
Abstract
Recent evidence shows that targeting NLRP3 inflammasome activation is an important means to treat inflammasome-driven diseases. Scoparone, a natural compound isolated from the Chinese herb Artemisia capillaris Thunb, has anti-inflammatory activity. In this study we investigated the effect of scoparone on NLRP3 inflammasome activation in inflammatory diseases. In LPS-primed, ATP or nigericin-stimulated mouse macrophage J774A.1 cells and bone marrow-derived macrophages (BMDMs), pretreatment with scoparone (50 μM) markedly restrained canonical and noncanonical NLRP3 inflammasome activation, evidenced by suppressed caspase-1 cleavage, GSDMD-mediated pyroptosis, mature IL-1β secretion and the formation of ASC specks. We then conducted a transcriptome analysis in scoparone-pretreated BMDMs, and found that the differentially expressed genes were significantly enriched in mitochondrial reactive oxygen species (ROS) metabolic process, mitochondrial translation and assembly process, as well as in inflammatory response. We demonstrated in J774A.1 cells and BMDMs that scoparone promoted mitophagy, a well-characterized mechanism to control mitochondrial quality and reduce ROS production and subsequent NLRP3 inflammasome activation. Mitophagy blockade by 3-methyladenine (3-MA, 5 mM) reversed the protective effects of scoparone on mitochondrial damage and inflammation in the murine macrophages. Moreover, administration of scoparone (50 mg/kg) exerted significant preventive effects via inhibition of NLRP3 activation in mouse models of bacterial enteritis and septic shock. Collectively, scoparone displays potent anti-inflammatory effects via blocking NLRP3 inflammasome activation through enhancing mitophagy, highlighting a potential action mechanism in treating inflammasome-related diseases for further clinical investigation.
Similar content being viewed by others


Introduction
Inflammasomes drive the activation of inflammatory caspases in response to invading pathogens and host-derived danger signals, leading to the release of cytokines and the induction of cell death. This process is critical in the initiation of inflammation and the development of immune responses [1, 2]. One of the most well-characterized inflammasomes is the NLR family pyrin domain containing 3 (NLRP3) inflammasome, which contains the receptor protein NLRP3, the adaptor protein apoptosis-associated speck-like protein containing a CARD (ASC) and the effector protein caspase-1 [3, 4]. Two signals are required to fully activate the NLRP3 inflammasome. The first (the priming signal) is triggered when pathogen-associated molecular patterns (PAMPs) bind to pattern recognition receptors (PRRs), leading to the expression of inflammasome components, including NLRP3, and proinflammatory cytokines, such as pro-interleukin (IL)-1β and pro-IL-18 [5, 6]. A second signal provided by various stimulators, including damage associated molecular patterns (DAMPs) such as extracellular ATP and nigericin, can fully activate the inflammasome [7, 8]. Upon stimulation, NLRP3 undergoes oligomerization and exposes its pyrin domain, which recruits ASC to form a large platform for pro-caspase-1 binding, triggering the autocatalytic cleavage and activation of caspase-1, which mediates the maturation of IL-1β and IL-18 [9,10,11]. Additionally, caspase-1 can cleave gasdermin D (GSDMD) to form an N-terminal cleavage product (GSDMD-NT), which executes pyroptosis by forming plasma membrane pores [12]. NLRP3 inflammasome activation results in a robust inflammatory response and is critical in defending against bacterial, fungal and viral infections. It has been well documented that persistent and aberrant inflammasome activation is involved in the initiation of various metabolic and inflammatory diseases [13, 14]; thus, targeting NLRP3 inflammasome activation is an important strategy to treat inflammasome-driven diseases.
How structurally diverse danger signals trigger NLRP3 inflammasome activation remains elusive. Although several models of NLRP3 activation have been proposed, many of them may be mutually exclusive, and no integrated, unified model has been produced. Of note, mitochondria have been described as the most likely intracellular intermediates that are crucial for NLRP3 activation [15,16,17]. Mitochondrial damage and the release of mitochondrial-derived DAMPs and mtROS are the key upstream events culminating in NLRP3 inflammasome activation [18, 19]. Many NLRP3 activators markedly induce increased mtROS production, and preincubation with certain antioxidants can subdue NLRP3 inflammasome overactivation [20, 21]. In addition, mitochondria serve as a platform for the recruitment and assembly of NLRP3 and the adaptor ASC, amplifying its effects on the pathophysiology of inflammasome-related diseases [22, 23]. Mitophagy is therefore necessary for maintaining mitochondrial quality and regulating NLRP3 activation through the removal of excessive or aberrant mitochondria and elimination of mtROS [24,25,26]. The process of mitophagy begins with the formation of an autophagosome around a mitochondrion that then fuses with a lysosome, resulting in degradation of the target mitochondrion [27]. LC3-decorated autophagosomes are recruited to mitochondria by mitochondria-specific receptors containing an LIR motif [28, 29]. It has been proposed that promoting mitophagy can negatively regulate NLRP3 inflammasome activation [27, 30]. Interestingly, the mitochondrial damage triggered by NLRP3 activators is different from that caused by proapoptotic BCL2 proteins, which enables activation of the apoptosis protease activating factor to activate caspase-3 rather than caspase-1 [31]. To this end, NLRP3 inflammasome blockers in association with mitophagy induction are gaining increasing attention as good candidates for further investigation.
Scoparone, also known as 6,7-dimethoxycoumarin (Fig. 1a), is the major compound isolated from the Chinese herb Artemisia capillaries Thunb (also known as Yinchenhao), which has long been used in traditional Chinese medicine formulas (e.g., Yin-Zhi-Huang decoction, Yin-Chen-Hao decoction) for the treatment of hepatic and cholestatic disorders or other inflammatory diseases. Although the anti-inflammatory effect of scoparone has been investigated [32, 33], whether scoparone has an inhibitory effect on the NLRP3 inflammasome remains unclear owing to the role of the NLRP3 inflammasome in inflammatory diseases. In this study, our results showed that scoparone markedly restrained canonical and noncanonical NLRP3 inflammasome activation and decreased pyroptotic cell death. Further experiments revealed that scoparone inhibited oxidative stress, ROS and inflammatory signals in ATP-activated BMDMs. This inhibitory effect of scoparone was mediated by mitophagy. Moreover, scoparone administration alleviated NLRP3-mediated diseases, including Escherichia coli (E. coli)-induced septic shock and Citrobacter rodentium (C. rodentium)-induced enteritis in mice, thereby improving animal survival. These findings suggest that scoparone could be a good candidate treatment for NLRP3 inflammasome-mediated diseases.

