
Abstract
Cyclin-dependent kinase (CDK) 9 associates mainly with cyclin T1 and forms the positive transcription elongation factor b (p-TEFb) complex responsible for transcriptional regulation. It has been shown that CDK9 modulates the expression and activity of oncogenes, such as MYC and murine double minute 4 (MDM4), and it also plays an important role in development and/or maintenance of the malignant cell phenotype. Malfunction of CDK9 is frequently observed in numerous cancers. Recent studies have highlighted the function of CDK9 through a variety of mechanisms in cancers, including the formation of new complexes and epigenetic alterations. Due to the importance of CDK9 activation in cancer cells, CDK9 inhibitors have emerged as promising candidates for cancer therapy. Natural product-derived and chemically synthesized CDK9 inhibitors are being examined in preclinical and clinical research. In this review, we summarize the current knowledge on the role of CDK9 in transcriptional regulation, epigenetic regulation, and different cellular factor interactions, focusing on new advances. We show the importance of CDK9 in mediating tumorigenesis and tumor progression. Then, we provide an overview of some CDK9 inhibitors supported by multiple oncologic preclinical and clinical investigations. Finally, we discuss the perspective and challenge of CDK9 modulation in cancer.
Similar content being viewed by others

Introduction
Cyclin-dependent kinase (CDK) 9 is a prominent member of the transcriptional CDK subfamily [1]. Associated with cyclin T1/T2, CDK9 forms the catalytic subunit of the positive transcription elongation factor b (p-TEFb) that is required for the phosphorylation of the RNA polymerase II (RNA Pol II) C-terminal domain (CTD) and transcription elongation [1, 2]. It has been shown that CDK9 modulates the expression and activity of oncogenes, such as MYC [3] and murine double minute 4 (MDM4) [4], and plays an important role in the development and/or maintenance of the malignant cell phenotype [5,6,7]. Dysregulation of the CDK9 pathway has been observed in a range of human tumors, such as acute myeloid leukemia [8], prostate cancer [9], colorectal cancer [10], and pediatric soft tissue sarcomas [11]. Due to its essential role in the development and progress of malignancy, CDK9 has emerged as a novel prognostic marker and an appealing target for cancer therapy [12,13,14,15]. Small-molecule inhibitors and natural compounds targeting CDK9 are being investigated in preclinical and clinical studies because they exert effective antiapoptotic and antitumor activity [16, 17]. In this review, we provide an updated overview of the biological characteristics of CDK9, focus on representative CDK9 modulators, and provide new insight into the roles of CDK9 in cancer.
Cyclin-dependent kinase 9: a key transcriptional regulator
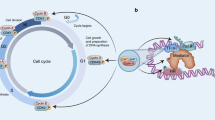
CDK9 is the catalytic subunit of p-TEFb that, in association with cyclin T, modulates RNA transcriptional elongation [1]. In the majority of active mammalian genes, RNA Pol II transcribes 30-60 nucleotides before elongation is interrupted by a regulated pause in the promoter-proximal regions [18]. The pausing is mainly regulated by two pause-promoting factors, the DRB sensitivity inducing factor (DSIF) and the negative elongation factor (NELF) [1]. Then, the active p-TEFb/CDK9 complex is recruited by the bromodomain-containing protein 4 (BRD4) and the super elongation complex (SEC). In this process, CDK9 phosphorylates three main substrates: Ser2 of RNA Pol II CTD, the DSIF, and the NELF, finally releasing the RNA Pol II from the pause status and assuring productive initiation of RNA synthesis [11, 15, 17] (Fig. 1).

CDK9 associates with cyclin T1 forming the p-TEFb complex. RNA Pol II is paused after adding a few dozen nucleotides to the transcript due to the DSIF and NELF influence. After activation and recruitment, the p-TEFb complex phosphorylates three main substrates: Ser2 of RNA Pol II CTD, the DSIF and the NELF, to release RNA Pol II from pause status and activate the productive elongation phase of transcription.
Novel links between CDK9 function and transcription were provided by several recent studies. It was discovered that small ubiquitin-like modifier (SUMO) regulates cell transcription by controlling the sumoylation of CDK9 and the regulation of transcriptional elongation, while MYC amplifies gene expression by antagonizing the sumoylation of CDK9 [19]. The results obtained by Ma et al. indicate that in human immunodeficiency virus-1 (HIV-1) infection CDK9 is significantly sumoylated by TRIM28, which inhibits the CDK9 kinase activity and p-TEFb assembly, subsequently suppressing viral transcription and contributing to HIV-1 latency [20]. Furthermore, it was demonstrated that histone deacetylase sirtuin 6 (SIRT6) binds to RNA Pol II and prevents the release of NELF, while deficiency of SIRT6 activates CDK9, enriches histone H3 lysine 9 acetylation (H3K9ac) and histone H3 lysine 56 acetylation (H3K56ac), and facilitates the recruitment of elongation-promoting factors (including MYC and BRD4), the SEC factors AF4/FMR2 family member 4 (AFF4) and elongation factor for RNA polymerase II 2 (ELL2) [21]. CDK9-mediated TAT-interacting protein 60 (TIP60) phosphorylation regulates the affinity of TIP60 to histone 3 and RNA Pol II, while the lysine acetyltransferase (KAT) activity of TIP60 is mainly regulated by the phosphorylation on S86, which is also dependent on the phosphorylation of S90 [22]. CDK7-mediated CDK9 Ser175 phosphorylation promotes the assembly of p-TEFb with trans-activator of transcription (TAT) and provirus HIV reactivation [23]. Moreover, CDK9-dependent co-and/or post-transcriptional events involving Spt5 and infectious cell culture protein 27 (ICP27) play crucial roles in the expression of herpes simplex virus 1 replication-dependent late genes [24].
