
Abstract
Articular cartilage serves as a low-friction, load-bearing tissue without the support with blood vessels, lymphatics and nerves, making its repair a big challenge. Transforming growth factor-beta 3 (TGF-β3), a vital member of the highly conserved TGF-β superfamily, plays a versatile role in cartilage physiology and pathology. TGF-β3 influences the whole life cycle of chondrocytes and mediates a series of cellular responses, including cell survival, proliferation, migration, and differentiation. Since TGF-β3 is involved in maintaining the balance between chondrogenic differentiation and chondrocyte hypertrophy, its regulatory role is especially important to cartilage development. Increased TGF-β3 plays a dual role: in healthy tissues, it can facilitate chondrocyte viability, but in osteoarthritic chondrocytes, it can accelerate the progression of disease. Recently, TGF-β3 has been recognized as a potential therapeutic target for osteoarthritis (OA) owing to its protective effect, which it confers by enhancing the recruitment of autologous mesenchymal stem cells (MSCs) to damaged cartilage. However, the biological mechanism of TGF-β3 action in cartilage development and OA is not well understood. In this review, we systematically summarize recent progress in the research on TGF-β3 in cartilage physiology and pathology, providing up-to-date strategies for cartilage repair and preventive treatment.
Similar content being viewed by others
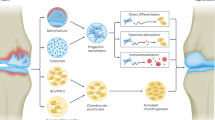


Introduction
The transforming growth factor-beta (TGF-β) superfamily includes TGF-β isoforms, activins/inhibins, nodal, anti-Müllerian hormone proteins, growth differentiation factors (GDFs), and bone morphogenetic proteins (BMPs), which play critical roles in embryogenesis and adult tissue homeostasis.1,2 The roles played by TGF-β isoforms, especially TGF-β1 and TGF-β2 in chondrocytes, have been extensively studied. However, few studies have focused on TGF-β3, which is equally important to cell proliferation, differentiation, migration and apoptosis.3 TGF-β3 is a 25-kDa homodimeric protein with a structure similar to its two homologous dimer-forming isoforms, TGF-β1 and TGF-β2.4 These three isoforms are potent agonists of chondrocyte differentiation and are generally applied in the field of tissue engineering and regenerative medicine. Moreover, they have specialized and overlapping effects on cartilage physiology and pathology.5
Articular cartilage, a smooth and wear-resistant tissue covering the surface of joints, supports and distributes applied loads. It is characterized by viscoelastic, anisotropic, tension–compression, and nonlinear properties, endowing cartilage with the ability to withstand extremely high mechanical stress.6 These characteristics rely on a set of cells composed of a small percentage of chondrocytes and an intricately organized matrix. The extracellular matrix (ECM) of hyaline cartilage constitutes as much as 98% of the cartilage volume and comprises primarily hyaluronan (HA), proteoglycans and collagens. Type II collagen accounts for 90% of all types of collagens in hyaline cartilage and confers tensile strength to the tissue.7 Proteoglycans form large aggregates by binding to spontaneously aggregating HA networks, endowing cartilage with compressive strength.8 Terminally differentiated chondrocytes constitute the only cell type in this tissue and account for ~5% of the total tissue volume. A small number of chondrocytes are sparsely dispersed within the matrix network and regulate cartilage health by controlling the dynamic balance between the anabolic and catabolic processes of ECM components.9 Without nerves, blood vessels, or a lymphatic system, cartilage fails to self-repair, and even minor lesions can result in progressive damage.10 The degradation of the cartilage structure ultimately leads to degenerative joint diseases such as osteoarthritis (OA), a painful condition that exerts a profound influence on individual physical health, including disability, the health care system, and society.11 Moreover, the repair of damaged cartilage with existing clinical techniques has been a challenge due to the extremely poor regenerative ability of this tissue.12 Therefore, it is essential to seek novel therapeutic targets for defective cartilage by elucidating the specific roles of various cytokines and growth factors in cartilage pathophysiology. In recent years, the profound roles of TGF-β3 in the anabolic processes of articular cartilage have been noticed by numerous researchers and may lead to new approaches to the restoration and reconstruction of cartilage defects.13
TGF-β3 contributes to cartilage physiology and pathology via the Smad-dependent pathway and a non-Smad pathway (herein, the Smad-independent pathway).14 The Smad family includes intracellular mediators that mediate TGF-β superfamily member signaling. Based on their structural and functional features, Smads are categorized into three major classes: receptor-regulated Smads (R-Smads), common mediator Smads (Co-Smads), and inhibitory Smads (I-Smads).15 As direct substrates of receptor kinases, R-Smads can be further classified into two subcategories, the Smad2/3 group and the Smad1/5/8 group, which each playing antagonistic roles during different stages of OA.16 The Smad-independent pathway is mediated by mitogen-activated protein kinases (MAPKs) (including extracellular regulated kinase (ERK), p38 kinase, c-Jun N-terminal kinase (JNK)), phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR), and Rho-like GTPase.17 Overall, the Smad-dependent and Smad-independent pathways function independently but interact with each other to regulate cartilage development and maintenance. In summary, we review the recent progress in the understanding of TGF-β3 in cartilage development and the occurrence/progression of OA that is providing new targets for the treatment of cartilage diseases.
Transforming growth factor-β3 overview
In the 1970s, polypeptides in the TGF-β family were first isolated and named sarcoma growth factor (SGF) by de Larco and Todaro.18 SGF is a mixture of two different compounds, TGF-β and transforming growth factor-alpha (TGF-α), that induces the malignant transformation of rat kidney fibroblasts.19,20 To date, the TGF-β superfamily primarily includes TGF-β isoforms, activins/inhibins, nodal, anti-Müllerian hormone proteins, GDFs and BMPs.
Three TGF-β isoforms are encoded by distinct genes located on different chromosomes. The active form is a homodimer consisting of two polypeptide chains connected by a disulfide bond.21,22 The biological activities of the three TGF-β isoforms are precisely modulated via four steps: synthesis, proteolytic processing, secretion, and activation (Fig. 1). Generally, TGF-β3 mRNA is produced in the nucleus and transported to the cytoplasm. The first polypeptide synthesized is pre-pro-TGF-β3, which carries N-terminal signal peptide (SP), a proregion called the latency-associated peptide (LAP), and a C-terminus. After SP removal and the dimerization of two polypeptides, a pro-TGF-β3 homodimer connected via three disulfide bonds is formed in the endoplasmic reticulum (ER) and is subsequently transported to the trans-Golgi network. In the Golgi apparatus, the LAP chains in pro-TGF-β3 are removed from the TGF-β3 molecule by a paired basic amino acid cleaving enzyme (PACE, also known as furin). Then, LAP and TGF-β3 are connected via noncovalent bonding, resulting in the formation of a small latent TGF-β3 complex (SLC). The SLC connects with the latent TGF-β3 binding protein (LTBP) to form the mature protein, the large latent TGF-β3 complex (LLC), which is transported from the cis pole of the Golgi apparatus secreted from a cell. After secretion, the trimeric structure of LLC prevents the interaction of TGF-β3 with its receptor, maintaining TGF-β3 in the inactive form.23,24 In addition, the TGF-β3 trimeric complex is attached to ECM components or the cell surface facilitated LTBP and glycoprotein-A repetitions predominant. The C-terminal end of LGBP interacts with fibrylin-1, and its N-terminal end covalently binds to fibronectin as catalyzed by a transglutaminase enzyme, providing a alternative mechanism to maintain TGF-β3 in the inactive form.25 Active TGF-β is released from LLC cells through the action of numerous ECM molecules, including proteases such as matrix metalloproteinase 2 (MMP2, also known as gelatinase A), matrix metalloproteinase 9 (MMP9, also known as gelatinase B), fibroblast growth factor 2 (FGF2), and reactive oxygen species.26,27,28 Hence, illuminating the activation mechanisms of latent TGF-β3 is of great importance. The RGD (Arg-Gly-Asp sequence) motif of TGF-β3 is recognized by αv integrins, which are necessary for integrin-mediated TGF-β3 activation.29 Once binding to integrins, LLC is conformationally changed via a nonproteolytic manner, leading to the inhibitory release and activation of mature TGF-β3.30,31 Among the members of the integrin family, integrin αvβ6 and αvβ8 are able of binding to LLC and activating TGF-β3.32

Schematic diagram showing the synthesis, proteolytic processing, secretion and activation of transforming growth factor-beta 3 (TGF-β3). TGF-β3 is produced through mRNA transcription and is transported to the cytoplasm. A pro-TGF-β3 homodimer consisting of the latency-associated peptide (LAP) and TGF-β3 is generated in the endoplasmic reticulum (ER) and then transported to the trans-Golgi network. In the Golgi apparatus, pro-TGF-β3 is further processed to form a small latent TGF-β3 complex (SLC). The SLC connects with the latent TGF-β3-binding protein (LTBP) and forms the resultant protein, the large latent TGF-β3 complex (LLC). LLC is secreted outside the cell and binds to various molecules in the extracellular matrix. Finally, the conformation of the LLC is change by modulating factors, leading to the release of the activated and mature TGF-β3. Activated TGF-β3 binds to corresponding receptors on the cell membrane
Since the processes of TGF-β3 synthesis, processing, secretion, and activation are elaborately regulated in chondrocytes, revealing the specific characteristics of TGF-β3 may lead to insights into cartilage repair and regeneration. Indeed, compared to other TGF-β isoforms, TGF-β3 shows subtle differences in the abovementioned process. For example, the precursor sequences of the TGF-β isoforms are largely dissimilar. The TGF-β3 precursor carries four potential N-glycosylation sites, while the precursors of TGF-β1 and TGF-β2 carry three of these sites, two of which are conserved in all three isoforms. In addition, the RGD peptide, which is recognized by αv integrins, is carried only by the TGF-β1 and TGF-β3 precursors. Therefore, αvβ6 integrin can cleave TGF-β3 ligand from LLC to activate TGF-β3.33 In addition, the three precursors differ in the number of cysteine residues; there are five in TGF-β3, six in TGF-β2, and three in TGF-β1 (which are conserved in all three isoforms).34 In addition, different TGF-β isoforms bind to different LTBPs. Evidence based on genetically mutated mice has shown that the association of TGF-β1 with LTBP is pivotal to extracellular TGF-β1 activity.35 Four isoforms of LTBP constitute a group of glycoproteins that interact with fibrillin microfibrils.36 LTBP-1 and LTBP-3 are thought to interact with all three TGF-β isoforms. LTBP-2 does not bind to any TGF-β isoform. LTBP-4 binds only to the LAP of TGF-β1.37 However, the specific role of each LTBP in the secretory mechanics of different TGF-β isoforms has not been clearly demonstrated.38
TGF-β3 receptors
TGF-β receptors (TβRs) are mediators that mediate TGF-β signaling from the ECM to the intracellular space. TβRs are classified as a TGF-β type I receptor (TβRI), TGF-β type II receptor (TβRII), or TGF-β type III receptor (TβRIII) based on their sequence characteristics. TβRI proteins include activin receptor-like kinase 1–7 (ALK1-7). The TβRII protein subfamily consist of TβRII, the bone morphogenetic protein 2 (BMP2) receptor, activin type II receptor (ActRII), activin type I receptor (ActRI/Ib), and anti-Müllerian hormone receptor type 2 (AMHRII). TβRIII molecules include β-glucan and endoglin.39,40,41,42 TβRI and TβRII are structurally related transmembrane glycoproteins with three main sections: an extracellular ligand-binding site, a transmembrane sequence, and an intracellular kinase domain with serine/threonine kinase activity.43 In contrast to TβRI and TβRII, membrane-anchored TβRIII is not a typical receptor that can transduce extracellular signals because it lacks of a domain with serine/threonine activity. It acts primarily as a coreceptor to regulate TGF-β access to TβRI and TβRII.44 The type of TGF-β ligand and TβRIII molecule determines binding affinity. β-Glycans, well-characterized members of the TβRIII subfamily, show affinity for all three TGF-β isoforms, with the highest affinity for TGF-β2.45 Endoglin, another member of the TβRIII subfamily, selectively binds TGF-β1 and TGF-β3.44
Signal transmission efficiency depends on the abundance and availability of ligands and their inhibitors, the number of receptors on the cell surface, and the regulation of kinase activity. TGF-β3-induced signaling pathways are primarily associated with TβRI and TβRII. Once active TGF-β3 is released from an LLC protein, it interacts with a heterotetrameric receptor complex carrying two TβRI subunits and two TβRII subunits.46 TβRII proteins are first activated in response to the binding of TGF-β3; then, constitutively active TβRII proteins phosphorylate the juxtamembrane regions of the cytoplasmic domains of TβRI and transmit the signal further into the intracellular space. The heterotetrameric TβRI–TβRII complex is essential for the initiation of TGF-β3 signaling.47 A previous study analyzed a synthetic TGF-β3 dimer composed of a wild-type protomer and a receptor-binding-deficient mutant to demonstrate that each pair of TβRI–TβRII heterodimers was necessary for signaling.46 TβRII can bind TGF-β ligands directly, while TβRI cannot bind ligands but forms a complex with TβRII in the presence of TGF-β ligands. Moreover, the direct association between TGF-β ligands and TβRII molecules differs among TGF-β isoforms. TGF-β1 and TGF-β3 bind to TβRII even without TβRI. TGF-β2 interacts very weakly with TβRII, which depends on TβRI or TβRIII.48
TGF-β3 signaling
TGF-β3 participates in physiological and pathological functions by elaborately regulating cell metabolism in all tissues of the human body. Intracellular signaling pathways are initiated when ligands activate heterotetrameric TβRI-TβRII complexes. TGF-β3 activates R-Smad-dependent signaling pathways, the canonical pathway of TGF-β family members, and R-Smad-independent signaling pathways.