a Scoparone (Sco) structure. b J774A.1 cells were treated with scoparone (50–400 μM, 12/24 h), and then the cytotoxicity of the compound was determined using a CCK-8 assay (n = 3 per group). c J774A.1 cells or d, e BMDMs were pretreated with various doses of scoparone (25, 50, 100 μM), primed with LPS (500 ng/ml for J774A.1 cells, 100 ng/ml for BMDMs) for 4 h and then stimulated with ATP (4 mM) for 1 h or nigericin (10 μM) for 1 h. Western blot analysis of mature IL-1β and caspase-1 (p10) protein in supernatants (Sup.), and NLRP3, caspase-1 (p45), pro-IL-1β and ASC proteins in whole-cell lysates (Lys.). f, g IL-1β production was analyzed by ELISA in LPS (4 h)- and ATP (1 h)- or nigericin (1 h)-treated BMDMs in the presence or absence of scoparone (50 μM) (n = 3 per group). h Pam3CSK4-primed BMDMs were pretreated with scoparone (50 μM) and then stimulated with LPS. ELISA was used to measure IL-1β secretion in supernatants (n = 3 per group). i, j Quantification of ASC speck formation in five random images (n = 5 per group). k, l Representative immunofluorescence images of ASC speck (green) formation in BMDMs treated as in f, g. Hoechst 33342 (blue) was used to stain nuclei. The scale bars represent 20 μm. The data are shown as the mean ± SD, ***P < 0.001.
Materials and methods
Reagents and antibodies
Scoparone (C11H10O4, MW: 206.20, >98% purity) was purchased from Chengdu Push Biotech (Chengdu, China); ultrapure Lipopolysaccharides (LPS), Adenosine 5′-triphosphate disodium salt hydrate (ATP), nigericin, propidium iodide (PI), 3-MA, Carbonyl cyanide 3-chlorophenylhydrazone (CCCP), dimethyl sulfoxide (DMSO), and Hoechst 33342 were purchased from Sigma–Aldrich (Munich, Germany). Cell culture reagents, Pam3CSK4, MitoSOX, MitoTracker Deep red, MitoTracker Green, and BCA assay kit were obtained from Invitrogen (Carlsbad, CA, USA). Murine M-CSF was obtained from PeproTech (Rocky Hill, NJ). The FuGENE HD transfection reagent was purchased from Promega. Mouse IL-1β and TNF-α ELISA Kits were purchased from Proteintech (Chicago, IL, USA). Cytotoxicity Lactate Dehydrogenase (LDH) and Cell Counting Kit-8 were purchased from Dojindo Laboratories (Kumamoto, Japan). C. rodentium was supplied by Dr. Chen Dong from Tsinghua University. The information for primary antibodies (name, supplier, catalog number, molecular weight) is provided in Supplementary Table 1.
RNA extraction, library preparation and sequencing
The cellular RNA was extracted from BMDMs using Trizol reagent (Invitrogen, Carlsbad, CA, USA). Testing for RNA integrity was conducted with an Agilent 2100 BioAnalyzer (Santa Clara, CA, USA) and a Qubit Fluorometer (Invitrogen, Carlsbad, CA, USA). To prepare the libraries for sequencing, NEB’s Next Ultra RNA Library Prep Kit for Illumina (NEB, Ipswich, MA, USA) was used. Poly(A)-tailed mRNA was enriched from 1 μg total RNA using NEB Next Poly(A) mRNA Magnetic Isolation Module kit (NEB, Ipswich, MA, USA). In order to amplify the library DNA, products were purified and enriched by polymerase chain reaction (PCR). Quantification of the final libraries was performed with an Agilent 2100 Bioanalyzer and the KAPA Library Quantification Kit (KAPA Biosystems, South Africa). After RT-qPCR validation, libraries were sequenced using an Illumina NovaSeq sequencer and 150 bp paired-end reads were established. CapitalBio Ltd constructed and sequenced the library.
Mice
The mice were all C57BL/6J except as otherwise noted. The male mice (weight, 20–22 g; age, 8 weeks) were obtained from SPF Biotechnology Co., Ltd (Beijing, China). All mice were fed with a standard mouse chow diet and held in ventilated cages in a pathogen-free facility with a 12 h dark/light cycle at 22 ± 2 °C. Experimental procedures and animal welfare were conducted strictly according to the Guidelines of Laboratory Animals Care and Use and with the approval of the Animal Ethics Committee of Beijing University of Chinese Medicine (protocol No. BUCM-4-2022041304-2017).
Macrophage culture
Using male C57BL/6 mice, bone marrow-derived macrophages (BMDMs) were isolated from bone marrow and cultured in DMEM with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin (P/S) and 50 ng/ml murine macrophage colony stimulating factor (M-CSF) for 7 days. With CD11b and F4/80 antibodies, flow cytometry was utilized to assess the BMDM purity.
Mouse macrophage line J774A.1 and human THP-1 cells were obtained from the Chinese Academy of Sciences (Kunming, China). J774A.1 cells were cultured in DMEM supplemented with 10% FBS and 1% P/S. Human THP-1 cells were cultured in RPMI-1640 medium supplemented with 10% FBS and 1% P/S. All of the cultured cells were kept in a humidified atmosphere with 5% CO2 at 37 °C.
Bacterial culture
C. rodentium and E. coli were grown in (lysogeny broth) LB media at 37 °C overnight. We collected viable bacteria by centrifuging at 3000 × g for 10 min, washing with PBS, and then re-suspending in the appropriate volume of PBS. Aliquots of C. rodentium and E. coli were serially diluted and plated in LB media agar for 18 h at 37 °C. Bacterial counts were measured by counting colony-forming units (CFU).
Inflammasomes activation
BMDMs and J774A.1 cells were seeded in 12-well plates at a density of 1 × 106 cells/ml and 5 × 105 cells/ml, respectively. Scoparone was added to the cell culture medium containing FBS at the indicated concentration. After 2 h, the medium was replaced with fresh media and the cells were primed with LPS (500 ng/ml for J774A.1 cells, 100 ng/ml for BMDMs) for 4 h. Next, the cells were exposed to an NLRP3 inflammasome activator, 4 mM ATP for 1 h, 10 μM nigericin for 1 h, 320 μg/ml alum for 1 h, or 2 μg/ml Poly (I:C) was transfected with FuGENE for 6 h. To activate noncanonical inflammasome, we primed the BMDMs with Pam3CSK4 (100 ng/ml) for 4 h, then removed the medium and replaced it with Opti-MEM. A dose of 1 μg/ml LPS was transfected for 16 h with FuGENE HD transfection reagent. ELISA kits were used to analyze the supernatants following the manufacturers’ instructions. Western blot analysis was performed on precipitated supernatants and cell lysates. To study the effects of scoparone on NLRP3 inflammasome priming, THP-1 cells were incubated with scoparone (50 µM) for 2 h and then stimulated with LPS (500 ng/ml) for 4, 8, and 12 h. J774A.1 cells were incubated with scoparone (50 μM) for 2 h and then stimulated with LPS for 5 h (Sco before LPS), or J774A.1 cells were treated with LPS (500 ng/ml) for 4 h and then stimulated with scoparone (50 µM) for 2 h (Sco after LPS).
