
As described above, the widespread applications of organoid models have been promised, but many challenges still prevent the clinical applications of organoid technologies. In the following section, we review the significant challenges involved in developing pharmaceutical applications and discuss future perspectives.
Clinical relevance (diagnostic potential)
Although the drug efficacy/toxicity in organoid models is different from that in 2D culture models, only a small set of data has confirmed that the drug efficacy/toxicity in organoid models resemble clinical data [35]. Identifying clinically relevant genetic signals (biomarkers) for drug efficacy/toxicity in organoid models are critical to rationalize and enhance the drug discovery and development process. In support, organoid-based modeling of drug-induced liver injury revealed critical correlation between organoid and patient’s phenotype, thus informing novel polygenic signatures behind the disease susceptibility [30]. It is not only critical to demonstrate the correlative functional characteristics in the patient-derived organoids, but also establish measurable clinically relevant phenotypes in long-term culture condition before being translated as diagnostic and/or prognostic tools.
Additional complexity
Advancing organoid technologies is pivotal for recapitulating more complex microanatomy in vitro to achieve higher-order functions [45]. Recent organoid models allow for co-differentiation or co-culture of epithelial and mesenchymal component for modeling diseases such as inflammatory disease [55]. However, recapitulation of full complexity in a dish remain highly challenging such includes immune, neural, and endocrine interactions found in homeostatic conditions [7]. As found in extra-digestive organs such as kidney–ureter–bladder and brain–spinal cord–peripheral tissues, it is obvious that the emergence of organ function will critically require inter-organ connections. To this aim, an iPSC-derived three-organoid system (hepato-biliary-pancreatic organoid) that connected functionally between organoids has been reported [75]. As such, establishing interconnection between adjacent organs and their functional interaction will improve in vitro modeling of diseases associated with multi-organ interactions such as biliary atresia.
Reproducibility and consistency
Large scale manufacturing of organoids could benefit a variety of fields including drug discovery/development, and transplantation therapy [76]. Major hurdle includes its limited reproducibility and consistency, which increases batch to batch variation in assays and makes it difficult to detect/quantify the disease-relevant phenotypes and accomplish large scale evaluation of drug candidates. Therefore, development and/or optimization of massive and reproducible production system using devices such as spinner flask [77] and microwell-array [9] that can yield homogeneous organoids with same phenotypic traits (e.g., size, shape, cellular composition and 3D architecture) is needed. Synthetic matrices with minimal batch to batch variation that can control organoid formation efficiency and proliferation by tuning the stiffness could expedite the development of organoid production and subsequent disease modeling [78]. In addition, development of a quality control system such by image-guided and biochemical analyses is required [79]. These systems will make the organoid-based drug discovery/development more realistic.
High-throughput screening
Many 3D cell models, such as organoids, have more complex morphology and function than 2D cultured cells, causing difficulties for their systematic assessments. This presents challenges in standardization of culture and assay protocols, phenotypes, and output data for analysis. Also, realistic drug screening applications require higher-throughput phenotypic readouts that are currently not available in organoid technologies. Through the convergence of several technologies including single cell analysis [80], imaging [81, 82], bioengineering, and organ-on-a-chip/microfluidics [83, 84], detection of complex diseases phenotypes and subsequent development of high-throughput phenotypic assays will improve disease modeling and facilitate the discovery of effective treatments against incurable diseases [85].
Conclusion and perspective
In vitro modeling of digestive tissues is pivotal for an understanding of the disease mechanisms and drug discovery/development. Challenges remain to achieve full functionality in organoid systems, however, recent advances in generating the disease-specific organoids from patient-derived cells hold great promise for recapitulating complex pathogenesis thereby expediting personalized medicine applications. Multidisciplinary efforts including from biologists, bioengineers, and professionals from other emerging areas will help to engineer increased complexity in organoids that is not possible today and facilitate digestive organoid medicine approaches to bring a cure for the patients with intractable disease.
Change history
27 November 2020
This article originally published with a minor typos/markings in figs 1 and 4, which have now been corrected/removed.