The R-Smad-dependent TGF-β3 signaling pathway and its role in cartilage
Smad proteins are canonical downstream mediators of TGF-β3-induced signaling pathways. After activation of TβRs induced by TGF-β3, intracellular Smad proteins transduce signals from the plasma membrane to the nucleus, ultimately eliciting transcriptional responses of target genes.49 To date, three types of Smads have been identified: including R-Smads (Smad1, Smad2, Smad3, Smad5 and Smad8/9), a co-Smad (Smad4), and I-Smads (Smad6 and Smad7).
R-Smads and the co-Smad have a unique structure consisting of highly conserved Mad homology 1 (MH1) in the N-terminus and Mad homology 2 (MH2) in the C-terminus end connected via a proline-rich linker region.50 The MH1 domain is critical for regulating transcriptional activity by binding with DNA, and the MH2 domain interacts with other proteins and directs the oligomerization of Smad proteins.51 Some cofactors that influence gene expression also function by binding to the MH2 domain. R-Smads are further classified into two groups on the basis of structures and functions. One group comprises Smad2 and Smad3, and another group comprises Smad1, Smad5 and Smad8/9. The two different groups of R-Smads direct the behaviors of chondrogenic and osteogenic cells antagonistically by activating and/or blocking the transcription of Runt-related transcription factor 2 (Runx2).52 In addition, Smad2/3 can be phosphorylated by TGF-β isoforms, Nodal, and activin. Smad1, 5 and 8 are activated by BMP signaling.15 These two different Smad groups are activated by different TβRI proteins and bind to different DNA sequences. Specifically, ALK1/2/3/6 proteins phosphorylate Smad1/5/9, and Smad1/5/9/4 complexes bind to the GCCGCGC sequence. ALK4/5/7 proteins phosphorylate Smad2/3, and Smad2/3/4 complexes, which bind to the GTCTAGAC sequence.53 I-Smad proteins inhibit TGF-β3-triggered R-Smad signaling pathways by directly binding to R-Smads and blocking their modification, including phosphorylation and threonylation. Smad6 generally participates in the inhibition of Smad1/5/9, and Smad7 always inactivates Smad2/3.54 The I-Smad proteins play inhibitory roles via three mechanisms: dephosphorylation and degradation of TβRs to inhibit the activation of R-Smads, inhibition of the association between R-Smads and specific DNA motifs, and inhibition of the formation of R-Smad/co-Smad complexes.50
Similar to signal transduction mediated by TGF-β1, TGF-β3 can drive the R-Smad-dependent signaling pathway, as demonstrated by several reports (Fig. 2). Activated TβRI typically induces the phosphorylation of the C-terminal Ser-Ser-X-Ser (SSXS) sequence (a motif of the MH2 domain) downstream of R-Smad proteins after the formation of the heterotetrameric complex. Then, co-Smad is recruited and facilitates the formation of a heterotrimer consisting of two phosphorylated R-Smads (Smad2 and Smad3) and the co-Smad.55 The complex enters the nucleus to bind with a DNA strand via the MH1 domain, which carries a nuclear localization signal, and influences gene expression.56 Whereas the Smad complex alone interacts weakly with DNA, the affinity is enhanced by transcription factors and cofactors. In collaboration with other nuclear cofactor proteins, the R-Smad/co-Smad complexes regulate gene transcription.57 These transcriptional cofactors, whether coactivators or corepressors, are regulated by other signaling pathways that participate in modifying initial Smad signaling, thereby allowing pleiotropic responses to TGF-β3. In cartilage, TGF-β3 increases the production of type II collagen and decreases the production of type I collagen via Smad2/3-mediated signaling. TGF-β3-induced R-Smad-dependent signaling pathways has been confirmed to collaborate with the transcription factor SRY‑box transcription factor 9 (SOX9) in modulating the gene expression of chondrocyte-specific markers, including type II collagen and aggrecan.56 Further studies have indicated that SOX9-dependent chondrogenesis are promoted by an increased association between SOX9 and Smad3/4. Moreover, transcription coactivators/factors, such as p300, FoxO (FoxO1, 3, and 4), Sp1, and cJun/c-Fos, facilitate the formation and stabilization of the SOX9–Smad3/4 complex.58,59

Schematic diagram showing the transforming growth factor-beta 3 (TGF-β3)-mediated R-Smad-dependent signaling pathway. Smad proteins transduce signals from the cell membrane to the nucleus and regulate gene expression after TGF-β3 binding to a TGF-β receptor (TβR). TGF-β3 plays a dual role in articular cartilage. It is involved in cartilaginous homeostasis via the classical Smad2/3 pathway and chondrocyte hypertrophy via the Smad1/5/8 pathway. The partner common to both pathways, Smad4, and inhibitory Smads (Smad6 and Smad7) play important roles. BMP bone morphogenetic protein, ALK1 activin receptor-like-kinase 1, ALK5 activin receptor-like kinase 5
The noncanonical TGF-β3 signaling pathway and its role in cartilage
TGF-β3 activates the noncanonical pathway (the R-Smad-independent pathway) mediated by MAPKs (ERK1/2, JNK, p38), PI3K/AKT/mTOR, and Rho-like GTPase (Ras, RhoA, Rac1, CDC42) (Fig. 3). TGF-β3-induced microRNAs (miRNAs), such as miRNA-410 and miRNA-495, also regulate cellular metabolism at the posttranscriptional level.48,60

Schematic diagram showing the noncanonical transforming growth factor-beta 3 (TGF-β3) signaling pathway. TGF-β3 activates R-Smad-independent pathways mediated by mitogen-activated protein kinases (MAPKs) (including extracellular regulated kinase (ERK), p38 kinase, and c-Jun N-terminal kinase (JNK)), phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR), and Rho-like GTPase (Ras, RhoA, Rac1, CDC42). These pathways function independently but interact with each other to regulate cartilage development and maintenance. TAK1 transforming growth factor-β-activated kinase 1, MKK3/4/6/7 mitogen-activated protein kinase 3/4/6/7, ROCK Rho-associated protein kinase, S6K S6 kinase, PP2A protein phosphatase 2A, MEK mitogen-activated protein kinase
The MAPK family primarily include ERKs (ERK1 and ERK2), JNKs (JNK1, JNK2, and JNK3), p38 isoforms (p38α, p38β, p38γ, and p38δ) and a fourth MAPK, ERK5. Each MAPK family member is activated by a specific upstream MAPK kinase (MKK), which is highly selective in phosphorylating a specific MAPK. For instance, ERK1 is the phosphorylation substrate of MKK1, and JNK1/ERK5/p38 can be selectively phosphorylated by MKK4/MKK5/MKK6.61 Before phosphorylating MAPK, TβRs first activates MAPK kinase kinase and MKK.62 TGF-β3-mediated MAPK pathways play significant roles in the regulation of chondrocyte behaviors, including their proliferation, maturation, differentiation, and death.63 Different MAPK members play disparate roles in regulating chondrocyte metabolism. For example, p38 and ERK play essential but opposing roles in chondrogenesis, but the effect that JNK exerts on chondrogenic differentiation is less profound than that of p38 and ERK.64 During chondrogenesis, the phosphorylation of p38 is enhanced, which is consistent with the increase in Smad2 phosphorylation. In contrast, the phosphorylation of ERK is rapid, transient, and diminished. Therefore, p38 is necessary for chondrogenesis and is slow acting, playing a prolonged role in the process, and ERK signaling may repress chondrogenesis.65 Another study pointed out that p38 promoted chondrogenesis by stabilizing SOX9 mRNA in chondrocytes.66 However, there are conflicting reports about the effect of p38 on the chondrocyte phenotype. Previous studies have suggested that p38 is involved in chondrocyte phenotype maintenance.67,68 Another report put forward a different viewpoint, suggesting that p38 signaling is associated with primary chondrocyte dedifferentiation.69 To date, the precise role of p38 in chondrocyte phenotype acquisition remains is unclear. In addition to regulating chondrogenesis and chondrocyte phenotype, p38 plays roles in stress- and inflammation-related activity in chondrocytes.70 Transforming growth factor-β-activated protein kinase 1 (TAK1), a signaling molecule upstream of p38, also plays important roles in cartilage physiology.71 TAK1 promotes the expression of Type II collagen in chondrocytes by upregulating SOX9 expression.72 Studies on mice have shown that the absence of TAK1 affects the proliferation and maturation of chondrocytes, leading to severe cartilage diseases and death.73 Similarly, the role of ERK is intricate and hinges on various intracellular and extracellular conditions. Multiple studies have reported that ERK plays a dual role by protecting and inhibiting in cartilage growth and development. Even though ERK is an inhibitor of chondrogenesis, ERK plays a positive and protective role in mature chondrocytes. Elevated ERK levels lead to TGF-β3-induced chondrocyte proliferation and the gene expression of aggrecan and Type II collagen.74
PI3K/AKT/mTOR and RhoA/Rho-associated protein kinase (ROCK) are also in the R-Smad-independent signaling pathway. mTOR, a factor downstream of PI3K, is associated with chondroblast growth and proliferation. mTOR is upregulated during OA but downregulated during chondroprotective autophagy.75,76 Regarding the RhoA/ROCK pathway, previous studies have found that RhoA-activated ROCK inhibits the mRNA expression of SOX9, Type II collagen, and aggrecan in primary chondrocytes.77 In contrast to the results of those on two-dimensional (2D) in vitro cultures, studies based on three-dimensional (3D) micromass cultures have shown that ROCK inhibition increased SOX9 mRNA expression but decreased the expression of SOX9-targeted aggrecan, leading to the conclusion that RhoA/ROCK regulates SOX9 and cartilage ECM proteins in a context-dependent manner.78 Another study reported that TGF-β treatment resulted in a ROCK-dependent increase in the phosphorylation and nuclear accumulation of SOX9, and this increase was achieved via the direct interaction between ROCK and SOX9.79
Taken together, the data suggest that TGF-β3 can activate a variety of noncanonical intracellular signaling pathways independent of Smads. These Smad-independent signaling molecules interact with the canonical TGF-β3 signaling pathway in a cell-type-specific manner, contributing to the formation of a complex network of signaling molecules engaged in crosstalk. In cartilaginous cells, the interaction between Smads and MAPKs is common and complicated. TGF-β3-induced MAPK activation may result from cellular responses triggered by Smad-dependent transcription. On the other hand, activated MAPKs are involved in Smad-mediated transcriptional regulation by directly phosphorylating Smad proteins and/or influencing their downstream molecules.80 For example, TGF-β3 increased the expression of chondroitin sulfate synthase 1 (CHSY1), an enzyme indispensable for the synthesis of proteoglycans, through the activation of Smad2/3 in rat nucleus pulposus cells. Moreover, the three major MAPK members, namely, ERK, p38 and JNK, were all markedly stimulated upon TGF-β3 treatment. Subsequently, MAPK-activated AP1 and Sp1 transcription factors selectively interacted with the nuclear translocated Smad2 and facilitated the transcriptional activation of CHSY1.81
MiRNAs are noncoding RNAs 18–24 nucleotides in length. They can recognize and bind specific sequences of the 3ʹ-untranslated region (3ʹ-UTR) of a target mRNA.82 TGF-β3-induced signaling influences the expression of numerous miRNAs, which can also mediate crosstalk between TGF-β3 signaling and other pathways, collectively directing chondrogenesis and chondrogenic differentiation (Table 1).83 For example, elevated miRNA-410 participates in the TGF-β3-induced chondrogenic differentiation of mesenchymal stem cells (MSCs) by binding to the 3ʹ-UTR of the Wnt3a gene, thus regulating the Wnt signaling pathway and increasing the expression of multiple chondrogenic markers, including Type II collagen, SOX9, aggrecan, and hyaluronan synthase 2.84 In contrast, miRNA-495 plays opposite roles in chondrogenic differentiation by directly binding to the SOX9 gene and downregulating the expression of Type II collagen and aggrecan.85
The basal role of TGF-β3 in the life cycle of chondrocytes
Chondrocytes are classified into articular chondrocytes located in articular cartilage and growth plate chondrocytes residing in growth plate cartilage based on the degree of terminal differentiation (Fig. 4).