Cell viability assay
As per the manufacturer’s instruction, the CCK-8 kit (Dojindo, CK04) was used to analyze J774A.1 cells for viability. Briefly, the J774A.1 cells were seeded in a 96-well plate overnight at 1 × 105 cells/well, followed by scoparone treatment for 12/24 h, then incubated for 30 min at 37 °C with 10 μl of CCK-8 solution. The absorbance of the mixture was measured at the wavelength of 450 nm.
Enzyme-linked immunosorbent assay (ELISA)
Supernatants from cell culture, serum and peritoneal cavity fluid samples were used to determine the levels of mouse IL-1β, IL-18, and TNF-α following the manufacturers’ instructions.
Western blot analysis
The cells were lysed in a lysis solution containing a protease inhibitor cocktail (Roche, 11836153001). BCA Protein Assay Kit was used to quantitate protein concentrations. The protein samples were separated through SDS-PAGE electrophoresis, and transferred to nitrocellulose membranes. The membranes were then sealed in 5% skim milk for an hour at room temperature. After overnight incubation at 4 °C with specific primary antibodies, the membranes were then incubated with the appropriate HPR-conjugated antibodies, and developed with enhanced chemiluminescence.
Precipitation of soluble proteins in supernatants
Equal volume of medium was collected and precipitated for detection of soluble proteins in the culture supernatants using trichloroacetic acid (TCA) precipitation methods as previously described [34]. After precipitation, the pellets were washed twice with cold acetone (acetone kept at −20 °C for 30 min), and resuspended in 2 × SDS-PAGE loading buffer.
Pyroptosis assay
As described previously, pyroptosis was detected using PI incorporation [35]. In short, the cells were plated overnight in a 24-well plate, followed by scoparone treatment for 2 h. After that, the cells were primed with LPS for 4 h, followed by ATP or nigericin stimulation, and then stained for 10 min with PI (2 µg/ml) and Hoechst 33342 (5 µg/ml). The stained cells were analyzed using the INCell Analyzer 2500HS with High Content Analysis (GE). At least ten randomly selected fields of images were used for each set of data.
The levels of LDH released into the culture supernatants were measured by a LDH cytotoxicity assay kit as directed by the manufacturer. The absorbance signal of 490 nm was measured with a Microplate Reader.
DNA and RNA isolation and real time quantitative(q) PCR
In accordance with manufacturer’s instructions, the cellular DNA was purified with DNeasy kits (Sangon Biotech, China). To quantify the mtDNA/nDNA ratio, mtDNA was quantified by qPCR using mitochondrial D-loop-specific primers [15]. Normalization was performed using nuclear DNA encoding Tert.
The cellular RNA was extracted using Trizol reagent (Invitrogen, Carlsbad, CA, USA). PrimeScript RT reagent kit (Accurate, Hunan, China) was used to reverse transcribe RNA. Equal quantities of cDNA were subjected to qRT-PCR with SYBR Green Master Mix (Beyotime Biotechnology). In Supplementary Table 2, primers used for qPCR were listed.
Mitochondrial membrane potential
BMDMs were seeded in a 6-well plate, drug intervention and stimulation were performed as described above. The JC-1 fluorescent probe was assembled following the kit’ instructions and images were acquired using an Olympus FV1000 confocal microscope. A strong red fluorescence was observed for the polymer (Ex = 585 nm, Em = 590 nm), and green fluorescence was observed for the monomers (Ex = 514 nm, Em = 529 nm). Analyses were conducted on the fluorescence rate of monomers/polymers.
Flow cytometric analyses
Mitochondrial mass was determined through staining with Mitotracker deep red and Mitotracker green at 50 nM for 30 min at 37 °C. Mitochondrial ROS levels were determined by staining cells with 5 μM MitoSOX for 30 min at 37 °C. We then washed the cells with PBS solution and resuspended them in cold PBS solution with 2% FBS for FACS analysis.
Cells from peritoneum were collected. A mixture of CD11b-PE and F4/80-APC antibodies was added to the cells and incubated at 4 °C in the dark for 30 min. The cells were then washed, fixed with paraformaldehyde, and analyzed by flow cytometry.
Transmission electron microscopy
BMDMs were grown and treated as described above. Using 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer, cells were fixed overnight at 4 °C, and then processed as described [36] by the Public Technology Platform of the school of Medicine, Tsinghua University. The samples were observed under an electron microscope (Hitachi, Japan).
Immunofluorescent staining and confocal microscopy
BMDMs were primed with LPS and then treated with NLRP3 agonists. Cells were then fixed, permeabilized, and blocked. After incubating with primary antibodies overnight, secondary fluorescent antibodies (Alexa-488, Alexa-594) or Lysotracker (200 nM) were added and DAPI was used to counterstain nucleus. Confocal microscopy analyses were carried out using an Olympus FV1000 confocal microscope.
E. coli-induced septic shock in vivo
The bacterial infection animal model was established in accordance with a previous report [37]. In brief, the mice were split into three groups (n = 6) that were intraperitoneally (i.p.) injected with either the vehicle (2% Tween-80 in PBS) or scoparone (50 mg/kg). Twelve hours later, E. coli (2 × 109 CFU/mouse) was injected into the peritoneal cavity of each mouse. The survival of mice was monitored every 2 h.
In parallel to this experiment, mice were treated similarly and were anesthetized and enucleated for blood collection 6 h after bacterial infection. Cytokine levels in the sera were measure by ELISA. Following washing the peritoneal cavity with PBS, the pelleted cells were stained with antibodies and analyzed using flow cytometry. The liver, lung and colon were removed for histopathological analysis.
C. rodentium-induced enteritis in vivo
Mice were intragastrically (i.g.) injected with either the vehicle (2% Tween-80 in PBS) or scoparone (50 mg/kg). The dosage of scoparone was designed according to the previous studies [38,39,40]. One hour later, C. rodentium infection was performed using 5 × 109 colony-forming units (CFU) as described [41]. Throughout the following 9 days, mice were weighed daily. The plasma samples of mice were collected for the assessment of AST, ALT, blood urea nitrogen (BUN) and creatinine (CRE) levels, as specified by the manufacturer. MLN, liver and spleen was homogenized in PBS for enumeration of bacterial CFU. Serially diluted tissue homogenates were plated onto LB agar and incubated at 37 °C for 24 h. Colon length and cytokine production were analyzed as described above.