The role of CDK9 in cancer
Clinical implications of CDK9 in cancer
Genomic alterations of CDK9 are relatively uncommon in human cancers and, involve mutations, amplifications, deep deletions and multiple alterations (Fig. 2). Due to its low frequency, CDK9 amplification as a primary criterion for inhibitor sensitivity is not available. Previous studies have indicated that dysregulation in CDK9 and the CDK9 pathway are related to oncogenesis and progression in multiple tumor types, such as osteosarcoma [13], ovarian cancer [25], synovial sarcoma [26], pancreatic cancer [27], and endometrial cancer [28]. McLaughlin et al. found that high expression of POLR2A (RNA-II), the essential downstream target of CDK9, is significantly correlated with poor metastasis-free survival in estrogen receptor-negative (ER-neg) breast cancer patients (n = 123), although CDK9 expression level is not [29]. Parvathareddy et al. evaluated CDK9 expression in 441 epithelial ovarian cancer (EOC) samples, and found that CDK9 is over-expressed in 56.2% (248/441) samples, associated with adverse clinic-pathological characteristics, such as distant metastasis (P < 0.0001), stage IV tumors (P < 0.0001), tumor recurrence (P = 0.0105) and high Ki-67 index (P < 0.0001) [30]. Moreover, patients with CDK9high and CDK9low status have shown disease-free survival (DFS) of 12 months and 20 months respectively, suggesting that CDK9 could be an independent predictor of poor DFS [30]. In addition, the association between CDK9 and drug resistance has been recently reported, including neoadjuvant chemotherapy [13], CDK4/6 inhibitors [6], EGFR-TKIs [29] and BCL2 inhibitor [31,32,33].

The data were obtained from www.cbioportal.org, under the set of sample quantity >100 and alteration frequency >1% (accessed on 16 August 2021).
Transcriptional regulation of CDK9 in cancer
To date, multiple CDK9 inhibitors are available and are undergoing clinical trials, and their anti-tumoral effects are mainly exerted via transcriptional regulation of different oncogenic target genes, involved in cell proliferation, apoptosis, migration, angiogenesis, metabolism and stemness characteristics (Fig. 3).

Activated CDK9 in the p-TEFb complex promotes cancer cell proliferation, anti-apoptosis, migration, angiogenesis, metabolism and stemness characteristics via transcriptional regulation of target genes.
In terms of proliferation and apoptosis, it has been determined that CDK9 inhibitors suppress cell proliferation and induce apoptosis across broad cancer cell line panels. Studies also showed reduced tumor burden and satisfactory tolerability upon CDK9 inhibition in vivo. Acute inhibition of CDK9 results in transient transcriptional suppression and preferential depletion of proteins with short half-lives, such as MCL-1, MYC, and CCND1. Therefore, most preclinical studies have attributed the mechanism of action of CDK9 inhibitors predominantly to depletion of MCL-1 and MYC, which are determinants for cancer cell survival and apoptosis. BFL-1 inhibitor alone is insufficient to overcome BFL-1 overexpression associated with intrinsic venetoclax resistance in lymphomas, while CDK9 inhibitor sufficient to downregulate both BFL-1 and MCL-1 [31]. In addition, several studies have revealed that CDK9 has a role in the recovery of WT-p53 function in cancer. It has been reported that CDK9 phosphorylates the p53-induced RING-H2 (PIRH2) protein and attenuates p53 protein degradation in primary microglia and astrocytes [34]. In colorectal tumor cells, CDK9 inhibitors decrease the transcription of inhibitor of apoptosis stimulating protein of p53 (iASPP), promoting p53’s transcriptional activity and its tumor-suppressive activities [35]. Yao et al. demonstrated not only that mouse double minute 2 (MDM2) mediates p53 degradation, but also that sirtuin 1 (SIRT1) activates p53 and its target gene transcription [36]. CDK9 inhibitor suppresses both MDM2 and SIRT1 to increase p53 stability and prevent hepatocellular carcinoma progression [36]. Štětková et al. found that CDK9 inhibitors can also activate p53 in melanoma and breast carcinoma cells by diminishing the MDM4 level [4].