Schematic diagram showing the basic structure of cartilage and the distribution of transforming growth factor-beta 3 (TGF-β3). Chondrocytes are divided into articular chondrocytes located in articular cartilage and growth plate chondrocytes residing in growth plate cartilage based on the degree of their terminal differentiation. Growth plate cartilage comprises the resting zone, proliferative zone, pre-hypertrophic zone and hypertrophic zone. TGF-β3 is mainly expressed in chondrocytes located in the proliferative and hypertrophic zones. Articular cartilage consists of five distinct zones: the lamina splendens, superficial zone, middle zone, deep zone and calcified zone. TGF-β3 is involved in chondrogenic differentiation and chondrocyte hypertrophy through the Smad2/3 pathway and Smad1/5/8 pathway, which play antagonistic roles during different stages of cartilage differentiation
The lubricated surface of articular cartilage is reversibly deformed and moves with minimal friction. The histological structure of cartilage allows it to disseminate shear, tensile, and compressive forces.86 Based on structural differences, cartilage can be divided into five unique zones: the lamina splendens, superficial zone, middle zone, deep zone, and calcified zone.87 Each zone exhibits its own histological and functional features. At the surface of articular cartilage, the lamina splendens comprise a cell-free layer. All collagen fibers in this zone are oriented parallel to the surface, which enables it to resist pressure. In addition, it contains a large amount of HA and proteoglycan 4 (PRG4), maintaining low friction and boundary lubrication.88 Below the superficial zone is underneath the lamina splendens, and it also contains high levels of HA and PRG4, with tightly packed collagen fibrils oriented parallel to the surface. In contrast to lamina splendens, however, the superficial zone contains a relatively high number of cells, which have a flattened morphology and are oriented parallel to the surface.89 The middle zone is underneath the superficial zone and accounts for 40%–60% of the total cartilage volume and comprises spherical and rare chondrocytes. Collagen fibers are oriented in oblique orientation to the cartilage surface, endowing this zone with low stress tolerance. Functionally, the middle zone is considered the first layer that resists compressive forces.87 A tidemark distinguishes the two basal zones: the deep zone and the calcified zone. Chondrocytes in the deep zone proliferate unidirectionally and are organized in columns oriented vertical to the cartilage surface and parallel to the collagen fibrils. This zone exhibits the highest proteoglycan level and is critical for providing the greatest resistance to compressive forces.90 The calcified zone, underneath the tidemark, forms the interface between bone and cartilage by anchoring the collagen fibrils in the deep zone to subchondral bone. This zone comprises hypertrophic-like chondrocytes undergoing endochondral ossification.91
Growth plate cartilage comprises the resting zone, proliferative zone, pre-hypertrophic zone, and hypertrophic zone.12,90,92 Cells in the resting zone are small, uniform, compact and chondrocytes critical for protein synthesis and exist as individual cells or in pairs. In the proliferative zone, flat chondrocytes are clearly divided into longitudinal columns and rapidly duplicate. A true germinal layer is found only at the base of columns and produces Type II and Type XI collagen.93 Underneath the proliferative zone, in the pre-hypertrophic zone and hypertrophic zone, chondrocytes terminally differentiate into pre-hypertrophic chondrocytes and hypertrophic chondrocytes, respectively, and exhibit a relatively large and swollen morphology.94 Once terminal differentiation is initiated, mature chondrocytes produce vascular endothelial growth factor to induce blood vessel invasion and matrix metalloproteinases (MMPs) to promote the degradation of the cartilage ECM by chondroclasts, contributing to the formation of a primary ossification center.95 Growth plate chondrocytes eventually undergo apoptosis and are replaced by bone cells during endochondral ossification. Correspondingly, the cartilage matrix is gradually converted into bone matrix.96,97
Chondrocytes exhibit a life cycle of proliferation, differentiation, maturation, and apoptosis.98 TGF-β3 plays crucial regulatory roles in these processes by interacting directly with TβRs on the outer cell membrane and triggering a cascade of molecular alterations involving SOX9. Then, the expression of multiple cartilage ECM proteins, such as Type II collagen and aggrecan, is further regulated.17 TGF-β3 is expressed in chondrocytes located in proliferative and hypertrophic zones. Notably, TGF-β3 is widely distributed at sites of intramembranous ossification.99 TβRI and TβRII, the two TβRs for which TGF-β3 shows strong affinity, are distributed in the hypertrophic zone and the calcified zone of the neonatal rib as well as in the resting zone and proliferative zone of a developing osteophyte.100 The variance in its distribution indicates that TGF-β3 plays a dual function.
Chondrocyte proliferation and migration
The limited repair capacity of cartilage is directly related to chondrocyte proliferation and migration into a wound site. In addition to its association with cartilage repair and regeneration, cell proliferation is important for the growth and development of cartilage. Therefore, evaluating the role played by TGF-β3 in chondrocyte proliferation may help us optimize a treatment strategy for cartilage defects. Sefat et al. reported that the proliferation rate of primary chondrocytes decreased, with more cells presenting with a fibroblastic morphology after treatment with TGF-β3.101 These variations were caused by the positive effect of TGF-β3 on chondrocyte dedifferentiation, a rapid change in phenotype and gene expression generally observed in vitro, particularly cultured chondrocyte monolayers. The inhibitory role played by TGF-β3 on cell proliferation has also been verified during the chondrogenesis of chick leg bud mesenchymal cells.102 However, Martinez-Alvernia et al. put forward a contradictory opinion by suggesting that exogenous TGF-β3 increases chondrocyte proliferation.103 James et al. also observed that TGF-β3 significantly increased cell proliferation in posterior frontal suture-derived mesenchymal cells.104 In addition, cell proliferation was promoted when chondrocytes were cocultured with adipose-derived stem cells (ADSCs) in medium supplemented with TGF-β3.105 In summary, the recent research findings are contradictory, and no unanimous conclusions can be drawn.
Nevertheless, TGF-β3 is widely used in cell-based cartilage tissue engineering owing to its other functions, such as the promotion of cell migration. Makhijani et al. reported that all three TGF-β isoforms stimulate chemotaxis in C3H10T1/2 cells.106 The migration of OP9 MSCs has also been efficiently induced by TGF-β3.107 The chemotactic role of TGF-β3 has been confirmed via in vivo experiments. After humeral head resection, rabbits were implanted with TGF-β3-infused bioscaffolds or TGF-β3-free bioscaffolds. Then, the regenerated cartilage samples were collected and analyzed, and the results revealed that compared with the number of cells that spontaneously migrated without TGF-β3 stimulation, ~130% more cells were recruited to the regenerated cartilage in the rabbits treated with TGF-β3.108 Brittberg et al. found that a suitable dose of TGF-β3 induced the migration of MSCs to cartilage lesions and the formation of hyaline-like grafts.109 With advancements in tissue engineering and regenerative medicine, some cofactors have also been applied, such as BMPs and dexamethasone, which play synergistic roles.110,111
Chondrocyte differentiation
Chondrogenic differentiation, a prerequisite for cartilage formation and endochondral ossification, begins with cell proliferation, precartilage condensation, and mesenchymal cell differentiation into chondrocytes, contributing to the formation of a cartilaginous template for future bone development.112,113 The cartilaginous template undergoes cell proliferation, hypertrophic differentiation, calcification, and apoptosis and is eventually replaced by bone.
Chondrogenic differentiation
MSCs residing in the bone marrow of adult humans retain the capacity to proliferate and differentiate into multiple cell lineages, including chondrocytes. Generally, the role played by TGF-β3 in chondrogenesis is cell type specific. Through the activation of Notch signaling, TGF-β3 plays an inhibitory role on chondrocytes in limb bud mesenchymal cells by repressing their proliferation and precartilage condensation.102 Similarly, TGF-β3 treatment inhibits chondrogenesis of chick leg bud mesenchymal cells by downregulating connexin 43 and integrin beta4 mediated by the suppression of protein kinase C-alpha (PKC-α) and the activation of ERK.114 Jin et al. demonstrated that TGF-β3 stimulated chondrogenic differentiation of wing bud mesenchymal cells through the activation of the PKC-α and p38 MAPK pathways.115 They hypothesized that the disparity is attributable to the multiple functions of TGF-β3. Specifically, TGF-β3 showed stimulated chick wing chondroblast differentiation into chondrocytes and inhibitory effects on chick leg chondroblast differentiation, thus establishing fore- and hind-limb identity.114
Despite its mixed inductive and inhibitory effects, TGF-β3 is considered to induce greater chondrogenic effects than TGF-β1, but TGF-β3 is not sufficient for promoting chondrogenic differentiation in monolayers.116,117 James et al. explained the different effects of TGF-β1 and TGF-β3 on chondrogenic differentiation in posterior frontal suture-derived mesenchymal cells.104 Notably, TGF-β1 promotes precartilage condensation and chondrocyte differentiation, while TGF-β3 may predominantly induce chondrogenesis via the significant expansion of chondroprogenitor cells. Currently, TGF-β3 is used as a standard additive that is encapsulated in a variety of scaffolds to induce the chondrogenesis of MSCs.118 The successfulness of this application has been supported by numerous in vitro, animal and human in vivo studies. For example, chondrogenic differentiation of human MSCs was induced in pellet culture after the medium was supplemented with dexamethasone, bone morphogenetic protein 6 (BMP6) and TGF-β3. This optimized medium enhanced the weight of the cartilage pellets approximately tenfold. In addition, the production of proteoglycans and the gene expression of Type II procollagen and Type X collagen were both increased.119 Sasaki et al. developed a model of meniscus radial tears and found that injection of ADSCs encapsulated in a TGF-β3-preloaded photo-cross-linkable hydrogel stimulated robust matrix-sulfated proteoglycan deposition and enhanced healing of meniscal radial tears. This outcome shows a novel strategy to repair meniscal radial tears and minimize further osteoarthritic joint degeneration.120 Similar results were reported for another in vitro experiment in which human MSCs seeded in silk-elastin-like polymer (SELP) scaffolds containing TGF-β3 promoted significant increases in glycosaminoglycan and total collagen content.121 TGF-β3 may also be delivered via direct encapsulation within HA hydrogels. TGF-β3 retained within a hydrogel altered chondrogenic gene expression of encapsulated MSCs when implanted in mice.122
Terminal hypertrophic differentiation
Most of the skeleton, such as the long bones in the limbs and vertebral columns, is formed through endochondral ossification.96 The condensation of MSCs is a prerequisite for endochondral bone formation. MSCs differentiate into chondrocytes, which are characterized by the production and accumulation of cartilage-specific proteins, such as Type II collagen and aggrecan. Apparently, unrestricted differentiation of precursor cells into the chondrocyte lineage leads to the formation of bones but not permanent cartilage, which develops only when this route is blocked in articular cartilage.91 In contrast to permanent cartilage, the temporary hyaline cartilage-like growth plate is ultimately replaced by bone. In growth plates, maturing chondrocytes produce a cartilaginous matrix and then exit the cell cycle, undergo hypertrophic differentiation, produce Type X collagen, and show alkaline phosphatase activity.123 Once hypertrophic chondrocytes have terminally differentiated, vascularization and focal calcification are initiated; finally, the cartilaginous matrix is mineralized, and the cells undergo apoptosis.95,124 Terminal hypertrophic differentiation is mainly controlled by the two transcription factors SOX9 and Runx2. SOX9 is a negative regulator of hypertrophic differentiation, cartilage angiogenesis, and bone marrow formation.125 Runx2 is a positive regulator of hypertrophic differentiation and cartilage vascularization.