HE staining
Sections of tissue were prepared, dewaxed with xylenes, rehydrated with a gradient of alcohol, then stained for 10 min with hematoxylin, followed by 3 min with eosin. In a gradient ethanol series, sections were dehydrated, and then transparent with dimethylbenzene. After sealing with neutral gum, the pathological changes of colon, jejunum, liver and lung tissue were observed under a super-resolution automatic scanning microscope.
Statistical analysis
Experimental data were indicated by mean ± standard deviation. Statistical comparison between two groups was performed via Student’s t test. Statistical comparison among multiple groups was carried out by one-way analysis of variance (one-way ANOVA) followed by LSD test (data with homogeneity of variance) in SAS 8.2. Correlation analysis was performed by Pearson chi-square test. P < 0.05 was considered statistically significant.
Results
Scoparone inhibits NLRP3 inflammasome activation in J774A.1 cells and BMDMs
To investigate the influence of scoparone on the survival of J774A.1 cells, different concentrations of scoparone were applied to cells for 12/24 h, and the cytotoxicity of the compound was determined using a CCK-8 assay. As shown in Fig. 1b, scoparone treatment did not affect the viability of J774A.1 cells below 350 μM in comparison with untreated controls. To determine the effect of scoparone on NLRP3 inflammasome activation, we pretreated J774A.1 cells and BMDMs with or without scoparone and then activated the inflammasome in LPS-primed cells with NLRP3 inflammasome activators (ATP or nigericin) (Fig. 1c–e). Western blot results showed that pretreatment with scoparone for 2 h significantly inhibited caspase-1 cleavage and mature IL-1β secretion but did not affect the expression of NLRP3 inflammasome complex proteins, including NLRP3, pro-caspase-1, pro-IL-1β and ASC, in whole-cell lysates (Fig. 1c–e). Consistent with the Western blot analysis, ELISA demonstrated that scoparone reduced the release of IL-1β and IL-18 from BMDMs induced by ATP, nigericin, or poly(I:C) (Fig. 1f, g and Supplementary Fig. 1b, c). Immunofluorescence microscopy revealed that scoparone efficiently inhibited the formation of ASC specks in LPS-primed BMDMs under stimulatory conditions (Fig. 1i–l), which is another indicator of inflammasome activation, suggesting that scoparone is a broad-spectrum inhibitor of the NLRP3 inflammasome. Intracellular LPS is recognized by a noncanonical activation pathway of NLRP3 inflammasomes, which mediates the maturation and secretion of pro-IL-1β based on caspase-11 activation [42, 43]. We then pretreated BMDMs with the TLR1/2 agonist Pam3CSK4 and transfected them with LPS to activate the noncanonical NLRP3 inflammasome, and we found that scoparone also markedly reduced IL-1β secretion (Fig. 1h). Taken together, these results demonstrate that scoparone could be a multitargeting inhibitor that markedly restrains canonical, noncanonical NLRP3 inflammasome activation, IL-1β release and ASC speck formation in macrophages.
Studies have found that scoparone inactivates the TLR-4/NF-κB pathway to prevent inflammation and apoptosis [32]. We then examined whether scoparone affects LPS-induced priming for inflammasome activation. qPCR results revealed that LPS stimulation sharply increased proinflammatory cytokine expression, whereas scoparone did not inhibit the relative mRNA levels of IL1B and TNF in THP-1 cells in response to LPS treatment (Supplementary Fig. 1d, e). Moreover, scoparone treatment did not affect NLRP3, ASC, CASP-1, p-IKK-β and IKK-β protein expression, as determined by Western blot analysis (Supplementary Fig. 1f). Similar results were obtained with J774A.1 cells. When J774A.1 cells were stimulated with scoparone before or after LPS treatment, scoparone did not suppress LPS-induced Il1b and Tnf mRNA expression (Supplementary Fig. 1g, h). In addition, Western blotting showed that scoparone treatment did not affect the protein expression of NLRP3, CASP-1, pro-IL-1β, ASC, p-IKBα, or IKBα (Supplementary Fig. 1i). Overall, these findings indicate that the effective doses at which scoparone restrains activation of the NLRP3 inflammasome are not sufficient to affect LPS-induced NLPR3 priming signaling.
Scoparone represses ATP- or nigericin-induced pyroptosis in macrophages
In addition to cleaving pro-IL-1β, activated caspase-1 also cleaves GSDMD to generate the N-terminal cleavage product (GSDMD-NT), which causes pyroptosis via the formation of plasma membrane pores [12]. Pyroptotic cell death is a form of necrosis that can be visualized with PI staining. We therefore investigated whether scoparone could inhibit pyroptosis induced by either ATP or nigericin. Consistent with blocked NLRP3 inflammasome activation, scoparone decreased the number of PI-positive cells as well as the release of LDH in LPS-primed J774A.1 cells and BMDMs upon ATP or nigericin stimulation (Fig. 2a–f and Supplementary Fig. 2a–d). Additionally, the number of dying cells exhibiting swelling and membrane ballooning, which reflect the morphological attributes of pyroptosis, was decreased in scoparone-treated BMDMs (Fig. 2g, h and Supplementary Fig. 2e, f). The Western blot results revealed that scoparone suppressed GSDMD-NT production in a dose-dependent manner in J774A.1 cells compared to ATP-stimulated cells (Fig. 2i). A similar inhibitory effect was observed with scoparone pretreatment on ATP-/nigericin-induced GSDMD-NT formation in BMDMs (Fig. 2j, k). Therefore, these results suggest that scoparone can protect murine macrophages from pyroptosis upon NLRP3 inflammasome activation.

J774A.1 cells (a, c) and BMDMs (b, d) were treated as in Fig. 1c–e. After treatment, representative immunofluorescence images of cell death were obtained by staining with PI (2 μg/ml, red; staining dead cells) and Hoechst 33342 (5 μg/ml, blue; staining all cells). An analysis was performed on five randomly chosen fields to determine the percentage of PI-positive cells relative to all. Scale bars represent 20 μm. e, f LDH release was measured in the supernatant of BMDMs (n = 3 per group). g, h The morphology and number of pyroptotic BMDMs were observed under a microscope (n = 5 per group). Scale bars represent 20 μm. i–k Western blot analysis of the expression of GSDMD protein in whole-cell lysates. The data are shown as the mean ± SD. ***P < 0.001.