In terms of migration and invasion, it has been shown that inhibition of CDK9 suppresses migration activity in ovarian cancer cells and osteosarcoma cells [13, 25]. CDK9 regulates the expression of matrix metalloproteinase (MMP)-9, but not MMP-2, although both engaged in the digestion of extracellular matrices in cancer cells [37]. Zhang et al. also verified that CDK9 inhibitor partially decreases MMP-9 levels by inhibiting YAP [38]. In the same study, intrasplenic uveal melanoma cell-injected mice displayed decreased metastatic nodules in the livers after being treated with CDK9 inhibitor [38]. Mechanistically, CDK9 transcriptionally regulates stemness-related protein Krüppel-like factor 4 (KLF4) and c-Myc-dependent RhoA, and thereby inhibites migration and invasion of uveal melanoma cells [38].
In terms of angiogenesis, it was reported that CDK9 inhibitors inhibit three-dimensional capillary network formations of endothelial cells, and can be rescued by recombinant vascular endothelial growth factor (VEGF), the most potent tumor angiogenic factor [39]. Moreover, CDK9 inhibition also suppresses intercellular adhesion molecule-1 (ICAM-1) expression, which is important for the activity of endothelial cells and their interaction with tumor cells [40].
In terms of metabolism, Su et al. showed that CDK9 inhibition induces mitochondrial dysfunction in glioblastoma cells via reducing MCL-1 and BIRC5 expression, which induces mitochondrial-mediated apoptosis in glioblastoma cells [41]. CDK9 inhibition downregulates most of the genes involved in respiratory complexes I, III, IV, and V, especially in complex I, as well as disrupted mitochondrial membrane integrity and released cytochrome c from mitochondria [41]. The study also showed that combination regimens of CDK9 inhibitor and temozolomide are lethal to glioblastoma in vivo and in vitro, suggesting a candidate combination for glioblastoma therapy [41]. Carte et al. discovered that MCL-1 regulates metabolic pathways, including the tricarboxylic acid cycle, glycolysis, and pentose phosphate pathway and modulates cell adhesion proteins and leukemia-stromal interactions, which are independent of apoptosis induction by MCL-1 [32]. Inhibition of MCL-1 by the CDK9 inhibitor AZD4573 sensitizes the BCL-2 inhibitor venetoclax, thereby extending the survival of patient-derived xenograft (PDX) mice established from venetoclax-resistant acute myeloid leukemia patients [32]. Mechanically, Huang et al. found that CDK9 inhibitor restrains the glycolysis of B-cell acute lymphocytic leukemia cells via downregulation of glucose transporter type 1 (GLUT1) and the key rate-limiting enzymes of glycolysis, such as hexokinase 2 (HK2) and lactate dehydrogenase A (LDHA) [42].
In terms of stemness characteristics, Wang et al. synthesized a series of CDK9 inhibitors and found that the most potential candidate (named 21e) downregulates NSCLC cells’ sphere formation ability, side population characteristics and stemness markers abundance, which includes SRY (sex-determining region Y)-box 2 (SOX2), octamer-binding transcription factor 4 (OCT4), KLF4, and Nanog [5]. NOTCH signaling is a promising target in glioblastoma due to its function in stem cell fate and self-renewal [43]. However, NOTCH antagonists have demonstrated limited efficacy in clinical trials [44]. Xie et al. proposed that recombining binding protein suppressor of hairless (RBPJ) can be a better target in glioblastoma with higher pharmacologic action [45]. It was reported that RBPJ binds to CDK9 to promote target gene transcription elongation [45]. Moreover, CDK9 targeting decreases the proliferation and self-renewal of brain tumor-initiating cells [45]. These results reveal the important role of CDK9 in maintaining stem cell phenotypes, which are associated with tumorigenesis and drug resistance.
Novel interactions between cellular factors and CDK9 in cancer cells
CDK9 associates with cyclin T1/T2 to form the catalytic subunit of p-TEFb, which is a two-unit molecule required for the proper function of polymerase II [46]. Recent studies have identified other cellular factors that interact with CDK9, including mammalian target of rapamycin (mTOR), anti-silencing function of 1B histone chaperone (ASF1B), signal transducer and transcription 3 (STAT3), and BRD4, suggesting that CDK9 target inhibition may impair the interactions between CDK9 and other cellular factors and cause subsequent antitumor properties (Fig. 4).

Except for cyclin T, recent studies have identified other cellular factors that interact with CDK9. For example, mTOR, ASF1B and STAT3 combine with CDK9 to form complexes that promote tumorigenesis and progression.

The formulas were obtained from previous studies or the PubChem database (https://pubchem.ncbi.nlm.nih.gov/) (accessed on 2 September 2021).
Two novel mTOR-like (CTOR) complexes (CTORC) formed by CDK9 and mTOR play key roles in mRNA transcription and translation of mitogenic genes [47]. In the cytoplasm, in association with the rapamycin-insensitive companion of mammalian target of rapamycin (RICTOR), mammalian stress-activated protein kinase-interacting protein 1 (mSIN1), and mammalian lethal with sec-13 protein 8 (mLST8), CDK9 forms the CTORC2 complex, which is associated with and/or impacted by the phosphorylation of proteins functioning in ribosomes that control mRNA translation. In the nucleus, CDK9, RAPTOR, and mLST8 combine to form the CTORC1 complex, which is found in gene promoter sites to promote the transcription of certain oncogenes. CDK9 inhibition impaires CTORC complex formation and suppresses the malignant phenotype of acute myelogenous leukemia (AML) in vitro and in vivo, and synergizes with cytarabine, a key component of standard chemotherapy for AML patients [47].