The role for TGF-β3 in chondrocyte hypertrophic differentiation has been reported. Initial research revealed that the terminal differentiation of cranial chondrocytes was arrested by TGF-β3 and FGF2, which synergistically suppressed the expression of terminal differentiation markers such as Type X collagen, matrix metalloproteinase 13 (MMP13, also known as collagenase 3) and Runx2.126 However, some studies put forward different opinions, suggesting that TGF-β3 upregulates the expression of MMP13, a marker of OA cartilage.127 This regulation is mediated by the altered expression of inflammatory cytokines, such as interleukin-1 (IL-1).128 In addition, Hennig et al. observed an induced hypertrophic phenotype in ADSCs after successful chondrogenic induction via TGF-β3 and BMP6.129 Similar results showing that differentiating MSCs express hypertrophic markers under standard chondrogenic conditions have also been reported by Mueller et al;116 this group pointed out that the hypertrophy observed in an in vitro model was most significantly increased after 14 days of chondrogenic predifferentiation. Indeed, TGF-β3 not only acts as a chondro-inductive growth factor but also predisposes MSCs to hypertrophy. The dual role of TGF-β3 in the terminal hypertrophic differentiation of chondrocytes is mediated by the Smad2/3 and Smad1/5/8 pathways. TGF-β3-induced Smad2/3 signaling induced via the activation of ALK5 helps to preserve the phenotype of chondrocytes by inhibiting hypertrophy and terminal differentiation.130 In contrast, TGF-β3-induced Smad1/5/8 signaling mediated via the activation of ALK1 facilitates chondrocyte hypertrophy.131
Chondrocyte death
Dynamic and accurate regulation of cell death in chondrocytes is pivotal in skeletal development, maintenance, and repair as well as in the pathological course of OA and osteoporosis (OP).132,133 Different types of cell death in cartilage have been reported, including three types of physiological/programmed cell death (apoptosis, chondroptosis, and paraptosis) and one type of pathological cell death (necrosis).134 Apoptosis is a highly regulated, “active” cell death (as opposed to “passive”) modality associated with homeostasis and senescence.135 It is an enzyme-dependent biochemical process that causes minimal damage to the surrounding microenvironment. The initiation of apoptosis relies on the activation of a series of caspases.136 Apoptosis has been reported to be critical to the regulation of cartilage development. Mrugala et al. used DNA microarrays and performed gene expression profiling of human MSCs in the different phases of chondrogenic differentiation induced by TGF-β3/BMP2.137 They found that apoptosis-related genes were highly expressed at an early stage, which is characterized by cell attachment, and the later stage, which is characterized by chondrocyte hypertrophy. Dysregulation of apoptosis results in developmental abnormalities, degenerative diseases, and cancer. Increasing evidence has suggested that programmed cell death is an outcome of multiple modalities, not merely apoptosis, and can involve different pathways of active self-destruction, such as those in autophagic cell death.138 Autophagy is a catabolic process in all eukaryotic cells that removes waste and damaged cellular components via lysosomal degradation.139 Autophagic cell death, which may be an alternative to classical apoptosis (classified as Type I cell death), is classified as Type II cell death.138 It is mediated by lysosomal proteinases, especially cathepsins B and D, but not caspases.140 Autophagy provides the timely regulation of chondrocyte maturation and ECM formation. Chondrocyte hypertrophy may be promoted in the absence of autophagy.141 Necrosis, however, is uncontrolled cell death and usually follows severe damage and subsequently affects tissue surrounding a lesion.136
The regulatory role played TGF-β1 in chondrocyte death has been extensively explored. Downstream signaling pathways that govern this process have also been identified.133 However, studies on the interaction between TGF-β3 and chondrocyte apoptosis/necrosis/autophagy are rare and primarily phenomenological. Among those who published articles that we retrieved and analyzed, most researchers paid focused on the role played TGF-β3 in chondrocyte apoptosis. Exogenous TGF-β3 has been found to decrease the apoptosis rate during chondrogenic differentiation of chick leg bud mesenchymal cells through the suppression of integrin β4 expression, activation of ERK, and suppression of PKC-α activation.114 An in vivo study reported that TGF-β2 and TGF-β3 played roles in tissue shaping during limb formation by regulating apoptosis. The mesenchyme of interdigital spaces shows chondrogenic potential, and interdigital cells need to undergo apoptotic cell death during limb development.142 Dünker et al. found that the apoptosis rate in the interdigital spaces of forelimbs and hindlimbs was significantly reduced in TGF-β2−/−TGF-β3−/− double-knockout mice as well as in TGF-β2−/−TGF-β3+/− mutants. Moreover, large hypertrophied chondrocytes were observed within the interdigital region of the two mutants, which meant chondrogenesis had been accelerated, and an advanced stage of enchondral ossification had been reached; in contrast, wild-type mouse digits contained small proliferating chondrocytes.143 Similarly, Cheah et al. knocked down TGF-β3 in zebrafish and found that normal chondrogenesis in the craniofacial region was dependent on the tight regulation of TGF-β3. They proposed that both TGF-β3 suppression and overexpression led to reduced chondrocyte formation but to different degrees and through different mechanisms. TGF-β3 dysregulation-induced apoptosis was detected in both situations and was considered to be the common mediating factor.144 In summary, the regulatory networks and mechanisms through which TGF-β3 affects chondrocyte death still need to be elucidated through further study.
Potential therapeutic roles of TGF-β3 in osteoarthritis (OA)
Basic characteristics of OA
Of all the joint diseases, OA is the most common degenerative disorder, and it is highly associated with age, exerting extensive influence on an individual physical health, leading to disability, and on the health care system and society.11 Debilitating and chronic joint disorders mainly affect weight-bearing joints, such as hips and knee.145 Etiological factors, such as abnormal joint loading, degenerative aging, and genetic predisposition, may irreversibly contribute to cartilage degradation.146 To date, effective reparative approaches to these conditions have not been established, as the precise pathogenesis of OA remains unclear.12,86,147 The current pathophysiological concept suggests that OA as a degenerative disease of the whole joint and that all components of the diarthrodial joint involving the synovium, subchondral bone and cartilage contribute to pathologic abnormalities.148 More specifically, osteophyte formation, subchondral bone sclerosis underneath eroded cartilage, and disruption of the tidemark, which is accompanied by angiogenesis at the osteochondral junction, are the characteristics of OA. In addition, ossification at the sites of ligament, tendon, and joint capsule insertion into bone is also observed, ultimately leading to joint asymmetry and malalignment.149,150
Cartilage degeneration, characterized by progressive loss of aggrecan and Type II collagen, had been previously considered to be the central pathological feature of OA. Elevated expression levels of multiple MMPs, such as MMP2, MMP9, and MMP13, are involved in this degenerative process.151 In recent years, however, alterations in the underlying subchondral bone have garnered increasing interest. Immoderate bone proliferation contributes to an eburnated subchondral plate and a profound increase in its thickness, increasing stiffness, altering the mechanical property of subchondral bone and changing the distribution of articular cartilage stress.152 The synovium is also involved in several stages of this pathological process. In the early phase of OA, catabolic cytokines released from the synovium induce transient chondrocyte proliferation as well as increased synthesis of type II collagen and aggrecan in an attempt to initiate repair.153 Subsequent synovial inflammation, commonly attributed to abnormal molecular signaling in damaged cartilage, leads to the constant production of multiple MMPs and ultimately to greater cartilage destruction.154
Involvement of TGF-β3 in the pathology of OA
The involvement of TGF‑β3 in OA has been confirmed with human pathological clinical data. Kapetanakis et al. reported that the serum protein level of TGF-β3 was markedly higher in patients with knee OA than in controls, as determined via enzyme-linked immunosorbent assay. This increase in TGF-β3 was strongly correlated with pain, functionality, and radiographic staging in OA.155 To adequately evaluate and refine joint lesion characterization, a better understanding of the pathophysiologic mechanisms is necessary. For that purpose, we focus on the role of TGF-β3 in the context of OA pathology (Fig. 5).

Schematic diagram showing the role of transforming growth factor-beta 3 (TGF-β3) in osteoarthritic and healthy joints. In healthy articular cartilage, low concentrations of TGF-β3 promote metabolic balance through the Smad2/3 pathway. Chondrocyte autophagy also plays a protective role. TGF-β3 is also involved in many aspects of osteoarthritis (OA) pathology mediated through the Smad1/5/8 pathway. High concentrations of TGF-β3 in osteoarthritic joints induce the production of catabolic factors and chondrocyte hypertrophy, ultimately resulting in cartilage matrix degradation, osteophyte formation, and synovial fibrosis. In addition, high levels of TGF-β3 upregulate the expression of Runt-related transcription factor 2 (Runx2) via the Smad1/5/8 pathway, leading to aberrant bone remodeling and further subchondral bone sclerosis. In addition, chondrocyte switching from autophagy to apoptosis has been implicated in OA progression. IL-1 interleukin-1, MMP13 matrix metalloproteinase 13, MMP2 matrix metalloproteinase 2, MMP9 matrix metalloproteinase 9
TGF-β3-mediated Smad2/3 signaling plays antihypertrophic and anti‑inflammatory roles in young and healthy cartilage. In contrast, TGF-β3-induced Smad1/5/8 signaling is associated with pro-hypertrophic control in pathological cartilage.131,156 The activation of these two differential pathways hinges on the concentration of active TGF‑β3. A high concentration of TGF-β3 preferentially stimulates Smad1/5/8 signaling, while a low concentration predominantly stimulates Smad2/3 signaling.157 In healthy joints, low levels of active TGF‑β3 stimulate chondrocyte proliferation and induce the deposition of Type II collagen and aggrecan via the Smad2/3 signaling pathway.158 This pathway is important in the regulation of joint homeostasis and counteracts pathological chondrocyte hypertrophy in OA, thereby preserving cartilage integrity.159 High levels of TGF-β3 in OA joints upregulate the expression of Runx2 via the Smad1/5/8 pathway, which induces further production of MMP13 and leads to chondrocyte hypertrophy, ultimately resulting in ECM degradation, osteophyte formation, synovial fibrosis and chondrocyte apoptosis.160 The interaction between the TGF-β3 signaling pathway and catabolic cytokines contributes to in OA cartilage pathology. For example, IL-1β, one of the most potent cytokines triggering an irreversibly destructive cascade of articular cartilage in OA, may inhibit Smad2/3 signaling by downregulating TβRII expression and upregulating Smad7 expression.161
Increasing evidence shows an association between OA-induced cartilage degradation and chondrocyte death.162 Hwang et al. suggested that chondrocyte death and matrix degradation may form a vicious cycle, with the progression of one aggravating the other.163 Among the multiple types of cell death, apoptotic chondrocyte death is drawing considerable attention from researchers exploring the pathogenesis of OA. There is a clear correlation between the degree of cartilage degradation and the chondrocyte apoptosis rate.164 Whether chondrocyte apoptosis leads to OA or cartilage degeneration leads to chondrocyte apoptosis has been widely debated, and the cause-and-effect relationship between OA and chondrocyte apoptosis has been difficult to clarify. Autophagy, leading to another form of chondrocyte death, is changed in OA. A switch from chondrocyte autophagy to apoptosis has been implicated in OA progression. Since autophagy inhibits inflammation and reduces chondrocyte apoptosis in OA, promoting chondrocyte autophagy and inhibiting apoptosis may become a therapeutic strategy to cure OA.165 In summary, we have mentioned explained that TGF-β3 is involved in the regulation of chondrocyte apoptosis and that both TGF-β3 and chondrocyte death are associated with OA pathogenesis. Therefore, it is reasonable to consider that there might be a relationship between TGF-β3 and chondrocyte death in OA, but we did not find an article discussing this possible interaction.