Scoparone affects mitochondrial signals in response to NLRP3 inflammasome activation
To explore the underlying pharmacological mechanisms and potential molecular targets of scoparone’s effects on NLRP3 inflammasome activation, transcriptomic analysis was carried out using RNA-sequencing (RNA-seq) on LPS/ATP-activated BMDMs with or without scoparone pretreatment. As shown in the volcano plot (Fig. 3a), a total of 606 differentially expressed genes (DEGs, P < 0.05) were identified in the scoparone-pretreated group under LPS and ATP stimulation compared to the stimulation group, with 165 genes upregulated and 441 genes downregulated. Moreover, GSEA was employed to identify the enrichment of scoparone-related DEGs in inflammation-related pathways. Notably, the GSEA results revealed that DEGs upregulated in the scoparone-pretreated group were significantly enriched in mitochondrial translation and assembly pathways with normalized enrichment scores of 1.87 and 1.56, respectively. Additionally, interleukin signaling and interferon-gamma signaling pathways were negatively enriched (Fig. 3b). Consistent with GSEA, GO biological process (GO: BP) analysis of these DEGs showed significant downregulation of inflammatory response- and ROS metabolism-related genes following scoparone pretreatment (Fig. 3c). According to the GSEA results, the relative expression levels of genes involved in mitochondrial function and homeostasis, including antioxidative stress, oxidative stress and mitophagy, are presented as a heatmap in Fig. 3d. As shown, scoparone markedly enhanced antioxidative stress-related gene expression but exerted the opposite effect on oxidative stress-related genes. Genes involved in mitophagy were also significantly regulated by scoparone. These observations suggest that mitochondria, which participate in oxidative stress metabolic processes and mitophagy regulation, might play critical roles in the inhibitory effect of scoparone on the NLRP3 inflammasome and might be promising therapeutic targets.

a Volcano plot: transcriptomes of BMDMs with and without scoparone (50 µM) pretreatment and LPS/ATP activation. b Gene set enrichment analysis (GSEA) plot for KEGG entry. NES normalized enrichment score. c GO pathway enrichment analysis (BP: biological process) based on DEGs. d The heatmap displays the differentially expressed genes (DEGs) obtained using hierarchical clustering analysis.
Scoparone promotes mitophagy to alleviate mitochondrial damage upon NLRP3 inflammasome stimulation
Previous reports have indicated that inflammasome activation might be triggered by signals associated with damaged mitochondria, which can be degraded within lysosomes via mitophagy [18]. Therefore, we further investigated whether and how scoparone influenced NLRP3 activator-induced mitochondrial damage. LPS and ATP treatment resulted in low mitochondrial membrane potential, and JC-1 was unable to aggregate in the mitochondrial matrix but existed as a monomer, producing green fluorescence, thus implying impaired mitochondria. Scoparone pretreatment for 2 h gradually changed the fluorescence color to red, indicating an increase in mitochondrial membrane potential (Fig. 4a, b). Similar results were obtained from two other types of mitochondria-specific labels that distinguish respiring mitochondria (MitoTracker Deep Red) or total mitochondria (MitoTracker Green). Flow cytometric analyses showed that LPS and ATP stimulation increased the percentage of damaged mitochondria (MitoTracker Green positive and MitoTracker Deep Red negative) from 6.13% to 20.9%; such a difference was also shown upon LPS and nigericin stimulation, while scoparone-pretreated macrophages exhibited decreased accumulation of damaged mitochondria upon stimulation with NLRP3 activators (ATP or nigericin) (Fig. 4c–e). Moreover, the addition of LPS and ATP resulted in robust ROS production and decreased mitochondrial DNA (mtDNA) levels, similar to treatment with CCCP, even though the damage caused by LPS and ATP was milder than that caused by CCCP. Scoparone pretreatment suppressed the release of mtROS and increased the levels of mtDNA in J774A.1 cells (Fig. 4f–i). To prevent cellular damage, ROS-producing mitochondria are constantly eliminated through mitophagy, a specialized form of autophagy. As expected, scoparone increased mitochondrial colocalization with LC3B in BMDMs (Fig. 4j, k), which is consistent with the mitochondrial disposal mechanism. The Western blot results also showed that scoparone enhanced the expression of microtubule-associated protein light chain 3 (LC3) in J774A.1 cells stimulated with ATP (Fig. 4o). To verify that mitochondria were indeed degraded by lysosomes, we costained for the mitochondrial protein TOM20 and the lysosome marker LysoTracker in BMDMs by using confocal microscopy and found that the colocalization of mitochondria and lysosomes increased significantly (Fig. 4l, m). Moreover, transmission electron microscopy revealed that LPS- and ATP-treated cells exhibited mitochondria with broken cristae enclosed by characteristic double-membrane autophagosomes, while scoparone significantly improved the quality and morphology of mitochondria (Fig. 4n). Collectively, the above findings suggest that scoparone promotes mitophagy to alleviate mitochondrial damage upon NLRP3 inflammasome stimulation.

a, b Analysis of mitochondrial membrane potential levels in LPS-(4 h)- and nigericin- (1 h)-activated BMDMs by JC-1 fluorescence (n = 6 per group). c J774A.1 cells were treated as in Fig. 1c and then stained with MitoTracker green and MitoTracker deep red (c–e) or MitoSOX (f, g) for 30 min before being analyzed by flow cytometry (n = 3 per group). h Mitochondrial mass was determined based on qPCR analysis of the mitochondrial DNA (mtDNA)/nuclear DNA (nDNA) ratio in BMDMs treated as in a or treated with CCCP for 2 h (i) (n = 3 per group). j Confocal microscopy was used to visualize the colocalization of LC3B (red) with mitochondria (green) in BMDMs with or without scoparone pretreatment and LPS/ATP activation. The scale bars represent 20 μm. k Quantification of Pearson’s colocalization coefficient between LC3B and mitochondria. l The colocalization of TOM20 (green) and LysoTracker (red) in BMDMs treated as in j (n = 6 per group). The scale bars represent 20 μm. m Quantification of Pearson’s colocalization coefficient between mitochondrial proteins and lysosomes (n = 6 per group). n Representative electron micrographs of mitochondrial morphology (boxes) in BMDMs with or without scoparone pretreatment and LPS/ATP activation. o Western blot analysis of LC3B in J774A.1 cells treated as in a. The data are shown as the mean ± SD. **P < 0.01, ***P < 0.001.