References
Takebe T, Wells JM. Organoids by design. Science. 2019;364:956–9.
Zorn AM, Wells JM. Vertebrate endoderm development and organ formation. Annu Rev Cell Dev Biol. 2009;25:221–51.
Shin D, Monga SP. Cellular and molecular basis of liver development. Compr Physiol. 2013;3:799–815. https://doi.org/10.1002/cphy.c120022.
Han L, Chaturvedi P, Kishimoto K, Koike H, Nasr T, Iwasawa K, et al. Single cell transcriptomics identifies a signaling network coordinating endoderm and mesoderm diversification during foregut organogenesis. Nat Commun. 2020;11:1–16.
Trisno SL, Philo KED, McCracken KW, Cata EM, Ruiz-Torres S, Rankin SA, et al. Esophageal organoids from human pluripotent stem cells delineate Sox2 functions during esophageal specification. Cell Stem Cell. 2018;23:501–15.e7.
Zhang Y, Yang Y, Jiang M, Huang SX, Zhang W, Al Alam D, et al. 3D modeling of esophageal development using human PSC-derived basal progenitors reveals a critical role for notch signaling. Cell Stem Cell. 2018;23:516–29.e5.
Takebe T, Sekine K, Enomura M, Koike H, Kimura M, Ogaeri T, et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature. 2013;499:481–4.
Takebe T, Enomura M, Yoshizawa E, Kimura M, Koike H, Ueno Y, et al. Vascularized and complex organ buds from diverse tissues via mesenchymal cell-driven condensation. Cell Stem Cell. 2015;16:556–65.
Takebe T, Sekine K, Kimura M, Yoshizawa E, Ayano S, Koido M, et al. Massive and reproducible production of liver buds entirely from human pluripotent stem cells. Cell Rep. 2017;21:2661–70.
McCracken KW, Aihara E, Martin B, Crawford CM, Broda T, Treguier J, et al. Wnt/beta-catenin promotes gastric fundus specification in mice and humans. Nature. 2017;541:182–7.
Broda TR, McCracken KW, Wells JM. Generation of human antral and fundic gastric organoids from pluripotent stem cells. Nat Protoc. 2019;14:28–50.
Munera JO, Sundaram N, Rankin SA, Hill D, Watson C, Mahe M, et al. Differentiation of human pluripotent stem cells into colonic organoids via transient activation of BMP signaling. Cell Stem Cell. 2017;21:51–64.e6.
Kim Y, Kim H, Ko UH, Oh Y, Lim A, Sohn JW, et al. Islet-like organoids derived from human pluripotent stem cells efficiently function in the glucose responsiveness in vitro and in vivo. Sci Rep. 2016;6:35145.
Spence JR, Mayhew CN, Rankin SA, Kuhar MF, Vallance JE, Tolle K, et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature. 2011;470:105–9.
Nair GG, Liu JS, Russ HA, Tran S, Saxton MS, Chen R, et al. Recapitulating endocrine cell clustering in culture promotes maturation of human stem-cell-derived beta cells. Nat Cell Biol. 2019;21:263–74.
Finkbeiner SR, Hill DR, Altheim CH, Dedhia PH, Taylor MJ, Tsai YH, et al. Transcriptome-wide analysis reveals hallmarks of human intestine development and maturation in vitro and in vivo. Stem Cell Rep. 2015;4:1140–1155.
Dedhia PH, Bertaux-Skeirik N, Zavros Y, Spence JR. Organoid models of human gastrointestinal development and disease. Gastroenterology. 2016;150:1098–1112.
Jung KB, Lee H, Son YS, Lee M, Kim Y, Oh SJ, et al. Interleukin-2 induces the in vitro maturation of human pluripotent stem cell-derived intestinal organoids. Nat Commun. 2018;9:3039 https://doi.org/10.1038/s41467-018-05450-8.
Sato T, Stange DE, Ferrante M, Vries RG, Van Es JH, Van den Brink S, et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology. 2011;141:1762–72.