The application of TGF-β3 in cartilage tissue engineering
Conventional treatments for OA only relieve symptoms and do not prevent or cure the disease. Nonsurgical treatments include physical therapy and CS supplementation. Some surgical treatments, such as abrasive methods (microfracturing by drilling small holes), are expected to lead to partial recovery of the articular cartilage structure through the migration of progenitor cells derived from the bone marrow to the lesion site following subchondral fracturing.166 Although it shows some shortcomings, the technology is used because it works quickly. Autologous chondrocyte implantation (ACI) is an alternative treatment that involves the collection, cultivation, and expansion of autologous chondrocytes. Preliminary cell modification has optimized the technology, but the long-term effects of this treatment method remain controversial.167 With the development of tissue engineering and regenerative medicine, the era of cell-based tissue engineered constructs (CTECs) used to repair cartilage damage has been initiated. Pretreated MSCs are placed on a biodegradable scaffold, and growth factors are added before implantation into defective cartilage.168 The primary goal of CTEC is to obtain cells in culture that can proliferate and form transparent grafts without causing degradation. This technique significantly reduces the early migration of cells from the defect site because biodegradable scaffolds protect cells from heavy mechanical loads. In addition, the 3D scaffold prolongs the release time of the chondrogenesis activator by ensuring a uniform distribution of stem cells throughout the scaffold.169 Some growth factors that regulate chondrogenesis and help to restore damaged cartilage surfaces via gene modulation have recently attracted attention; one of these factors is TGF-β3. Herein, we summarize thirteen papers to demonstrate the application of TGF-β3 using different delivery scaffolds in cartilage tissue engineering (Table 2).
Biodegradable scaffolds with immobilized TGF-β3 are effective for constructing tissue-engineered cartilage. Suitable doses170 and scaffold carriers171 of TGF-β3 are important factors influencing the efficacy of the chondrogenic stimulation. Previous in vitro evidence has suggested that TGF-β3 at concentrations of 10, 20, and 60 ng·mL−1 exerted a dose- and time-dependent effect on the gene expression of chondrogenic markers in human MSCs.111 However, high doses of TGF-β (>900 ng·mL−1) may lead to undesired side effects such as synovitis, pannus formation, and cartilage erosion.172 Another in vivo study on a large animal model verified the effect of TGF-β3 and provided a reference for its suitable dose. Two hundred milliliters of blood from the jugular vein of sheep in a 25 mL suspension containing chitosan (45 mg), TGF-β3 (50 ng), and autologous ovine MSCs isolated from bone marrow to form a fibrin clot was delivered. Then, the clot was used to fill partial-thickness defects in the inner region of the patella. After 9 weeks, histological tests indicated the presence of chondrocyte‑like cells surrounded by a hyaline-like cartilaginous matrix that had wholly integrated into native cartilage.173 Notably, the repair results were greatly influenced by the mode of delivery. A variety of scaffolds have been explored and refined with the aim of providing compatible and optimal conditions to better mimic the natural microenvironment, which is beneficial to direct chondrogenesis. These biomaterials include poly-(lactic-co-glycolic acid) (PLGA), various hydrogels, atelocollagen, alginate beads, poly-glycolic acid (PGA), and poly-L-lactic acid (PLA).63 Synthetic polymers, such as PLGA, PGA and PLA, have distinct advantages in terms of shape manipulation, surface morphology, and biomechanical and biochemical properties.174 However, most of these polymers induce inflammatory responses in vivo.175 Therefore, researchers have adopted natural polymers to modify the structure of the previously used synthetic polymers. For example, Fan et al. fabricated a TGF-β3-immobilized PLGA-gelatin/chondroitin sulfate/hyaluronic acid (PLGA-GCH) hybrid scaffold and confirmed its great potential for use in cartilage tissue engineering. After implantation of an MSC-seeded PLGA-GCH scaffold crosslinked with TGF-β3, impaired cartilage and subchondral bone were successfully regenerated. Histological observation showed that chondrocyte-like cells were uniformly located within the regenerated matrix, which showed extensive metachromatic staining. Advantages of the scaffold included reduced preimplantation culture time and decreased TGF-β3 consumption.176 Recently, emerging evidence has focused considerable interest on the use of natural silk-derived materials owing to their unique structure, biomechanical and biochemical properties, and biocompatible features.177 Human MSCs were encapsulated in a 3D aqueous-derived silk scaffold and cultured in chondrogenic medium supplemented with TGF-β3 and dexamethasone. After 3 weeks, the cell arrangement and Type II collagen distribution in the constructs were similar to those of native cartilage tissues.178 Another similar in vivo study supported this finding. Haider et al. used genetically engineered SELP-47K in an injectable scaffold for the delivery of human MSCs cultured with or without TGF-β3.121 The constructs exhibited a significant increase in the content of sulfated glycosaminoglycan and total collagen, which were increased by up to 65% and 300%, respectively, with the addition of TGF-β3. With the development of material scaffolds via tissue engineering, new scaffolds based on magnetic nanoparticles crosslinked with methacrylated gelatin (GelMA), combined with slow-release TGF-β3, may be a new method to repair cartilage.179
Other growth factors administered with TGF-β3 can lead to synergistic chondrogenesis. For example, scaffolds loaded with TGF-β3 and bone morphogenetic protein 7 (BMP7) provided a better environment for cartilage regeneration than scaffolds loaded with either TGF-β3 or BMP7 alone.180 The recruitment of chondrocytes cultured with both mechano growth factor (MGF) and TGF-β3 increased significantly, by 1.8- and 2-fold in vivo and in vitro, respectively, compared with that of cells cultured with TGF-β3 alone.181 TGF-β3 and BMP6 are both potent chondrogenic growth factors, and their ability to induce chondrogenesis was enhanced when combined. Several studies found synergy in the chondrogenic differentiation of ADSCs, bone marrow-derived mesenchymal stem cells (BMSCs), and periodontal ligament stem cells.182,183,184,185,186,187 In addition, Yoo et al. put forward an innovative idea, suggesting that a combination of TGF‑β3 and BMP6, which shows a synergistic effect on chondrogenesis, loaded into an efficient carrier such as exosomes, which themselves show a chondroprotective function, offers an effective approach to treat damaged cartilage in OA.13 In addition to growth factors, agents such as heparin and HA enhance chondrogenesis when used in combination with TGF-β3.188 The synergy between HA and TGF-β3 was reported in a study in which thermoreversible hydrogels containing TGF-β3, HA and chondrocytes were implanted into mice. The mice treated with both TGF-β3 and HA exhibited a twofold increase in the production of glycosaminoglycan and collagen compared with mice treated with hydrogels alone.189 External factors such as the mechanical load capacity and stiffness of the substrate contribute to the regulation of TGF-β3.190,191,192
Conclusion and perspective
Three TGF-β isoforms regulate many aspects of growth and development in a cell-type-specific manner. Although they play distinct biological roles, TGF-β3 isoforms have received particular attention because of their ability to regulate cartilage physiology and pathology. TGF-β3 is a pleiotropic cytokine involved in the whole life cycle of chondrocytes, including their proliferation, migration, differentiation, and death. Even though TGF-β3 exerts mixed inductive and inhibitory effects, TGF-β3 is considered to be show greater pro-chondrogenic effects than TGF-β1. Regulatory TGF-β3-mediated Smad2/3 and Smad1/5/8 signaling plays roles in the pathogenesis of OA. Stem cell-based tissue engineering is a relatively novel strategy to repair OA-induced cartilage lesions. TGF‑β3 is a promising therapeutic target for OA owing to its potential to regulate chondrogenesis and restore damaged cartilage. Considering recent results of various in vitro and in vivo experiments, we conclude that an optimal scaffold carrier containing stem cells and a suitable dose of TGF-β3 may form hyaline-like cartilaginous constructs that are entirely integrated into native cartilage, providing a novel approach to slowing the progression of OA.
Although considerable progress has been made in the exploration into the role of TGF-β3 in cartilage pathophysiology, some problems have yet to be solved. First, combinations of multiple growth factors/agents and scaffolds needs to be optimized. In addition, although a preclinical model has been identified, future investigations based on animal models are needed to refine this strategy, which seems a reasonable approach, and must be performed before clinical application. Furthermore, the possible complications and side effects of TGF-β3 supplementation should not be ignored. For example, high doses of TGF-β3 may lead to undesired problems such as fibrosis, synovitis, pannus formation, cartilage erosion, and osteophyte formation in cartilaginous and noncartilaginous tissues. In addition to faster and more prolific chondrogenic differentiation, supplementation with TGF-β3 is also associated with a higher susceptibility to chondrocyte hypertrophy, which may ultimately result in chondrocyte apoptosis, vascular invasion, and ossification of new tissue. Therefore, strategies to prevent hypertrophic differentiation and generate stable hyaline cartilage are absolutely necessary to cartilage tissue engineering.
References
Lafyatis, R. Transforming growth factor β–at the centre of systemic sclerosis. Nat. Rev. Rheumatol. 10, 706–719 (2014).
Pickup, M., Novitskiy, S. & Moses, H. L. The roles of TGFβ in the tumour microenvironment. Nat. Rev. Cancer 13, 788–799 (2013).
Occleston, N. L., Laverty, H. G., O’Kane, S. & Ferguson, M. W. J. Prevention and reduction of scarring in the skin by Transforming Growth Factor beta 3 (TGF beta 3): from laboratory discovery to clinical pharmaceutical. J. Biomater. Sci. Polym. Ed. 19, 1047–1063 (2008).
Moses, H. L., Roberts, A. B. & Derynck, R. The discovery and early days of TGF-beta: a historical perspective. Cold Spring Harb. Perspect. Biol. 8, a021865 (2016).
Grimaud, E., Heymann, D. & Redini, F. Recent advances in TGF-beta effects on chondrocyte metabolism. Potential therapeutic roles of TGF-beta in cartilage disorders. Cytokine Growth Factor Rev. 13, 241–257 (2002).
Moutos, F. T., Freed, L. E. & Guilak, F. A biomimetic three-dimensional woven composite scaffold for functional tissue engineering of cartilage. Nat. Mater. 6, 162–167 (2007).
Shoulders, M. D. & Raines, R. T. Collagen structure and stability. Annu. Rev. Biochem. 78, 929–958 (2009).
Kiani, C., Chen, L., Wu, Y. J., Yee, A. J. & Yang, B. B. Structure and function of aggrecan. Cell Res. 12, 19–32 (2002).
Cai, L. et al. Biomaterial stiffness guides cross-talk between chondrocytes: implications for a novel cellular response in cartilage tissue engineering. ACS Biomater. Sci. Eng. 6, 4476–4489 (2020).
Wei, J. et al. Osteoblasts induce glucose-derived ATP perturbations in chondrocytes through noncontact communication. Acta Biochim. Biophys. Sin. (Shanghai). 54, 625–636 (2022).
Hootman, J. M. & Helmick, C. G. Projections of US prevalence of arthritis and associated activity limitations. Arthritis Rheum. 54, 226–229 (2006).
Xie, J., Zhang, D., Lin, Y., Yuan, Q. & Zhou, X. Anterior cruciate ligament transection-induced cellular and extracellular events in menisci: implications for osteoarthritis. Am. J. Sports Med. 46, 1185–1198 (2018).
Yoo, K. H. et al. Transforming growth factor-beta family and stem cell-derived exosome therapeutic treatment in osteoarthritis (Review). Int. J. Mol. Med. 49, 62 (2022).
Wang, M. K. et al. Different roles of TGF-beta in the multi-lineage differentiation of stem cells. World J. Stem Cells 4, 28–34 (2012).
Wrana, J. L. & Attisano, L. The Smad pathway. Cytokine Growth Factor Rev. 11, 5–13 (2000).
Thielen, N. G. M., van der Kraan, P. M. & van Caam, A. P. M. TGFbeta/BMP signaling pathway in cartilage homeostasis. Cells 8, 969 (2019).
Jin, K. et al. TGF-β1-induced RAP2 regulates invasion in pancreatic cancer. Acta Biochim. Biophys. Sin. (Shanghai). 54, 361–369 (2022).
de Larco, J. E. & Todaro, G. J. Growth factors from murine sarcoma virus-transformed cells. Proc. Natl Acad. Sci. USA 75, 4001–4005 (1978).
Roberts, A. B., Anzano, M. A., Lamb, L. C., Smith, J. M. & Sporn, M. B. New class of transforming growth factors potentiated by epidermal growth factor: isolation from non-neoplastic tissues. Proc. Natl Acad. Sci. USA 78, 5339–5343 (1981).