Mitophagy inhibition reverses the protective effects of scoparone against mitochondrial damage and inflammation in murine macrophages
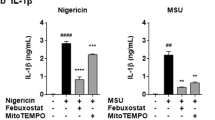
To further verify whether mitophagy is a major mechanism of scoparone’s inhibitory effects on NLRP3 activation, 3-methyladenine (3-MA), an inhibitor of mitophagy/autophagy, was added to BMDMs. As expected, 3-MA treatment resulted in the accumulation of damaged mitochondria and increases in mitochondrial ROS levels, as expected. More importantly, 3-MA reversed the effects of scoparone on mtDNA and ROS production (Fig. 5a–c). Moreover, the inhibitory effects of scoparone on Il1b1 and Il18 expression in BMDMs were significantly attenuated, while Il6 and Tnf were more inhibited by 3-MA (Fig. 5d–h). We also found that scoparone promoted mitophagy to alleviate mitochondrial damage, as evidenced by the colocalization of mitochondria with LC3B (Fig. 5i). 3-MA treatment significantly suppressed the expression of LC3B as determined by Western blot analysis (Fig. 5j). These findings collectively suggest that mitophagy induced by scoparone serves as a scavenger of mtROS by removing damaged mitochondria, which in turn suppresses NLRP3 activation.

a, b J774A.1 cells were treated with 3-MA (5 mM, 1 h) before scoparone (50 μM, 2 h) treatment, stimulated with LPS (500 ng/ml, 4 h) and ATP (4 mM, 1 h), stained with MitoSOX for 30 min and analyzed by flow cytometry (n = 3 per group). c Mitochondrial mass determined based on qPCR analysis of the mtDNA/nuclear DNA (nDNA) ratio in BMDMs treated as in a (n = 3 per group). d–g qPCR analysis of Il1b1, Il18, Il6 and Tnf mRNA expression in BMDMs treated as in a (n = 3 per group). h IL-1β production was analyzed by ELISA (n = 3 per group). i Confocal microscopy was used to visualize the colocalization of LC3B (red) with mitochondria (green) in BMDMs treated as in a. Scale bars, 20 μm. j Western blot analysis of LC3B protein expression in J774A.1 cells treated as in a. The data are shown as the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, NS not significant.
Scoparone mitigates E. coli-induced septic shock in vivo
To determine whether scoparone inhibits inflammasome activation in vivo, an NLRP3 inflammasome-dependent septic shock mouse model induced by E. coli was used to explore the functional relevance of scoparone in NLRP3 activation (Fig. 6a). Survival was assessed after mice were intraperitoneally injected with scoparone and then injected with viable E. coli 12 h later. According to our results, scoparone administration significantly improved the survival rate of mice infected with bacteria compared with that of the E. coli group (Fig. 6b). Additionally, we examined whether scoparone might prevent the production of inflammatory cytokines. As expected, scoparone significantly inhibited E. coli-induced IL-1β and TFN-α secretion in serum, suggesting that scoparone can block NLRP3 inflammasome activation in mice (Fig. 6c, d). Consistently, histopathological analysis showed overt infiltration of inflammatory cells in the lung, colon and liver following E. coli infection, whereas scoparone-treated mice did not display apparent infiltrated inflammatory cells (Fig. 6e–g). Collectively, our data indicate that scoparone effectively ameliorates bacteria-induced acute inflammation and systemic injury, possibly by inhibiting activation of the NLRP3 inflammasome in mice.

Male C57BL/6 mice were intraperitoneally (i.p.) injected with scoparone (50 mg/kg) or vehicle (2% Tween-80 in PBS). After 12 h, freshly prepared E. coli (2 × 109 CFU/mouse) was administered into the peritoneal cavity of each mouse. a Mouse model and work flow. b Observations of mouse survival were taken every 2 h (n = 8 mice per group). ***P < 0.001, scoparone (50 mg/kg) group vs. E. coli group. c, d A parallel experiment was conducted with mouse serum at 6 h post infection. IL-1β and TNF-α in the serum were evaluated by ELISA (n = 5 mice per group). e–g Representative histological images of the lung, colon and liver. Scale bars, 200 μm. The data are shown as the mean ± SD. ***P < 0.001.
Scoparone alleviates enteritis caused by C. rodentium infections in vivo
Studies have found that an alternative noncanonical pathway of NLRP3 inflammasome activation that additionally activates upstream caspase-11 is stimulated in response to gram-negative enteric pathogens such as C. rodentium [34]. Therefore, we determined whether scoparone influenced noncanonical NLRP3 inflammasome activation in vivo. Mice were intragastrically (i.g.) administered scoparone 1 h prior to oral gavage with C. rodentium daily (Fig. 7a). Serum biochemistry assays showed that only intragastric administration of scoparone (50 mg/kg) for 9 days did not induce liver and kidney injuries, as indicated by the unchanged levels of ALT, AST, BUN and CRE (Fig. 7b). Moreover, the body weight, colon length and spleen and liver indexes of mice showed that C. rodentium infection exacerbated enteritis pathogenesis, whereas scoparone treatment alleviated the degree of pathogenesis in mice (Fig. 7c–g). In addition, we found that bacterial loads in the liver, kidneys and lymph were reduced by scoparone (Fig. 7h–j). Histopathological observation revealed that scoparone obviously reduced the overt infiltration of inflammatory cells in the colon and jejunum tissues (Fig. 7k, l). Overall, these results indicate that scoparone can alleviate the severity of mouse pathological lesions caused by C. rodentium infection.

Male C57BL/6 mice were administered either vehicle (2% Tween-80 in PBS) or scoparone (50 mg/kg) intragastrically (i.g.). After 1 h, the mice were orally gavaged with freshly prepared C. rodentium (5 × 109 CFU/mouse in 0.2 ml of PBS). a Mouse model and work flow. b Serum ALT, AST, BUN and CRE activity levels (n = 6 mice per group). c Representative images of the colon 9 days post bacterial infection (n = 5 mice per group). d Relative body weights during mouse model development (n = 5 mice per group). e Colon length in each group (n = 5 mice per group). f, g Spleen and liver organ indices of mice (n = 5 mice per group). h–j Log10 CFU of C. rodentium in the liver, kidneys and lymph (n = 5 mice per group). k, l Representative histological images of the colon and jejunum. Scale bars, 200 μm.
Discussion
The NLRP3 inflammasome is quickly activated in response to various types of infection and stress signals and elicits a robust inflammatory response, and pharmacological molecules targeting the NLRP3 inflammasome are of considerable value in identifying potential therapeutic interventions [1, 44]. Mounting evidence has shown that scoparone has a wide spectrum of pharmacological properties, including anti-inflammatory, anti-hypertension, antitumour, hypolipidaemic, antioxidant, and anti-fibrotic properties [33]. However, whether scoparone influences the NLRP3 inflammasome and inflammasome-mediated infectious inflammatory diseases remains unknown. In this study, we found for the first time that scoparone exhibits a powerful inhibitory effect on the NLRP3 inflammasome. Mechanistically, our study suggests that scoparone inhibits inflammasome activation by promoting mitophagy to alleviate mitochondrial damage. Additionally, we have demonstrated that scoparone can prevent NLRP3-driven diseases, including septic shock and enteritis, in mouse models. We have also confirmed that scoparone (50 mg/kg) administered intragastrically for 9 days is well tolerated in mice (Fig. 7b). Consequently, the present study unravels a previously unknown mechanism of scoparone, which may be a safe and promising candidate treatment for NLRP3 inflammasome-induced diseases.