Kalabis J, Wong GS, Vega ME, Natsuizaka M, Robertson ES, Herlyn M, et al. Isolation and characterization of mouse and human esophageal epithelial cells in 3D organotypic culture. Nat Protoc. 2012;7:235–46.
Huch M, Dorrell C, Boj SF, van Es JH, Li VS, van de Wetering M, et al. In vitro expansion of single Lgr5+ liver stem cells induced by Wnt-driven regeneration. Nature. 2013;494:247–50.
Broutier L, Andersson-Rolf A, Hindley CJ, Boj SF, Clevers H, Koo BK, et al. Culture and establishment of self-renewing human and mouse adult liver and pancreas 3D organoids and their genetic manipulation. Nat Protoc. 2016;11:1724–43.
Huch M, Bonfanti P, Boj SF, Sato T, Loomans CJ, van de Wetering M, et al. Unlimited in vitro expansion of adult bi-potent pancreas progenitors through the Lgr5/R-spondin axis. EMBO J. 2013;32:2708–21.
Yui S, Nakamura T, Sato T, Nemoto Y, Mizutani T, Zheng X, et al. Functional engraftment of colon epithelium expanded in vitro from a single adult Lgr5+ stem cell. Nat Med. 2012;18:618–23.
Fujii M, Matano M, Toshimitsu K, Takano A, Mikami Y, Nishikori S, et al. Human intestinal organoids maintain self-renewal capacity and cellular diversity in niche-inspired culture condition. Cell Stem Cell. 2018;23:787–93.e6.
Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–5.
Aloia L, McKie MA, Vernaz G, Cordero-Espinoza L, Aleksieva N, Ameele J, et al. Epigenetic remodelling licences adult cholangiocytes for organoid formation and liver regeneration. Nat Cell Biol. 2019;21:1321–1333.
Rossi G, Manfrin A, Lutolf MP. Progress and potential in organoid research. Nat Rev Genet. 2018;19:671–687.
Lewis S, Nachun D, Martín MG, Horvath S, Coppola G, Jones L. DNA methylation analysis validates organoids as a viable model for studying human intestinal aging. Cell Mol Gastroenterol Hepatol. 2020;9:527–41.
Koido M, Kawakami E, Fukumura J, Noguchi Y, Ohori M, Nio Y, et al. Polygenic architecture informs potential vulnerability to drug-induced liver injury. Nature Med. 2020. https://doi.org/10.1038/s41591-020-1023-0.
Saint-Criq V, Gray MA. Role of CFTR in epithelial physiology. Cell Mol Life Sci. 2017;74:93–115.
Ferec C, Cutting GR. Assessing the disease-liability of mutations in CFTR. Cold Spring Harb Perspect Med 2012;2:a009480 https://doi.org/10.1101/cshperspect.a009480. Published 2012 Dec 1
Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, Cipolli M, et al. Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for phe508del CFTR. N Engl J Med. 2015;373:220–31.
Dekkers JF, Berkers G, Kruisselbrink E, Vonk A, de Jonge HR, Janssens HM, et al. Characterizing responses to CFTR-modulating drugs using rectal organoids derived from subjects with cystic fibrosis Sci. Transl. Med. 2016;8:344ra84. https://doi.org/10.1126/scitranslmed.aad8278.
Berkers G, van Mourik P, Vonk AM, Kruisselbrink E, Dekkers JF, de Winter-de Groot KM, et al. Rectal organoids enable personalized treatment of cystic fibrosis. Cell Rep. 2019;26:1701–8.e3.
Schwank G, Koo BK, Sasselli V, Dekkers JF, Heo I, Demircan T, et al. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell. 2013;13:653–8.
Vaidyanathan S, Salahudeen AA, Sellers ZM, Bravo DT, Choi SS, Batish A, et al. High-efficiency, selection-free gene repair in airway stem cells from cystic fibrosis patients rescues CFTR function in differentiated epithelia. Cell Stem Cell. 2020;26:161–171.e4. https://doi.org/10.1016/j.stem.2019.11.002.