Anzano, M. A. et al. Synergistic interaction of two classes of transforming growth factors from murine sarcoma cells. Cancer Res. 42, 4776–4778 (1982).
Herpin, A., Lelong, C. & Favrel, P. Transforming growth factor-beta-related proteins: an ancestral and widespread superfamily of cytokines in metazoans. Dev. Comp. Immunol. 28, 461–485 (2004).
Okamura, T., Yamamoto, K. & Fujio, K. Early growth response gene 2-expressing CD4(+)LAG3(+) regulatory T cells: the therapeutic potential for treating autoimmune diseases. Front. Immunol. 9, 340 (2018).
Annes, J. P., Munger, J. S. & Rifkin, D. B. Making sense of latent TGFbeta activation. J. Cell Sci. 116, 217–224 (2003).
Okamura, T. et al. Role of TGF-beta3 in the regulation of immune responses. Clin. Exp. Rheumatol. 33, S63–S69 (2015).
Chaudhry, S. S. et al. Fibrillin-1 regulates the bioavailability of TGFbeta1. J. Cell Biol. 176, 355–367 (2007).
Ge, G. & Greenspan, D. S. BMP1 controls TGFbeta1 activation via cleavage of latent TGFbeta-binding protein. J. Cell Biol. 175, 111–120 (2006).
Sengle, G., Ono, R. N., Sasaki, T. & Sakai, L. Y. Prodomains of transforming growth factor beta (TGFbeta) superfamily members specify different functions: extracellular matrix interactions and growth factor bioavailability. J. Biol. Chem. 286, 5087–5099 (2011).
Koli, K., Myllarniemi, M., Keski-Oja, J. & Kinnula, V. L. Transforming growth factor-beta activation in the lung: focus on fibrosis and reactive oxygen species. Antioxid. Redox Signal. 10, 333–342 (2008).
Shi, M. et al. Latent TGF-beta structure and activation. Nature 474, 343–349 (2011).
Annes, J. P., Chen, Y., Munger, J. S. & Rifkin, D. B. Integrin alphaVbeta6-mediated activation of latent TGF-beta requires the latent TGF-beta binding protein-1. J. Cell Biol. 165, 723–734 (2004).
Wipff, P. J. & Hinz, B. Integrins and the activation of latent transforming growth factor beta1 - an intimate relationship. Eur. J. Cell Biol. 87, 601–615 (2008).
Wang, J. et al. Atypical interactions of integrin alphaVbeta8 with pro-TGF-beta1. Proc. Natl. Acad. Sci. USA 114, E4168–E4174 (2017).
Annes, J. P., Rifkin, D. B. & Munger, J. S. The integrin alphaVbeta6 binds and activates latent TGFbeta3. FEBS Lett. 511, 65–68 (2002).
Cordeiro, M. F. Beyond Mitomycin: TGF-beta and wound healing. Prog. Retin. Eye Res. 21, 75–89 (2002).
Yoshinaga, K. et al. Perturbation of transforming growth factor (TGF)-beta1 association with latent TGF-beta binding protein yields inflammation and tumors. Proc. Natl. Acad. Sci. USA 105, 18758–18763 (2008).
Robertson, I. B. et al. Latent TGF-beta-binding proteins. Matrix Biol. 47, 44–53 (2015).
Saharinen, J. & Keski-Oja, J. Specific sequence motif of 8-Cys repeats of TGF-beta binding proteins, LTBPs, creates a hydrophobic interaction surface for binding of small latent TGF-beta. Mol. Biol. Cell 11, 2691–2704 (2000).
Rifkin, D. B., Rifkin, W. J. & Zilberberg, L. LTBPs in biology and medicine: LTBP diseases. Matrix Biol. 71-72, 90–99 (2018).
Zuo, W. et al. c-Cbl-mediated neddylation antagonizes ubiquitination and degradation of the TGF-beta type II receptor. Mol. Cell 49, 499–510 (2013).
Atfi, A. et al. The disintegrin and metalloproteinase ADAM12 contributes to TGF-beta signaling through interaction with the type II receptor. J. Cell Biol. 178, 201–208 (2007).
Kang, J. S., Liu, C. & Derynck, R. New regulatory mechanisms of TGF-beta receptor function. Trends Cell Biol. 19, 385–394 (2009).
Imamura, T., Oshima, Y. & Hikita, A. Regulation of TGF-beta family signalling by ubiquitination and deubiquitination. J. Biochem. 154, 481–489 (2013).
Hinck, A. P. Structural studies of the TGF-betas and their receptors - insights into evolution of the TGF-beta superfamily. FEBS Lett. 586, 1860–1870 (2012).
Goumans, M. J., Liu, Z. & ten Dijke, P. TGF-beta signaling in vascular biology and dysfunction. Cell Res. 19, 116–127 (2009).
Mitchell, E. J., Fitz-Gibbon, L. & O’Connor-McCourt, M. D. Subtypes of betaglycan and of type I and type II transforming growth factor-beta (TGF-beta) receptors with different affinities for TGF-beta 1 and TGF-beta 2 are exhibited by human placental trophoblast cells. J. Cell. Physiol. 150, 334–343 (1992).
Huang, T. et al. TGF-beta signalling is mediated by two autonomously functioning TbetaRI:TbetaRII pairs. EMBO J. 30, 1263–1276 (2011).
Tzavlaki, K. & Moustakas, A. TGF-beta signaling. Biomolecules 10, 487 (2020).
Derynck, R. & Zhang, Y. E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 425, 577–584 (2003).
Feng, X. H. & Derynck, R. Specificity and versatility in tgf-beta signaling through Smads. Annu. Rev. Cell Dev. Biol. 21, 659–693 (2005).
Gaarenstroom, T. & Hill, C. S. TGF-beta signaling to chromatin: how Smads regulate transcription during self-renewal and differentiation. Semin. Cell Dev. Biol. 32, 107–118 (2014).
Makkar, P., Metpally, R. P., Sangadala, S. & Reddy, B. V. Modeling and analysis of MH1 domain of Smads and their interaction with promoter DNA sequence motif. J. Mol. Graph. Model. 27, 803–812 (2009).
MacFarlane, E. G., Haupt, J., Dietz, H. C. & Shore, E. M. TGF-beta family signaling in connective tissue and skeletal diseases. Cold Spring Harb. Perspect. Biol. 9, a022269 (2017).
Derynck, R. & Budi, E. H. Specificity, versatility, and control of TGF-beta family signaling. Sci. Signal. 12, eaav5183 (2019).
Yano, M. et al. Smad7 inhibits differentiation and mineralization of mouse osteoblastic cells. Endocr. J. 59, 653–662 (2012).
Inman, G. J. Linking Smads and transcriptional activation. Biochem. J. 386, e1–e3 (2005).
Furumatsu, T., Tsuda, M., Taniguchi, N., Tajima, Y. & Asahara, H. Smad3 induces chondrogenesis through the activation of SOX9 via CREB-binding protein/p300 recruitment. J. Biol. Chem. 280, 8343–8350 (2005).
Inman, G. J., Nicolas, F. J. & Hill, C. S. Nucleocytoplasmic shuttling of Smads 2, 3, and 4 permits sensing of TGF-beta receptor activity. Mol. Cell 10, 283–294 (2002).
Massague, J., Seoane, J. & Wotton, D. Smad transcription factors. Genes Dev. 19, 2783–2810 (2005).
Hill, C. S. Transcriptional control by the SMADs. Cold Spring Harb. Perspect. Biol. 8, a022079 (2016).
Lee, M. K. et al. TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 26, 3957–3967 (2007).
Johnson, G. L. & Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 298, 1911–1912 (2002).
Yamashita, M. et al. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta. Mol. Cell 31, 918–924 (2008).
Tang, Q. O. et al. TGF-beta3: A potential biological therapy for enhancing chondrogenesis. Expert Opin. Biol. Ther. 9, 689–701 (2009).
Wang, Q. et al. The involvement of the ERK-MAPK pathway in TGF-β1-mediated connexin43-gap junction formation in chondrocytes. Connect. Tissue Res. 60, 477–486 (2019).
Yang, Y. et al. Lipid metabolism in cartilage and its diseases: a concise review of the research progress. Acta Biochim. Biophys. Sin. (Shanghai). 53, 517–527 (2021).
Tew, S. R. & Hardingham, T. E. Regulation of SOX9 mRNA in human articular chondrocytes involving p38 MAPK activation and mRNA stabilization. J. Biol. Chem. 281, 39471–39479 (2006).
Kim, S. J. et al. ERK-1/2 and p38 kinase oppositely regulate nitric oxide-induced apoptosis of chondrocytes in association with p53, caspase-3, and differentiation status. J. Biol. Chem. 277, 1332–1339 (2002).
Yoon, Y. M. et al. Maintenance of differentiated phenotype of articular chondrocytes by protein kinase C and extracellular signal-regulated protein kinase. J. Biol. Chem. 277, 8412–8420 (2002).
Rosenzweig, D. H., Ou, S. J. & Quinn, T. M. P38 mitogen-activated protein kinase promotes dedifferentiation of primary articular chondrocytes in monolayer culture. J. Cell. Mol. Med. 17, 508–517 (2013).
Hirose, N. et al. Protective effects of cilengitide on inflammation in chondrocytes under excessive mechanical stress. Cell Biol. Int. 44, 966–974 (2020).
Qiao, B., Padilla, S. R. & Benya, P. D. Transforming growth factor (TGF)-beta-activated kinase 1 mimics and mediates TGF-beta-induced stimulation of type II collagen synthesis in chondrocytes independent of Col2a1 transcription and Smad3 signaling. J. Biol. Chem. 280, 17562–17571 (2005).
Gunnell, L. M. et al. TAK1 regulates cartilage and joint development via the MAPK and BMP signaling pathways. J. Bone Miner. Res. 25, 1784–1797 (2010).
Shim, J. H. et al. TAK1 is an essential regulator of BMP signalling in cartilage. EMBO J. 28, 2028–2041 (2009).
Wang, Q. et al. TGF-β1 promotes gap junctions formation in chondrocytes via Smad3/Smad4 signalling. Cell Prolif. 52, e12544 (2019).
Zhang, Y. et al. Cartilage-specific deletion of mTOR upregulates autophagy and protects mice from osteoarthritis. Ann. Rheum. Dis. 74, 1432–1440 (2015).
Liu, Y. et al. PDGF-AA promotes cell-to-cell communication in osteocytes through PI3K/Akt signaling pathway. Acta Biochim. Biophys. Sin. (Shanghai). 53, 1640–1649 (2021).
Woods, A., Wang, G. & Beier, F. RhoA/ROCK signaling regulates Sox9 expression and actin organization during chondrogenesis. J. Biol. Chem. 280, 11626–11634 (2005).
Woods, A. & Beier, F. RhoA/ROCK signaling regulates chondrogenesis in a context-dependent manner. J. Biol. Chem. 281, 13134–13140 (2006).
Haudenschild, D. R., Chen, J., Pang, N., Lotz, M. K. & D’Lima, D. D. Rho kinase-dependent activation of SOX9 in chondrocytes. Arthritis Rheum. 62, 191–200 (2010).
Javelaud, D. & Mauviel, A. Crosstalk mechanisms between the mitogen-activated protein kinase pathways and Smad signaling downstream of TGF-beta: implications for carcinogenesis. Oncogene 24, 5742–5750 (2005).
Hu, B. et al. TGF-beta induces up-regulation of chondroitin sulfate synthase 1 (CHSY1) in nucleus pulposus cells through MAPK signaling. Cell. Physiol. Biochem. 37, 793–804 (2015).
Ratti, M. et al. MicroRNAs (miRNAs) and long non-coding RNAs (lncRNAs) as new tools for cancer therapy: first steps from bench to bedside. Target. Oncol. 15, 261–278 (2020).
Zhang, M. et al. miR-199b-5p promoted chondrogenic differentiation of C3H10T1/2 cells by regulating JAG1. J. Tissue Eng. Regen. Med. 14, 1618–1629 (2020).
Zhang, Y., Huang, X. & Yuan, Y. MicroRNA-410 promotes chondrogenic differentiation of human bone marrow mesenchymal stem cells through down-regulating Wnt3a. Am. J. Transl. Res. 9, 136–145 (2017).