Sandrine et al. provided the first evidence that STAT3 is involved in chromatin remodeling and transcriptional elongation by interacting with brahma-related gene 1 (BRG1) and CDK9 [48]. Interaction between STAT3 and BRG1 increases the accessibility of the p21waf1 promoter so that it can bind to RNA Pol II. Then, STAT3 recruits CDK9 to phosphorylate RNA Pol II Ser 2 and promotes the prolongation of cell transcription [48]. Hou et al. extended their previous study to show that the STAT3-CDK9 complex can stimulate gamma-fibrinogen (γ-FBG) gene recruitment and prolong transcription [48]. Acetyl-bufalin shows potent efficacy against non-small cell lung cancer (NSCLC) by impairing the formation of the CDK9-STAT3 complex, verifying that it can be used as a possible target for NSCLC and other cancers [49]. It was reported that ASF1B positively regulates CDK9 stabilization and forms stable complexes with CDK9, promoting cell proliferation and reducing apoptosis in cervical cancer [50]. Disruption of ASF1B by targeting CDK9 may elicit strong anti-cervical tumor responses [50]. Zhang’s results defined a function for sirtuin 2 (SIRT2) in regulating checkpoint pathways that respond to replication stress through the deacetylation of CDK9, providing insight into how SIRT functions in protecting genomic integrity, and even in cancer progression [51]. The SIRT2-mediated deacetylation of CDK9 promotes STAT1 signaling to regulate the transcription of interferon-stimulated genes and interferon-driven antiproliferative responses [52]. This study suggests that targeting the SIRT2-CDK9-STAT1 axis can provide therapeutic benefits for immune-related diseases and cancer [52].
BRD4 has emerged as a key epigenetic modulator for gene transcription and cancer development [53, 54]. Biochemical and immunofluorescence data identified an interaction between BRD4 and the active kinase form of CDK9 in the nuclei of human cells [55, 56]. Two regions of BRD4 directly bind to p-TEFb. The CTD interacts with cyclin T1 and CDK9, while the second bromodomain (BD2) region interacts with the acetylated region of cyclin T1. Previous publications indicated that the BRD4-p-TEFb interaction plays a central role in the rapid initiation of transcription after the exit from mitosis [57]. The coordination between BRD4 and CDK9 promotes TGF-β-induced Nox4 expression and myofibroblast transition, and inhibition of the CDK9-BRD4 pathway may be a useful strategy to limit hypertrophic scar formation [55]. Wang and colleagues discovered that the kinase activity of CDK9 in the p-TEFb complex stimulates BRD4 in NUT midline carcinoma [7]. Blocking the BRD4-CDK9 interaction reduces BRD4 hyperphosphorylation, represses expression of downstream oncogenes SOX2 and MYC, and abolishes cellular transformation [7]. Fisher et al. proposed a phosphorylation-dephosphorylation cycle whereby the p-TEFb complex catalyzes the inhibitory phosphorylation of PP1 and PP4 that is localized to the 3′ and 5′ ends of genes, respectively, to govern pause-release and the elongation-termination transition [58].
CDK9 epigenetically regulates genes in cancer
CDK9 has also been implicated in epigenetic regulation, expanding its functional repertoires. A tremendous number of studies in the last two decades have centered on the recognition that epigenetic alterations are associated with almost every step of tumor development and progression [59,60,61]. It is now widely recognized that genomic and epigenomic mutations, as well as environmental factors, work in concert to modify cellular and organismal pathobiology. Epigenetic modifications involve changes to the chromatin structure and result in differential gene expression rather than changes to the DNA sequence [61]. The major epigenetic regulators include DNA methylation enzymes, histone modifiers and readers, chromatin remodelers, non-coding RNAs, and other components of chromatin [62, 63].
Most recently, it was found that CDK9 provides guidance for a complex network of chromatin modifications and translational control. Previous work showed that the inhibition or knockdown of CDK9 resulted in a nearly complete loss of histone H2B mono-ubiquitylation (H2Bub1) in tumor cells, linking the unique cross-talk between CDK9 activity and histone code in the regulation of histone mRNA 3’-end processing, as the inhibition of CDK9 results in the insufficient formation of the 3’-end of histone messenger RNA [64]. Furthermore, Miriam et al. uncovered gene regulatory mechanisms dependent on CDK9 and H2Bub1 and revealed that CDK9 and H2Bub1 collaborate to suppress antisense transcription through the histone deacetylase Clr6-CII in fission yeast [65].