Geurts MH, de Poel E, Amatngalim GD, Oka R, Meijers FM, Kruisselbrink E, et al. CRISPR-based adenine editors correct nonsense mutations in a cystic fibrosis organoid biobank. Cell Stem Cell 2020;26:503–510.e7.
Sampaziotis F, de Brito MC, Madrigal P, Bertero A, Saeb-Parsy K, Soares FAC, et al. Cholangiocytes derived from human induced pluripotent stem cells for disease modeling and drug validation. Nat Biotechnol. 2015;33:845–52.
Huch M, Gehart H, van Boxtel R, Hamer K, Blokzijl F, Verstegen MM, et al. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell. 2015;160:299–312.
Nantasanti S, Spee B, Kruitwagen HS, Chen C, Geijsen N, Oosterhoff LA, et al. Disease modeling and gene therapy of copper storage disease in canine hepatic organoids. Stem Cell Rep. 2015;5:895–907.
Guan Y, Xu D, Garfin PM, Ehmer U, Hurwitz M, Enns G, et al. Human hepatic organoids for the analysis of human genetic diseases. JCI insight. 2017;2:e94954. https://doi.org/10.1172/jci.insight.94954.
Gomez-Mariano G, Matamala N, Martinez S, Justo I, Marcacuzco A, Jimenez C, et al. Liver organoids reproduce alpha-1 antitrypsin deficiency-related liver disease. Hepatol Int. 2020;14:127–37.
Heuckeroth RO. Hirschsprung disease - integrating basic science and clinical medicine to improve outcomes. Nature Rev Gastroenterol Hepatol. 2018;15:152–67.
Workman MJ, Mahe MM, Trisno S, Poling HM, Watson CL, Sundaram N, et al. Engineered human pluripotent-stem-cell-derived intestinal tissues with a functional enteric nervous system. Nat Med. 2017;23:49–59.
Peek RM. Helicobacter pylori infection and disease: from humans to animal models. Dis Models Mech. 2008;1:50–5.
McCracken KW, Cata EM, Crawford CM, Sinagoga KL, Schumacher M, Rockich BE, et al. Modelling human development and disease in pluripotent stem-cell-derived gastric organoids. Nature. 2014;516:400–4.
Bartfeld S, Bayram T, van de Wetering M, Huch M, Begthel H, Kujala P, et al. In vitro expansion of human gastric epithelial stem cells and their responses to bacterial infection. Gastroenterology. 2015;148:126–36.e6.
Min S, Kim S, Cho SW. Gastrointestinal tract modeling using organoids engineered with cellular and microbiota niches. Exp Mol Med. 2020;52:227–237.
Schumacher MA, Aihara E, Feng R, Engevik A, Shroyer NF, Ottemann KM, et al. The use of murine-derived fundic organoids in studies of gastric physiology. J Physiol. 2015;593:1809–1827.
Holokai L, Chakrabarti J, Broda T, Chang J, Hawkins JA, Sundaram N, et al. Increased programmed death-ligand 1 is an early epithelial cell response to Helicobacter pylori infection. PLoS Pathog. 2019;15:e1007468. Published 2019 Jan 31. doi:10.1371/journal.ppat.1007468
Nie YZ, Zheng YW, Miyakawa K, Murata S, Zhang RR, Sekine K, et al. Recapitulation of hepatitis B virus-host interactions in liver organoids from human induced pluripotent stem cells. EBioMedicine. 2018;35:114–23.
Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64:73–84.
Friedman SL, Ratziu V, Harrison SA, Abdelmalek MF, Aithal GP, Caballeria J, et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology. 2018;67:1754–1767.
Ouchi R, Togo S, Kimura M, Shinozawa T, Koido M, Koike H, et al. Modeling steatohepatitis in humans with pluripotent stem cell-derived organoids. Cell Metab. 2019;30:374–84.e6.
Ratziu V, Sanyal AJ, Loomba R, Rinella M, Harrison S, Anstee QM, et al. REGENERATE: design of a pivotal, randomised, phase 3 study evaluating the safety and efficacy of obeticholic acid in patients with fibrosis due to nonalcoholic steatohepatitis. Contemp Clin Trials. 2019;84:105803.