Lee, S. et al. microRNA-495 inhibits chondrogenic differentiation in human mesenchymal stem cells by targeting Sox9. Stem Cells Dev. 23, 1798–1808 (2014).
Zhou, C. et al. Runx1 protects against the pathological progression of osteoarthritis. Bone Res. 9, 50 (2021).
Duan, M., Wang, Q., Liu, Y. & Xie, J. The role of TGF-β2 in cartilage development and diseases. Bone Jt. Res. 10, 474–487 (2021).
Majd, S. E. et al. Both hyaluronan and collagen type II keep proteoglycan 4 (lubricin) at the cartilage surface in a condition that provides low friction during boundary lubrication. Langmuir 30, 14566–14572 (2014).
Rhee, D. K. et al. The secreted glycoprotein lubricin protects cartilage surfaces and inhibits synovial cell overgrowth. J. Clin. Investig. 115, 622–631 (2005).
Chen, H., Li, J., Zhang, D., Zhou, X. & Xie, J. Role of the fibroblast growth factor 19 in the skeletal system. Life Sci. 265, 118804 (2021).
van der Kraan, P. M. & van den Berg, W. B. Chondrocyte hypertrophy and osteoarthritis: role in initiation and progression of cartilage degeneration? Osteoarthr. Cartil. 20, 223–232 (2012).
Li, J., Chen, H., Zhang, D., Xie, J. & Zhou, X. The role of stromal cell-derived factor 1 on cartilage development and disease. Osteoarthr. Cartil. 29, 313–322 (2021).
Melrose, J., Smith, S. M., Smith, M. M. & Little, C. B. The use of Histochoice for histological examination of articular and growth plate cartilages, intervertebral disc and meniscus. Biotech. Histochem. 83, 47–53 (2008).
Burdan, F. et al. Morphology and physiology of the epiphyseal growth plate. Folia Histochem. Cytobiol. 47, 5–16 (2009).
Michigami, T. Regulatory mechanisms for the development of growth plate cartilage. Cell. Mol. Life Sci. 70, 4213–4221 (2013).
Kronenberg, H. M. Developmental regulation of the growth plate. Nature 423, 332–336 (2003).
Siebler, T., Shalet, S. M. & Robson, H. Effects of chemotherapy on bone metabolism and skeletal growth. Horm. Res. 58, 80–85 (2002).
Shum, L. & Nuckolls, G. The life cycle of chondrocytes in the developing skeleton. Arthritis Res. 4, 94–106 (2002).
Horner, A. et al. Expression and distribution of transforming growth factor-beta isoforms and their signaling receptors in growing human bone. Bone 23, 95–102 (1998).
Baardsnes, J., Hinck, C. S., Hinck, A. P. & O’Connor-McCourt, M. D. TbetaR-II discriminates the high- and low-affinity TGF-beta isoforms via two hydrogen-bonded ion pairs. Biochemistry 48, 2146–2155 (2009).
Sefat, F., Youseffi, M., Khaghani, S. A., Soon, C. F. & Javid, F. Effect of transforming growth factor-beta3 on mono and multilayer chondrocytes. Cytokine 83, 118–126 (2016).
Jin, E. J., Choi, Y. A., Sonn, J. K. & Kang, S. S. Suppression of ADAM 10-induced Delta-1 shedding inhibits cell proliferation during the chondro-inhibitory action of TGF-beta3. Mol. Cells 24, 139–147 (2007).
Martinez-Alvernia, E. A., Rudnick, J. A. & Mankarious, L. A. Transforming growth factor beta3 increases chondrocyte proliferation and decreases apoptosis in murine cricoid cartilage in vitro. Otolaryngol. Head. Neck Surg. 138, 435–440 (2008).
James, A. W., Xu, Y., Lee, J. K., Wang, R. & Longaker, M. T. Differential effects of TGF-beta1 and TGF-beta3 on chondrogenesis in posterofrontal cranial suture-derived mesenchymal cells in vitro. Plast. Reconstr. Surg. 123, 31–43 (2009).
Ma, X. F. et al. Co-culture of adipose-derived stem cells and chondrocytes with transforming growth factor-beta 3 promotes chondrogenic differentiation. J. Craniofac. Surg. 31, 2355–2359 (2020).
Makhijani, N. S., Bischoff, D. S. & Yamaguchi, D. T. Regulation of proliferation and migration in retinoic acid treated C3H10T1/2 cells by TGF-beta isoforms. J. Cell. Physiol. 202, 304–313 (2005).
Gao, J. et al. Characterization of OP9 as authentic mesenchymal stem cell line. J. Genet. Genom. 37, 475–482 (2010).
Lee, C. H. et al. Regeneration of the articular surface of the rabbit synovial joint by cell homing: a proof of concept study. Lancet 376, 440–448 (2010).
Brittberg, M. et al. Treatment of deep cartilage defects in the knee with autologous chondrocyte transplantation. N. Engl. J. Med. 331, 889–895 (1994).
Sekiya, I. et al. Dexamethasone enhances SOX9 expression in chondrocytes. J. Endocrinol. 169, 573–579 (2001).
Park, J. S., Yang, H. N., Woo, D. G., Jeon, S. Y. & Park, K. H. SOX9 gene plus heparinized TGF-beta 3 coated dexamethasone loaded PLGA microspheres for inducement of chondrogenesis of hMSCs. Biomaterials 33, 7151–7163 (2012).
Knudson, C. B. & Knudson, W. Cartilage proteoglycans. Semin. Cell Dev. Biol. 12, 69–78 (2001).
DeLise, A. M., Fischer, L. & Tuan, R. S. Cellular interactions and signaling in cartilage development. Osteoarthr. Cartil. 8, 309–334 (2000).
Jin, E. J., Lee, S. Y., Jung, J. C., Bang, O. S. & Kang, S. S. TGF-beta3 inhibits chondrogenesis of cultured chick leg bud mesenchymal cells via downregulation of connexin 43 and integrin beta4. J. Cell. Physiol. 214, 345–353 (2008).
Jin, E. J. et al. Wnt-5a is involved in TGF-beta3-stimulated chondrogenic differentiation of chick wing bud mesenchymal cells. Int. J. Biochem. Cell Biol. 38, 183–195 (2006).
Mueller, M. B. et al. Hypertrophy in mesenchymal stem cell chondrogenesis: effect of TGF-beta isoforms and chondrogenic conditioning. Cells Tissues Organs 192, 158–166 (2010).
Pittenger, M. F. et al. Multilineage potential of adult human mesenchymal stem cells. Science 284, 143–147 (1999).
Mehlhorn, A. T. et al. Mesenchymal stem cells maintain TGF-beta-mediated chondrogenic phenotype in alginate bead culture. Tissue Eng. 12, 1393–1403 (2006).
Sekiya, I., Colter, D. C. & Prockop, D. J. BMP-6 enhances chondrogenesis in a subpopulation of human marrow stromal cells. Biochem. Biophys. Res. Commun. 284, 411–418 (2001).
Sasaki, H. et al. In vitro repair of meniscal radial tear with hydrogels seeded with adipose stem cells and TGF-beta3. Am. J. Sports Med. 46, 2402–2413 (2018).
Haider, M., Cappello, J., Ghandehari, H. & Leong, K. W. In vitro chondrogenesis of mesenchymal stem cells in recombinant silk-elastinlike hydrogels. Pharm. Res. 25, 692–699 (2008).
Chung, C. & Burdick, J. A. Influence of three-dimensional hyaluronic acid microenvironments on mesenchymal stem cell chondrogenesis. Tissue Eng. Part A 15, 243–254 (2009).
Beier, F. Cell-cycle control and the cartilage growth plate. J. Cell. Physiol. 202, 1–8 (2005).
Li, B., Guan, G., Mei, L., Jiao, K. & Li, H. Pathological mechanism of chondrocytes and the surrounding environment during osteoarthritis of temporomandibular joint. J. Cell. Mol. Med. 25, 4902–4911 (2021).
Hattori, T. et al. SOX9 is a major negative regulator of cartilage vascularization, bone marrow formation and endochondral ossification. Development 137, 901–911 (2010).
Bohme, K., Winterhalter, K. H. & Bruckner, P. Terminal differentiation of chondrocytes in culture is a spontaneous process and is arrested by transforming growth factor-beta 2 and basic fibroblast growth factor in synergy. Exp. Cell Res. 216, 191–198 (1995).
Moldovan, F., Pelletier, J. P., Hambor, J., Cloutier, J. M. & Martel-Pelletier, J. Collagenase-3 (matrix metalloprotease 13) is preferentially localized in the deep layer of human arthritic cartilage in situ: in vitro mimicking effect by transforming growth factor beta. Arthritis Rheum. 40, 1653–1661 (1997).
Wilson, S. E. Interleukin-1 and transforming growth factor beta: commonly opposing, but sometimes supporting, master regulators of the corneal wound healing response to injury. Invest. Ophthalmol. Vis. Sci. 62, 8 (2021).
Hennig, T. et al. Reduced chondrogenic potential of adipose tissue derived stromal cells correlates with an altered TGFbeta receptor and BMP profile and is overcome by BMP-6. J. Cell. Physiol. 211, 682–691 (2007).
Yang, X. et al. TGF-beta/Smad3 signals repress chondrocyte hypertrophic differentiation and are required for maintaining articular cartilage. J. Cell Biol. 153, 35–46 (2001).
Blaney Davidson, E. N. et al. Increase in ALK1/ALK5 ratio as a cause for elevated MMP-13 expression in osteoarthritis in humans and mice. J. Immunol. 182, 7937–7945 (2009).
Komori, T. Cell death in chondrocytes, osteoblasts, and osteocytes. Int. J. Mol. Sci. 17, 2045 (2016).
Zhang, D. et al. Osteoporosis-decreased extracellular matrix stiffness impairs connexin 43-mediated gap junction intercellular communication in osteocytes. Acta Biochim. Biophys. Sin. (Shanghai). 52, 517–526 (2020).
Roach, H. I., Aigner, T. & Kouri, J. B. Chondroptosis: a variant of apoptotic cell death in chondrocytes? Apoptosis 9, 265–277 (2004).
Green, D. R. The coming decade of cell death research: five riddles. Cell 177, 1094–1107 (2019).
Elmore, S. Apoptosis: a review of programmed cell death. Toxicol. Pathol. 35, 495–516 (2007).
Mrugala, D. et al. Gene expression profile of multipotent mesenchymal stromal cells: Identification of pathways common to TGFbeta3/BMP2-induced chondrogenesis. Cloning Stem Cells 11, 61–76 (2009).
Bursch, W. The autophagosomal-lysosomal compartment in programmed cell death. Cell Death Differ. 8, 569–581 (2001).
Reggiori, F. & Klionsky, D. J. Autophagy in the eukaryotic cell. Eukaryot. Cell 1, 11–21 (2002).
Lockshin, R. A. & Zakeri, Z. Caspase-independent cell deaths. Curr. Opin. Cell Biol. 14, 727–733 (2002).
Ogier-Denis, E. & Codogno, P. Autophagy: a barrier or an adaptive response to cancer. Biochim. Biophys. Acta 1603, 113–128 (2003).
Garcia-Martinez, V. et al. Internucleosomal DNA fragmentation and programmed cell death (apoptosis) in the interdigital tissue of the embryonic chick leg bud. J. Cell Sci. 106, 201–208 (1993).
Dunker, N., Schmitt, K. & Krieglstein, K. TGF-beta is required for programmed cell death in interdigital webs of the developing mouse limb. Mech. Dev. 113, 111–120 (2002).
Cheah, F. S., Winkler, C., Jabs, E. W. & Chong, S. S. Tgfbeta3 regulation of chondrogenesis and osteogenesis in zebrafish is mediated through formation and survival of a subpopulation of the cranial neural crest. Mech. Dev. 127, 329–344 (2010).
Chen, H., Cui, Y., Zhang, D., Xie, J. & Zhou, X. The role of fibroblast growth factor 8 in cartilage development and disease. J. Cell Mol. Med. 26, 990–999 (2022).
Wu, Q. et al. Induction of an osteoarthritis-like phenotype and degradation of phosphorylated Smad3 by Smurf2 in transgenic mice. Arthritis Rheum. 58, 3132–3144 (2008).
Hawker, G. A., Mian, S., Bednis, K. & Stanaitis, I. Osteoarthritis year 2010 in review: non-pharmacologic therapy. Osteoarthr. Cartil. 19, 366–374 (2011).