In You’s study, BRD4 and the p-TEFb complex are enriched at the BRD4-NUT-induced histone-hyperacetylated chromatin domain, thus stimulating BRD4 hyperphosphorylation [7]. Using an inhibitor to disrupt the p-TEFb-BRD4 interaction in NUT midline carcinoma (NMC) cells led to the repression of BRD4 downstream oncogenes SOX2 and MYC and abrogation of NMC cellular transformation [7]. This finding revealed the mechanism of CDK9 dysregulating BRD4, which is an important epigenetic reader via phosphorylation that could lead to tumorigenesis. Surprisingly, recent data indicate that bromodomain and bromodomain extra-terminal (BET) proteins function as master transcription elongation factors independent of CDK9 recruitment [66]. Targeted BET degradation disruptes the CTD phosphorylation of RNA Pol II and global productive elongation, but no significant abrogation of chromatin-engaged CDK9 was observed [66]. However, researchers cannot rule out whether the subsequent recruitment of p-TEFb will affect this process [66]. Moreover, future studies are needed to address the complex connections between BRD4 and CDK9, as well as their independent functions in RNA Pol II phosphorylation and elongation control.
Jean et al. presented preliminary evidence that CDK9 is essential for maintaining gene silencing at heterochromatic loci [14]. First, through genetic inhibition and overexpression of CDK9, the researchers identified CDK9 as a novel epigenetic target that is required to maintain transcriptional repression at epigenetically silenced loci [14]. CDK9 inhibition has a bimodal effect, in that one gene subset initially undergoes downregulation, with subsequent upregulation for another. Most of these silenced or upregulated genes are highly hypermethylated in their promoters, indicating that CDK9 inhibition is irrelevant for the occurrence of DNA demethylation [14]. Second, they delved into the mechanism of CDK9-mediated repression and found that CDK9 directly mediates the phosphorylation of BRG1, which is a central component of the SWI/SNF family that regulates chromatin structure [67]. Global and diffuse relaxation of chromatin caused by CDK9 inhibition indicated that CDK9 is involved in chromatin remodeling. Based on the above, the group gave rise to the theory that CDK9-mediated phosphorylation of BRG1 leads to its release from chromatin [14]. Then, in vitro and in vivo experiments further demonstrated that specific CDK9 inhibition could hinder colorectal tumor proliferation and sensitize ovarian tumors to the immune checkpoint inhibitor α-PD-1 in mice [14]. A previous study reported that CDK9 mediates the phosphorylation of the SWI/SNF complex components in HIV-1-infected T-cells, and phosphorylation of the SWI/SNF component Baf53 leads to its release from DNA [68]. Given its mechanisms governing enhancer regulation (e.g., MYC) [17], it remains to be seen how CDK9 can maintain a balance between upregulating and silencing genes.
Several miRNAs are extensively involved in the process of tumorigenesis by regulating cell cycle, metastasis, angiogenesis, metabolism, and apoptosis in different types of cancer [69, 70], including pancreatic [71], ovarian [72], and lung cancer [73], and therefore represent a new molecular marker and therapeutic strategy to regulate gene expression through the epigenetic machinery [74, 75]. CDK9 not only controls myogenic transcription factors (MyoD and Srf), but also increases the expression of miRNA1 and miRNA206 and inhibits the expression of miRNA133, thereby modulating myocyte progenitor cell growth, differentiation, and apoptosis [76]. Additional studies that concentrate on elucidating the mechanism of how miRNA is regulated by CDK9 in tumor progression will provide valuable data.
Small molecule inhibitors targeting CDK9
According to the role of CDK9 in cancer development and progression, multiple CDK9 inhibitors are currently under development. However, most CDK9 inhibitors, especially those in clinical development, are broad-spectrum inhibitors due to the highly conserved structure of CDKs [77]. Although the antitumor activity of pan-CDK inhibitors has been clinically tested, few monotherapy developments have advanced beyond phase I/II trial evaluation due to narrow safety margins and poor pharmacokinetic properties [78]. Importantly, a combinational approach for CDK-targeted agents may be more effective than monotherapy, as some combination trials are currently ongoing. Natural compounds have been proved to be a rich source of target drugs, including CDK9 inhibitors, due to their diverse biological properties and chemical structures. Here we present several natural product-derived and chemically synthesized CDK9 inhibitors, especially those that have entered clinical trials (http://clinicaltrials.gov/) (Fig. 5, Table 1).