NLRP3 inflammasome signaling depends on priming and activation steps. Previous studies have found that scoparone inactivates NF-κB signaling activation [32]. However, by treating cells with scoparone and LPS, we were able to rule out any effects on the priming step (Supplementary Fig. 1d–i). Upon NLRP3 inflammasome activation, it has been found that the functional oligomeric inflammasome components NLRP3 and ASC are secreted out of the cell together with cleaved caspase-1 and mature IL-1β, amplifying the inflammatory response by activating caspase-1 intracellularly and extracellularly [45, 46]. However, the opposite observation has also been found: the expression levels of inflammasome complex proteins in cell lysates can remain unchanged [47,48,49]. In our study, the expression of NLRP3 inflammasome components was not affected by scoparone treatment, which was likely due to the different types of cells and the short duration of stimulation in our study.
To further clarify the mechanism by which scoparone inhibits NLRP3 inflammasome activation, we performed RNA-seq analysis and found a strong relationship between scoparone-mediated inhibition of the NLRP3 inflammasome and mitochondrial regulation. Mitochondrial involvement in NLRP3 inflammasome activation was first proposed by Tschopp and coworkers [18]. Subsequently, Arditi and colleagues showed that mtROS and ox-mtDNA (mitochondrial-derived DAMPs) from the process of apoptosis can bind NLRP3. Not only do mitochondria provide a scaffold for inflammasome complex assembly, but mitochondrial damage is also necessary for NLRP3 inflammasome activation [17]. Accumulating evidence supports the idea that mitophagy attenuates NLRP3 inflammasome activation by removing excess or damaged mitochondria after macrophage exposure to NLRP3 activators and regulates the quality of mitochondria and maintains mitochondrial homeostasis [16, 30, 50]. As mitophagy proceeds, the endoplasmic reticulum membrane wraps the abnormal mitochondria to form autophagosomes, and the subsequent modification of the LC3-I protein to LC3-II promotes their fusion with lysosomes for degradation [28, 51]. Our current results show that scoparone treatment alleviates mitochondrial damage, reduces the ROS accumulation caused by mitochondrial membrane potential depolarization and regulates mitochondrial quality by promoting mitophagy, thus maintaining mitochondrial homeostasis. In addition, mitophagy blockade is often accompanied by severe mitochondrial injury and ROS production and subsequent NLRP3 inflammasome activation [30, 52]. We sought to elucidate whether mitophagy plays any role in NLRP3 inflammasome suppression and found that specific inhibition of mitophagy with 3-MA, which blocks autophagosome formation, reversed the inhibitory effects of scoparone on ROS and IL-1β release from LPS/ATP-treated BMDMs, indicating that scoparone restricts NLRP3 inflammasome activation in a mitophagy-dependent manner.
Inflammasome activation is an essential defence mechanism of the innate immune system response against infections [53]. The outcomes of inflammasome activation include the maturation and release of IL-1β as well as other inflammatory cytokines. In addition to increasing phagocytes, these cytokines are also responsible for the recruitment of other immune cells, such as macrophages and neutrophils [54]. It is of interest to learn whether scoparone can also have an effect in vivo. Our research findings show that scoparone has definite therapeutic effects in mouse models of NLRP3-driven diseases, including bacterial enteritis and septic shock. Therefore, other diseases associated with the NLRP3 inflammasome may benefit from the therapeutic use of scoparone. This possibility remains to be further explored in the future. These findings add further insight into the concept that scoparone is a critical determinant of innate immune responses with profound anti-inflammatory effects.
Taken together, our findings provide strong evidence for the novel pharmacological effect of scoparone on inhibiting NLRP3 inflammasome activation in vitro and in vivo. Mechanistically, as illustrated in Fig. 8, scoparone induces mitophagy-mediated ROS elimination-dependent NLRP3 inflammasome inactivation, resulting in a reduction in NLRP3-/pyroptosis-driven IL1β release from macrophages.

Working model depicting scoparone as a mitophagy promoter to alleviate mitochondrial damage and suppress the release of mtROS, which inhibits inflammasome activation and reduces NLRP3-/pyroptosis-mediated IL-1β release, consequently preventing NLRP3 inflammasome-driven disease.
References
Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16:407–20.
Duncan JA, Canna SW. The NLRC4 inflammasome. Immunol Rev. 2018;281:115–23.
Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019;19:477–89.
Sharif H, Wang L, Wang WL, Magupalli VG, Andreeva L, Qiao Q, et al. Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature. 2019;570:338–43.
Zhong Z, Sanchez-Lopez E, Karin M. Autophagy, inflammation, and immunity: a troika governing cancer and its treatment. Cell. 2016;166:288–98.
Gross O, Thomas CJ, Guarda G, Tschopp J. The inflammasome: an integrated view. Immunol Rev. 2011;243:136–51.
Song N, Liu ZS, Xue W, Bai ZF, Wang QY, Dai J, et al. NLRP3 phosphorylation is an essential priming event for inflammasome activation. Mol Cell. 2017;68:185–97.
Chen J, Chen ZJ. PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature. 2018;564:71–6.
Chauhan D, Vande Walle L, Lamkanfi M. Therapeutic modulation of inflammasome pathways. Immunol Rev. 2020;297:123–38.
He Y, Hara H, Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. 2016;41:1012–21.
Kotas ME, Medzhitov R. Homeostasis, inflammation, and disease susceptibility. Cell. 2015;160:816–27.
Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–5.
Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21:677–87.
Heneka MT, McManus RM, Latz E. Inflammasome signalling in brain function and neurodegenerative disease. Nat Rev Neurosci. 2019;20:187.
Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ, et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature. 2018;560:198–203.
Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, et al. NF-κB restricts inflammasome activation via elimination of damaged mitochondria. Cell. 2016;164:896–910.
Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36:401–14.
Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;475:221–25.
Han Y, Xu X, Tang C, Gao P, Chen X, Xiong X, et al. Reactive oxygen species promote tubular injury in diabetic nephropathy: the role of the mitochondrial ros-txnip-nlrp3 biological axis. Redox Biol. 2018;16:32–46.