Funck-Brentano, T. & Cohen-Solal, M. Crosstalk between cartilage and bone: when bone cytokines matter. Cytokine Growth Factor Rev. 22, 91–97 (2011).
Rogers, J., Shepstone, L. & Dieppe, P. Is osteoarthritis a systemic disorder of bone? Arthritis Rheum. 50, 452–457 (2004).
Suri, S. & Walsh, D. A. Osteochondral alterations in osteoarthritis. Bone 51, 204–211 (2012).
Xie, J. et al. The effects of interleukin-1β in modulating osteoclast-conditioned medium’s influence on gelatinases in chondrocytes through mitogen-activated protein kinases. Int. J. Oral. Sci. 7, 220–231 (2015).
Kan, S., Duan, M., Liu, Y., Wang, C. & Xie, J. Role of mitochondria in physiology of chondrocytes and diseases of osteoarthritis and rheumatoid arthritis. Cartilage 13, 1102S–1121S (2021).
Aigner, T., Gluckert, K. & von der Mark, K. Activation of fibrillar collagen synthesis and phenotypic modulation of chondrocytes in early human osteoarthritic cartilage lesions. Osteoarthr. Cartil. 5, 183–189 (1997).
Goldring, M. B. & Otero, M. Inflammation in osteoarthritis. Curr. Opin. Rheumatol. 23, 471–478 (2011).
Kapetanakis, S. et al. Serum TGF-beta2 and TGF-beta3 are increased and positively correlated to pain, functionality, and radiographic staging in osteoarthritis. Orthopedics 33, https://doi.org/10.3928/01477447-20100625-09 (2010).
Finnson, K. W., Parker, W. L., ten Dijke, P., Thorikay, M. & Philip, A. ALK1 opposes ALK5/Smad3 signaling and expression of extracellular matrix components in human chondrocytes. J. Bone Miner. Res. 23, 896–906 (2008).
Remst, D. F. et al. TGF-ss induces Lysyl hydroxylase 2b in human synovial osteoarthritic fibroblasts through ALK5 signaling. Cell Tissue Res. 355, 163–171 (2014).
Wang, H. Effect of Sox9 on TGF-β1-mediated atrial fibrosis. Acta Biochim. Biophys. Sin. (Shanghai). 53, 1450–1458 (2021).
Sueyoshi, T., Yamamoto, K. & Akiyama, H. Conditional deletion of Tgfbr2 in hypertrophic chondrocytes delays terminal chondrocyte differentiation. Matrix Biol. 31, 352–359 (2012).
Wang, X. et al. Regulation of MMP-13 expression by RUNX2 and FGF2 in osteoarthritic cartilage. Osteoarthr. Cartil. 12, 963–973 (2004).
Bauge, C. et al. Interleukin-1beta impairment of transforming growth factor beta1 signaling by down-regulation of transforming growth factor beta receptor type II and up-regulation of Smad7 in human articular chondrocytes. Arthritis Rheum. 56, 3020–3032 (2007).
Mobasheri, A., Kalamegam, G., Musumeci, G. & Batt, M. E. Chondrocyte and mesenchymal stem cell-based therapies for cartilage repair in osteoarthritis and related orthopaedic conditions. Maturitas 78, 188–198 (2014).
Hwang, H. S. & Kim, H. A. Chondrocyte apoptosis in the pathogenesis of osteoarthritis. Int. J. Mol. Sci. 16, 26035–26054 (2015).
Zamli, Z. & Sharif, M. Chondrocyte apoptosis: a cause or consequence of osteoarthritis? Int. J. Rheum. Dis. 14, 159–166 (2011).
Zhong, G. et al. miRNA-335-5p relieves chondrocyte inflammation by activating autophagy in osteoarthritis. Life Sci. 226, 164–172 (2019).
Dhollander, A. A. et al. The use of scaffolds in the treatment of osteochondral lesions in the knee: current concepts and future trends. J. Knee Surg. 25, 179–186 (2012).
Xiang, Y., Bunpetch, V., Zhou, W. & Ouyang, H. Optimization strategies for ACI: A step-chronicle review. J. Orthop. Transl. 17, 3–14 (2019).
Ahmed, T. A. & Hincke, M. T. Mesenchymal stem cell-based tissue engineering strategies for repair of articular cartilage. Histol. Histopathol. 29, 669–689 (2014).
Crecente-Campo, J., Borrajo, E., Vidal, A. & Garcia-Fuentes, M. New scaffolds encapsulating TGF-beta3/BMP-7 combinations driving strong chondrogenic differentiation. Eur. J. Pharm. Biopharm. 114, 69–78 (2017).
Yang, Q. et al. Silk fibroin/cartilage extracellular matrix scaffolds with sequential delivery of TGF-beta3 for chondrogenic differentiation of adipose-derived stem cells. Int. J. Nanomed. 12, 6721–6733 (2017).
Kim, S. H., Kim, S. H. & Jung, Y. TGF-beta3 encapsulated PLCL scaffold by a supercritical CO2-HFIP co-solvent system for cartilage tissue engineering. J. Control. Release 206, 101–107 (2015).
Tang, Q. O. et al. Preclinical and clinical data for the use of mesenchymal stem cells in articular cartilage tissue engineering. Expert Opin. Biol. Ther. 12, 1361–1382 (2012).
Mrugala, D. et al. Phenotypic and functional characterisation of ovine mesenchymal stem cells: application to a cartilage defect model. Ann. Rheum. Dis. 67, 288–295 (2008).
Wei, W. & Dai, H. Articular cartilage and osteochondral tissue engineering techniques: recent advances and challenges. Bioact. Mater. 6, 4830–4855 (2021).
Cancedda, R., Dozin, B., Giannoni, P. & Quarto, R. Tissue engineering and cell therapy of cartilage and bone. Matrix Biol. 22, 81–91 (2003).
Fan, H. et al. TGF-beta3 immobilized PLGA-gelatin/chondroitin sulfate/hyaluronic acid hybrid scaffold for cartilage regeneration. J. Biomed. Mater. Res. A 95, 982–992 (2010).
Altman, G. H. et al. Silk-based biomaterials. Biomaterials 24, 401–416 (2003).
Wang, Y., Kim, U. J., Blasioli, D. J., Kim, H. J. & Kaplan, D. L. In vitro cartilage tissue engineering with 3D porous aqueous-derived silk scaffolds and mesenchymal stem cells. Biomaterials 26, 7082–7094 (2005).
Zhou, C. et al. Hydrogel platform with tunable stiffness based on magnetic nanoparticles cross-linked GelMA for cartilage regeneration and its intrinsic biomechanism. Bioact. Mater. https://doi.org/10.1016/j.bioactmat.2022.07.013.
Huang, X., Zhong, L., Post, J. N. & Karperien, M. Co-treatment of TGF-beta3 and BMP7 is superior in stimulating chondrocyte redifferentiation in both hypoxia and normoxia compared to single treatments. Sci. Rep. 8, 10251 (2018).
Luo, Z. et al. Mechano growth factor (MGF) and transforming growth factor (TGF)-beta3 functionalized silk scaffolds enhance articular hyaline cartilage regeneration in rabbit model. Biomaterials 52, 463–475 (2015).
Yang, Y. et al. Transforming growth factor-β1-induced N-cadherin drives cell-cell communication through connexin43 in osteoblast lineage. Int. J. Oral. Sci. 13, 15 (2021).
Diekman, B. O., Rowland, C. R., Lennon, D. P., Caplan, A. I. & Guilak, F. Chondrogenesis of adult stem cells from adipose tissue and bone marrow: induction by growth factors and cartilage-derived matrix. Tissue Eng. Part A 16, 523–533 (2010).
Ye, K. et al. Chondrogenesis of infrapatellar fat pad derived adipose stem cells in 3D printed chitosan scaffold. PLoS One 9, e99410 (2014).
Choi, S., Cho, T. J., Kwon, S. K., Lee, G. & Cho, J. Chondrogenesis of periodontal ligament stem cells by transforming growth factor-beta3 and bone morphogenetic protein-6 in a normal healthy impacted third molar. Int. J. Oral. Sci. 5, 7–13 (2013).
Ude, C. C. et al. The evaluation of cartilage differentiations using transforming growth factor beta3 alone and with combination of bone morphogenetic protein-6 on adult stem cells. Cell Tissue Bank 18, 355–367 (2017).
Hildner, F. et al. FGF-2 abolishes the chondrogenic effect of combined BMP-6 and TGF-beta in human adipose derived stem cells. J. Biomed. Mater. Res. A 94, 978–987 (2010).
Park, J. S. et al. Heparin-bound transforming growth factor-beta3 enhances neocartilage formation by rabbit mesenchymal stem cells. Transplantation 85, 589–596 (2008).
Na, K. et al. Synergistic effect of TGFbeta-3 on chondrogenic differentiation of rabbit chondrocytes in thermo-reversible hydrogel constructs blended with hyaluronic acid by in vivo test. J. Biotechnol. 128, 412–422 (2007).
Lima, E. G. et al. The beneficial effect of delayed compressive loading on tissue-engineered cartilage constructs cultured with TGF-beta3. Osteoarthr. Cartil. 15, 1025–1033 (2007).
Zhou, C. et al. Microenvironmental stiffness mediates cytoskeleton re-organization in chondrocytes through laminin-FAK mechanotransduction. Int. J. Oral. Sci. 14, 15 (2022).
Allen, J. L., Cooke, M. E. & Alliston, T. ECM stiffness primes the TGFbeta pathway to promote chondrocyte differentiation. Mol. Biol. Cell 23, 3731–3742 (2012).
Song, J. et al. MicroRNA-488 regulates zinc transporter SLC39A8/ZIP8 during pathogenesis of osteoarthritis. J. Biomed. Sci. 20, 31 (2013).
Song, J. et al. MicroRNA-181b regulates articular chondrocytes differentiation and cartilage integrity. Biochem. Biophys. Res. Commun. 431, 210–214 (2013).
Yang, B. et al. MicroRNA-145 regulates chondrogenic differentiation of mesenchymal stem cells by targeting Sox9. PLoS One 6, e21679 (2011).
Sun, B. et al. A 3D-Bioprinted dual growth factor-releasing intervertebral disc scaffold induces nucleus pulposus and annulus fibrosus reconstruction. Bioact. Mater. 6, 179–190 (2021).
Martin, A. R. et al. Nanofibrous hyaluronic acid scaffolds delivering TGF-beta3 and SDF-1alpha for articular cartilage repair in a large animal model. Acta Biomater. 126, 170–182 (2021).
Legemate, K., Tarafder, S., Jun, Y. & Lee, C. H. Engineering human TMJ discs with protein-releasing 3D-printed scaffolds. J. Dent. Res. 95, 800–807 (2016).
Merlin Rajesh Lal, L. P., Suraishkumar, G. K. & Nair, P. D. Chitosan-agarose scaffolds supports chondrogenesis of Human Wharton’s Jelly mesenchymal stem cells. J. Biomed. Mater. Res. A 105, 1845–1855 (2017).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81771047 to J.X., 81670978 and 81870754 to X.Z.) and the Sichuan Science & Technology Innovation Talent Project (2022JDRC0044).
Author information
Authors and Affiliations
Contributions
J.X. and X.Z. designed the concept of the study. X.D., L.C., and J.X. drafted the paper. X.Z. revised the paper. All the authors read the paper, and X.Z. approved the final version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Du, X., Cai, L., Xie, J. et al. The role of TGF-beta3 in cartilage development and osteoarthritis. Bone Res 11, 2 (2023). https://doi.org/10.1038/s41413-022-00239-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41413-022-00239-4
This article is cited by
-
Specific lipid magnetic sphere sorted CD146-positive bone marrow mesenchymal stem cells can better promote articular cartilage damage repair
BMC Musculoskeletal Disorders (2024)
-
Comparative Analysis of Hyaline Cartilage Characteristics and Chondrocyte Potential for Articular Cartilage Repair
Annals of Biomedical Engineering (2024)
-
Redifferentiation of genetically modified dedifferentiated chondrocytes in a microcavitary hydrogel
Biotechnology Letters (2024)
-
Annexin A5 derived from matrix vesicles protects against osteoporotic bone loss via mineralization
Bone Research (2023)
-
The role of exosomes and their enhancement strategies in the treatment of osteoarthritis
Human Cell (2023)