Representative natural product-derived CDK9 inhibitors
Flavopiridol, a synthetic flavonoid derived from rohitukine, inhibits multiple CDKs, including CDK1, CDK2, CDK4, CDK6, CDK7, and CDK9 [79]. Preclinical findings have demonstrated its ability against breast cancer, prostate cancer, colorectal cancer, and esophageal cancer, as well as leukemia and lymphoma [79]. Flavopiridol is the first CDK inhibitor entering clinical trials. However, most single-agent clinical trials have failed due to insignificant benefits or severe adverse effects, as reported in Table 1 [80,81,82,83]. Monotherapy has resulted in few objective responses in patients with relapsed chronic lymphocytic leukemia (NCT00058240) [84, 85]. In combination with other anti-cancer drugs, evidence for a positive therapeutic outcome has been mixed. When combined with cytosine arabinoside plus mitoxantrone, flavopiridol has passed safety and tolerability tests and shown encouraging clinical responses in patients with poor-risk AML in a phase I/II trial (NCT00016016) [86]. This combination has also shown promising results in newly diagnosed and relapsed AML patients, with complete remissions (CR) observed in 75% of patients [86]. In a phase II study (NCT00083122), a flavopiridol and cisplatin regimen displayed clinical activity and manageable toxicity in patients with platin-resistant ovarian and primary peritoneal carcinoma, with a 17.5% partial response (PR) + CR rate, median time to progression of 4.3 months, and overall survival of 16.1 months [87]. Doses of (irinotecan 100 mg/m2 + flavopiridol 60 mg/m2/irinotecan 125 mg/m2 + flavopiridol 50 mg/m2) have been proven to be safe, but with limited antitumor activity in a phase I trial (NCT00006485) [88]. Researchers also conducted a phase I trial (NCT00042874), demonstrating the combination efficacy with flavopiridol and oxaliplatin and fluorouracil/leucovorin (FOLFOX) in patients with advanced solid tumors [89]. Particularly, in platinum-refractory germ cell tumors, this combination has shown promising results with 33% partial response and 70% decrease in serum tumor markers [89]. However, when combined with docetaxel in metastatic breast cancer patients, dose-limiting toxicities frequently occurred either with 72-h continuous infusion or with dose escalation for 1-h in a phase I trial (NCT00020332) [90]. Several serious adverse reactions have been observed in the conducted trials, such as tumor lysis syndrome, secretory diarrhea, neutropenia, cytokine release syndrome, asthenia and vascular thrombosis. Due to the relatively high incidence of serious toxicity, enthusiasm for the research of flavopiridol has waned, even in combination therapy.
IIIM-290, derived from the natural product rohitukine, is an oral CDK2/9 inhibitor exhibiting therapeutic efficacy in colon, pancreatic, and leukemia xenograft models [91]. The IC50 values for in vitro inhibition of CDK2/A and CDK9/T1 are 16 ± 1 nM and 1.9 ± 1 nM, respectively [91]. Due to poor solubility, the efficacious antitumor dose in vivo is 50 mg/kg [91], although bio-pharmaceutical improvement is currently under investigation [92, 93]. Mubashir et al. found IIIM-290 induces p53-dependent mitochondrial apoptosis, including mitochondrial membrane potential impairment, reactive oxygen species (ROS) enhancement, PUMA expression, cytochrome c release and caspase activation [94].
Meriolin is a chemical structural hybrid synthesized by meridianins and variolins, both derived from marine invertebrates. Meijer et al. found meriolin 3 inhibits CDK1, CDK2, CDK4 and CDK9, and its cytotoxicity is validated in multiple cancer cell types [95]. The mechanism for meriolin 3‐triggered apoptosis related to CDK inhibition activity is as follows: CDK9 inhibition down-regulates short-live MCL-1 expression; CDK1 inhibition reduces the sequestration of proapoptotic protein MCL-1; and CDK1 and CDK2 inhibition blocks cell cycle transition [95, 96].
Oroxylin A, a methylated metabolite of baicalein, possesses antitumor activity against a wide range of human neoplasms, such as breast cancer, colorectal cancer, liver cancer, and myelogenous leukemia [97,98,99,100]. The tumor suppressor protein p53 coordinates key biological processes, such as cell death, DNA repair, metabolic homeostasis and immunomodulation, and thereby p53 activation is considered an attractive target for cancer therapy [101, 102]. Wei et al. proved oroxylin A blocks hepatocellular carcinoma cancer progression by inhibiting CDK9-associated MDM2 and SIRT1 signaling and stabilizing the level of WT-p53 [102]. Recovered WT-p53 functions as a genome guardian to suppress tumor progression. Due to their structural complexity, natural compounds always affect multi-targets and pathways. In another study, Yang et al. have reported oroxylin A impairs CDK7 stability, resulting in G2/M phase arrest [103]. Zhang et al. also found oroxyloside, a metabolite of oroxylin A, possesses CDK2/D1 inhibition properties, leading to cell cycle G0/G1 arrest [104]. Currently, no relevant clinical trial is ongoing.
Bufalin is a cardiotonic steroid and an active ingredient extracted from the Chinese medicine chansu. Yang et al. optimized bufalin into acetyl-bufalin for more optimal pharmacokinetics and lower toxicity [49]. Further experiments validated the increased antitumor activity of acetyl-bufalin on cell proliferation, apoptosis, migration, and tumor burden. Molecular docking, high throughput proteomics and Western blot demonstrated the binding interaction between acetyl-bufalin and CDK9. This study suggests that acetyl-bufalin, as a CDK9 inhibitor, might be a potential candidate for NSCLC therapy [49].