Liverani E, Scaioli E, Digby RJ, Bellanova M, Belluzzi A. How to predict clinical relapse in inflammatory bowel disease patients. World J Gastroenterol. 2016;22:1017–1033.
Torres J, Caprioli F, Katsanos KH, Lobaton T, Micic D, Zeroncio M, et al. Predicting outcomes to optimize disease management in inflammatory bowel diseases. J Crohns Colitis. 2016;10:1385–1394.
Ho SM, Lewis JD, Mayer EA, Plevy SE, Chuang E, Rappaport SM, et al. Challenges in IBD research: environmental triggers. Inflamm Bowel Dis. 2019;25:S13–S23. https://doi.org/10.1093/ibd/izz076.
Singh S, George J, Boland BS, Vande Casteele N, Sandborn WJ. Primary non-response to tumor necrosis factor antagonists is associated with inferior response to second-line biologics in patients with inflammatory bowel diseases: a systematic review and meta-analysis. J Crohns Colitis. 2018;12:635–643.
Smillie CS, Biton M, Ordovas-Montanes J, Sullivan KM, Burgin G, Graham DB, et al. Intra- and Inter-cellular rewiring of the human colon during ulcerative colitis. Cell. 2019;178:714–30.e22.
Nanki K, Fujii M, Shimokawa M, Matano M, Nishikori S, Date S, et al. Somatic inflammatory gene mutations in human ulcerative colitis epithelium. Nature. 2020;577:254–9.
Van de Wetering M, Francies Hayley E, Francis Joshua M, Bounova G, Iorio F, Pronk A, et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell. 2015;161:933–45.
Matano M, Date S, Shimokawa M, Takano A, Fujii M, Ohta Y, et al. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat Med. 2015;21:256–62.
Drost J, van Jaarsveld RH, Ponsioen B, Zimberlin C, van Boxtel R, Buijs A, et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature. 2015;521:43–7.
Nanki K, Toshimitsu K, Takano A, Fujii M, Shimokawa M, Ohta Y, et al. Divergent routes toward Wnt and R-spondin niche independency during human gastric carcinogenesis. Cell. 2018;174:856–69.e17.
Smith RC, Tabar V. Constructing and deconstructing cancers using human pluripotent stem cells and organoids. Cell Stem Cell. 2019;24:12–24.
Broutier L, Mastrogiovanni G, Verstegen MM, Francies HE, Gavarro LM, Bradshaw CR, et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat Med. 2017;23:1424–35.
Fujii M, Shimokawa M, Date S, Takano A, Matano M, Nanki K, et al. A colorectal tumor organoid library demonstrates progressive loss of niche factor requirements during tumorigenesis. Cell Stem Cell. 2016;18:827–38.
Boj SF, Hwang CI, Baker LA, Chio II, Engle DD, Corbo V, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell. 2015;160:324–38.
Seino T, Kawasaki S, Shimokawa M, Tamagawa H, Toshimitsu K, Fujii M, et al. Human pancreatic tumor organoids reveal loss of stem cell niche factor dependence during disease progression. Cell Stem Cell. 2018;22:454–67.e6.
Nuciforo S, Fofana I, Matter MS, Blumer T, Calabrese D, Boldanova T. Organoid models of human liver cancers derived from tumor needle biopsies. Cell Rep. 2018;24:1363–1376.
Crespo M, Vilar E, Tsai SY, Chang K, Amin S, Srinivasan T, et al. Colonic organoids derived from human induced pluripotent stem cells for modeling colorectal cancer and drug testing. Nat Med. 2017;23:878–84.
Ohlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med. 2017;214:579–96.
Koike H, Iwasawa K, Ouchi R, Maezawa M, Giesbrecht K, Saiki N, et al. Modelling human hepato-biliary-pancreatic organogenesis from the foregut-midgut boundary. Nature. 2019;574:112–6.