Abais JM, Xia M, Zhang Y, Boini KM, Li PL. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid Redox Signal. 2015;22:1111–29.
Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov. 2018;17:588–606.
Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell. 2013;153:348–61.
Yu JW, Lee MS. Mitochondria and the NLRP3 inflammasome: physiological and pathological relevance. Arch Pharm Res. 2016;39:1503–18.
Lee J, Giordano S, Zhang J. Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem J. 2012;441:523–40.
Kubli DA, Gustafsson ÅB. Mitochondria and mitophagy: the yin and yang of cell death control. Circ Res. 2012;111:1208–21.
Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 2014;157:65–75.
Kim MJ, Yoon JH, Ryu JH. Mitophagy: a balance regulator of NLRP3 inflammasome activation. BMB Rep. 2016;49:529–35.
Onishi M, Yamano K, Sato M, Matsuda N, Okamoto K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021;40:e104705.
Birgisdottir ÅB, Lamark T, Johansen T. The LIR motif—crucial for selective autophagy. J Cell Sci. 2013;126:3237–47.
Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12:222–30.
Elena-Real CA, Díaz-Quintana A, González-Arzola K, Velázquez-Campoy A, Orzáez M, López-Rivas A, et al. Cytochrome c speeds up caspase cascade activation by blocking 14-3-3ε-dependent Apaf-1 inhibition. Cell Death Dis. 2018;9:365.
Lu C, Li Y, Hu S, Cai Y, Yang Z, Peng K. Scoparone prevents IL-1β-induced inflammatory response in human osteoarthritis chondrocytes through the PI3K/Akt/NF-κB pathway. Biomed Pharmacother. 2018;106:1169–74.
Hui Y, Wang X, Yu Z, Fan X, Cui B, Zhao T, et al. Scoparone as a therapeutic drug in liver diseases: pharmacology, pharmacokinetics and molecular mechanisms of action. Pharmacol Res. 2020;160:105170.
Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–21.
Wang Y, Yang F, Zhong C, Ye X, He X, Ouyang D. Scutellarin inhibits caspase-11 activation and pyroptosis in macrophages via regulating PKA signaling. Acta Pharm Sin B. 2021;11:112–26.
Song Y, Ge X, Chen Y, Hussain T, Liang Z, Dong Y, et al. Mycobacterium bovis induces mitophagy to suppress host xenophagy for its intracellular survival. Autophagy. 2021;31:1–15.
Yang F, Ye XJ, Chen MY, Li HC, Wang YF, Zhong MY, et al. Inhibition of NLRP3 inflammasome activation and pyroptosis in macrophages by taraxasterol is associated with its regulation on mTOR signaling. Front Immunol. 2021;12:632606.
Lyu L, Chen J, Wang W, Yan T, Lin J, Gao H, et al. Scoparone alleviates Ang II-induced pathological myocardial hypertrophy in mice by inhibiting oxidative stress. J Cell Mol Med. 2021;25:3136–48.
Zhang A, Sun H, Wang X. Urinary metabolic profiling of rat models revealed protective function of scoparone against alcohol induced hepatotoxicity. Sci Rep. 2014;4:6768.
Gao Y, Xi B, Li J, Li Z, Xu J, Zhong M, et al. Scoparone alleviates hepatic fibrosis by inhibiting the TLR-4/NF-κB pathway. J Cell Physiol. 2020;236:3044–58.
Wang B, Lim JH, Kajikawa T, Li X, Vallance BA, Moutsopoulos NM, et al. Macrophage β2-integrins regulate IL-22 by ILC3s and protect from lethal citrobacter rodentium-induced colitis. Cell Rep. 2019;26:1614–26.
Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. 2013;341:1246–9.
Man SM, Kanneganti TD. Regulation of inflammasome activation. Immunol Rev. 2015;265:6–21.
Zhu YY, Ouyang ZJ, Du HJ, Wang MJ, Wang JJ, Sun HY, et al. New opportunities and challenges of natural products research: when target identification meets single-cell multiomics. Acta Pharm Sin B. 2022. https://doi.org/10.1016/j.apsb.2022.08.022.
Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, Engels G, et al. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat Immunol. 2014;15:727–37.
Baroja-Mazo A, Martín-Sánchez F, Gomez AI, Martínez CM, Amores-Iniesta J, Compan V, et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol. 2014;15:738–48.
Wang Z, Xu G, Gao Y, Zhan X, Qin N, Fu S, et al. Cardamonin from a medicinal herb protects against LPS-induced septic shock by suppressing NLRP3 inflammasome. Acta Pharm Sin B. 2019;9:734–44.
Shi W, Xu G, Zhan X, Gao Y, Wang Z, Fu S, et al. Carnosol inhibits inflammasome activation by directly targeting HSP90 to treat inflammasome-mediated diseases. Cell Death Dis. 2020;11:252.
He H, Jiang H, Chen Y, Ye J, Wang A, Wang C, et al. Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat Commun. 2018;9:2550.
Guo W, Sun Y, Liu W, Wu X, Guo L, Cai P, et al. Small molecule-driven mitophagy-mediated NLRP3 inflammasome inhibition is responsible for the prevention of colitis-associated cancer. Autophagy. 2014;10:972–85.
Palikaras K, Lionaki E, Tavernarakis N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol. 2018;20:1013–22.
Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264–8.
Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157:1013–22.
Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Förster I, et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity. 2011;34:213–23.
Acknowledgements
This work was supported by the National Natural Science Foundation (NNSF) of China (Nos. 91942301, 31900661 and 82001663), Young Elite Scientists Sponsorship Program by China Association for Science and Technology (No. 2020-QNRC1-03), National Key Research and Development Project (No. 2019YFC1710104), and Joint Fund of Beijing University of Traditional Chinese Medicine and USANA. We thank Dr. Chen Dong for the C. rodentium. We are indebted to all individuals who participated in or helped with this research project.
Author information
Authors and Affiliations
Contributions
ALX conceived the study; WDF and TL designed and performed most of the experiments and collected results. WDF analyzed, interpreted results and wrote the manuscript. YW and XJ designed research and instructed experiment. YW helped to analyze and interprete results, and revised the manuscript. CQC participated in H&E performance. HJW contributed to RNA-seq analysis. MQZ instructed article figure drawing. QQL, XJW, YYL and JYW helped to establish the animal model. GRH and TW oversaw a portion of the work and provided advice. ALX provided funding for the project, and supervised the project and revised/approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Feng, Wd., Wang, Y., Luo, T. et al. Scoparone suppresses mitophagy-mediated NLRP3 inflammasome activation in inflammatory diseases. Acta Pharmacol Sin 44, 1238–1251 (2023). https://doi.org/10.1038/s41401-022-01028-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-022-01028-9