Wogonin, a natural compound isolated from the plant Scutellaria baicalensis, can selectively inhibit CDK9 activity. Bian et al. synthesized and evaluated a series of wogonin-derived CDK9 inhibitors, the most specific named as 20b and LBJ-23 [105, 106]. 20b and LBJ-23 exhibit strong antiproliferative activity against a panel of tumor cell lines by inducing apoptotic cell death. They also synthesized CDK9-targeted proteolysis targeting chimera (PROTAC) 11c, which induces degradation of CDK9 protein and antiproliferation of CDK9-overexpressed tumor cell [107].
Lu et al. discovered a coumarin derivative acting as a CDK9 inhibitor, named 30i [108]. 30i displays 160 to 978-fold selectivity over CDK1/2/3/4/5/6 and 3250-fold selectivity over CDK7/8/19. Substituent coumarin moiety occupied the flexible hinge/aD region of CDK9, which shows the steric hindrance in other CDKs, explains the high CDK9 selectivity. Furthermore, 30i’s antitumor activity has been demonstrated both in vivo and in vitro [108].
Selective chemically synthesized CDK9 inhibitors
Bernard and colleague discovered AZD4573, a new selective CDK9 inhibitor effective against hematological malignancies [109]. Its known affinities in MCF7 cells encompass CDK 9 (IC50 = 0.014 μM), CDK1 (IC50 = 0.37 μM), CDK7 (IC50 = 1.1 μM), CDK4/6 (IC50 = 1.1 μM) and CDK2 (IC50 > 10 μM) [109]. Gene expression and proteomic analyses revealed that MCL-1 is the most significantly modulated gene in AZD4573-treated cells, while the other members of the BCL-2 family remain unchanged [16]. The combination of AZD4573 with the BCL-2 inhibitor venetoclax showed promising results in AZD4573-resistant models, raising the possibility of combination therapy [16]. Notably, a phase I trial (NCT03263637) and a phase I/II trial (NCT04630756) are being evaluated in hematological malignancy patients to assess the safety, tolerability, preliminary antitumor activity, and synergism with acalabrutinib. Clinical trials with AZD4573 are still in their infancy, and further efforts are urgently needed.
Vladimir et al. found a selective CDK9 inhibitor, designated CAN508, through further optimizing the pharmacophore in 4-arylazo-3,5-diamino-1H-pyrazoles [110]. CAN508 exhibits potent inhibitory activity against CDK1/B (IC50 = 44.02 ± 7 μM), CDK2/E (IC50 = 20 ± 6 μM), CDK2/A (IC50 = 69 ± 1 μM), CDK4/D1 (IC50 = 13.5 ± 3.1 μM), CDK7/H (IC50 = 26 ± 13 μM), CDK9/T1 (IC50 = 0.35 ± 0.04 μM) in the presence of 100 μM ATP [110]. CAN508 treatment induces apoptosis and suppressed proliferation in human colorectal cancer and breast cancer cells, as well as in esophageal adenocarcinoma cell xenograft models [40, 110, 111]. CAN508 also exerts anti-angiogenic effects by reducing transcription of vascular endothelial growth factor (VEGF) [40].
FIT-039 inhibits the kinase activity of CDK9/T1 (IC50 = 5.8 μM) and CDK4/D3 (IC50 = 30 μM), while it is inactive against other CDKs, including CDK2, CDK5, CDK6 and CDK7 [112]. FIT-039 inhibits HPV viral replication and expression of E6 and E7 viral oncogenes, reducing the tumor burden in HPV + cervical cancer xenografts [113]. FIT-039 has also been clinically evaluated in a phase I/II study of patients with verruca caused by the human papilloma virus (UMIN000029695). The safety and tolerability of FIT-039 patches have been evaluated in patients with cutaneous warts [114]. To date, there is no randomized clinical trial focusing on the safety and efficacy of FIT-039 in tumors.
Atuveciclib (BAY 1143572) was optimized to inhibit CDK9/T1 (IC50 = 6 μM) and showed over 100-fold selectivity for other CDK/cyclin complexes, such as CDK1/B (IC50 = 1100 μM), CDK2/E (IC50 = 1000 μM), CDK3/E (IC50 = 890 μM), CDK5/p53 (IC50 = 1600 μM), CDK6/D (IC50 > 10000 μM) and CDK7/H (IC50 > 10000 μM) [115]. Tumor regression and even remission were observed following daily oral administration of BAY 1143572 in human AML models in female NMRI nu/nu mice (MOLM-13) and athymic rats (MV4-11)[115]. Subsequent experiments by Narita et al. also proved its effectiveness and safety in adult T cell leukemia (ATL) cells in preclinical models [116]. Similarly, the utility of BAY 1143572 in the treatment of natural killer cell leukemia/lymphoma was also confirmed in vitro and in vivo [117]. In addition to hematological malignancies, BAY 1143572 suppresses cell proliferation and invasion in triple-negative breast cancer (TNBC) cells, suggesting CDK9 is a potential therapeutic target in CDK9-high TNBC [118]. Synergistic antitumor effects were observed when BAY 1143572 is combined with 5-fluorouracil [119], cisplatin [118], and the BCL2 inhibitor venetoclax [120]. Encouraged by a promising overall profile in vitro and in vivo, two phase I trials of BAY 1143572 have been completed using patients with advanced cancer and leukemia (NCT01938638, NCT02345382). However, both studies were terminated early due to severe adverse events (e.g., neutropenia) and the lack of clinical responses at any of the doses tested [121, 122].