Takahashi Y, Sekine K, Kin T, Takebe T, Taniguchi H. Self-condensation culture enables vascularization of tissue fragments for efficient therapeutic transplantation. Cell Rep. 2018;23:1620–9.
Schneeberger K, Sánchez-Romero N, Ye S, van Steenbeek FG, Oosterhoff LA, Pla Palacin I. Large-scale production of LGR5-positive bipotential human liver stem cells. Hepatology. 2020;72:257–70.
Sorrentino G, Rezakhani S, Yildiz E, Nuciforo S, Heim MH, Lutolf MP. Mechano-modulatory synthetic niches for liver organoid derivation. Nat Commun. 2020;11:1–10.
Kassis T, Hernandez-Gordillo V, Langer R, Griffith LG. OrgaQuant: human intestinal organoid localization and quantification using deep convolutional neural networks. Sci Rep. 2019;9:12479.
Hayashi T, Ozaki H, Sasagawa Y, Umeda M, Danno H, Nikaido I. Single-cell full-length total RNA sequencing uncovers dynamics of recursive splicing and enhancer RNAs. Nat Commun. 2018;9:619.
Booij TH, Price LS, Danen EHJ. 3D cell-based assays for drug screens: challenges in imaging, image analysis, and high-content analysis. SLAS Discov. 2019;24:615–27.
Dekkers JF, Alieva M, Wellens LM, Ariese HCR, Jamieson PR, Vonk AM, et al. High-resolution 3D imaging of fixed and cleared organoids. Nat Protoc. 2019;14:1756–71.
Takebe T, Zhang B, Radisic M. Synergistic engineering: organoids meet organs-on-a-chip. Cell Stem Cell. 2017;21:297–300.
Tomasi RF, Sart S, Champetier T, Baroud CN. Individual control and quantification of 3D spheroids in a high-density microfluidic droplet array. Cell Rep. 2020;31:107670.
Shinozawa T, Kimura M, Cai Y, Saik N, Yoneyama Y, Ouchi R, et al. High-Fidelity Drug Induced Liver Injury Screen Using Human PSC-derived Organoids. Gastroenterology. https://doi.org/10.1053/j.gastro.2020.10.002.
Acknowledgements
Editorial support, in the form of medical rewriting, assembling tables based on authors’ detailed directions, collating author comments, copyediting, fact checking, and referencing, was provided by Editage, Cactus Communications. Figures were illustrated by Asuka Kodaka and partly created with BioRender.com. This work was supported by a NIH Director’s New Innovator Award (DP2 DK128799-01), NIH grant UG3 DK119982, Cincinnati Center for Autoimmune Liver Disease Fellowship Award, PHS Grant P30 DK078392 (Integrative Morphology Core and Pluripotent Stem Cell and Organoid Core) of the Digestive Disease Research Core Center in Cincinnati, Takeda Science Foundation award, Mitsubishi Foundation award and AMED JP19fk0210037, JP19bm0704025, JP19fk0210060, JP19bm0404045, and JSPS JP18H02800, 19K22416. TT is a New York Stem Cell Foundation – Robertson Investigator.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
TT have served on scientific advisory boards for Healios inc.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by F. Pentimalli
Rights and permissions
About this article

Cite this article
Funata, M., Nio, Y., Erion, D.M. et al. The promise of human organoids in the digestive system. Cell Death Differ 28, 84–94 (2021). https://doi.org/10.1038/s41418-020-00661-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-020-00661-3
This article is cited by
-
Multiple Dimensions of using Mesenchymal Stem Cells for Treating Liver Diseases: From Bench to Beside
Stem Cell Reviews and Reports (2023)
-
Applications of human organoids in the personalized treatment for digestive diseases
Signal Transduction and Targeted Therapy (2022)
-
Understanding p53 tumour suppressor network
Biology Direct (2021)
-
Recapitulating pancreatic cell–cell interactions through bioengineering approaches: the momentous role of non-epithelial cells for diabetes cell therapy
Cellular and Molecular Life Sciences (2021)