SNS-032, an N-acyl-2-aminothiazole originally developed as a selective CDK2 inhibitor (IC50 = 38 nM), was later discovered to be a potent inhibitor of CDK7 (IC50 = 62 nM) and CDK9 (IC50 = 4 nM). This drug has been effective against leukemia cells [123, 124], while it has also inhibited tumor growth respectively in xenografts of uveal melanoma and breast cancer [38, 125]. It has also been demonstrated to sensitize non-small cell lung cancer to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis both in vitro and in vivo [126]. In preclinical settings, it reduces angiogenesis, hypoxia-mediated cell invasion, antiapoptotic proteins transcription and abrogated oncogene addiction [39, 127,128,129]. In particular, SNS-032 prevents cancer metastasis in uveal melanoma cell xenografts through transcriptional inhibition of stemness-associated proteins and C-MYC-dependent genes [38]. Considering that the current CDK9 inhibitors are reversible and require continuous target occupancy to maintain CDK9 inhibition, Olson and colleagues designed a hybrid compound, THAL-SNS-032, formed by the conjugation of SNS-032 to thalidomide [130]. This optimized compound exhibits higher selectivity for CDK9 in a cereblon (CRBN)-dependent fashion, and prolongs the inhibitory effect compared to traditional kinase inhibitors [130]. Two phase I clinical trials of SNS-032 (NCT00292864, NCT00446342) in patients with solid and hematologic malignancies have been completed. The results showed the tolerability and limited clinical efficacy of SNS-032, although the maximum administered dose (MAD) was not reached due to a decision made by the sponsor [131, 132].
KB-0742 is a novel and highly selective CDK9 inhibitor currently being investigated in phase I studies (NCT04718675). Richters’s group attempted to develop AR-variant activity modulators, and they discovered a potent and selective CDK9 inhibitor, KI-ARv-03 [133]. Optimization of this compound into KB-0742 increased its shape complementarity with the CDK9 ATP-binding pocket, resulting in an ultra-selective and single-digit nanomolar potent CDK9 inhibitor [133].
Considering the adverse events, Lücking et al. optimized atuveciclib into the CDK9 inhibitor BAY 1251152 (IC50 = 3 μM) with high selectivity against other CDKs, such as CDK2 (ratio of IC50 values 1033), and CDK7 (ratio of IC50 values > 5000) [134]. Pharmacokinetic studies in rats showed that BAY 1251152 has a low blood clearance (CLb: 1.1 L·h−1·kg−1), a moderate volume of distribution (Vss: 0.74 L/kg), and a short to moderate half-life (t1/2: 1.0 h) [134]. Furthermore, BAY 1251152 exhibited a blood/plasma ratio of approximately 1 and did not show significant inhibition of cytochrome P450 activity, with IC50 values > 20 μM [134]. In the MV4-11 xenograft model, BAY 1251152 (4.5 mg/kg, Q7D ×3, iv.) exhibits increased antitumor efficacy and safety compared with atuveciclib (12 mg/kg × 15 days, po) [134]. BAY 1251152 is currently being evaluated in phase I studies to determine the safety, tolerability, pharmacokinetics, and initial pharmacodynamic biomarker response in patients with advanced cancer (NCT02745743, NCT02635672). Although satisfactory safety and tolerability were observed, BAY 1251152 did not result in objective responses in AML patients in one phase I trial (NCT02745743) [135, 136].
Future perspectives
Since it was first identified in the early 1990s, CDK9 has been shown to be a primary participant in several physiological and pathological pathways [137]. Due to being a part of the major pathways responsible for the development of highly prevalent pathologies, such as cancer, CDK9 represents a new class of pharmaceutical targets. Although CDK9 traditionally acts in transcription elongation, recent studies revealed that it also interacts with a wide range of proteins involved in translation, RNA processing, translation termination, and epigenetic modifications [58, 138, 139]. Research efforts should continue to elucidate CDK9’s functions to understand the higher-order mechanism responsible for the development of malignancies.
Additional work on the development of CDK9 inhibitors will also be important, as the following reasons explain. (1) Searching for more potent and selective CDK9 inhibitors is still necessary. Most known CDK9 inhibitors lack high selectivity for the conserved structures of CDKs [77], and these CDKs are pivotal regulators of the cell cycle and transcription process under physiological conditions. An ideal CDK9-targeted agent should exhibit high affinity, low toxicity, and enhanced specificity and efficacy against tumor cells. In this review, we summarize a series of selective CDK9 inhibitors currently being investigated, some having demonstrated satisfactory antitumor activity. (2) Searching for optimal therapies mode. CDK9 is involved in extensive crosstalk with other signaling pathways in cancer cells. Thus, combination therapy may avoid potential drug resistance and elicit more powerful antitumor responses. Preclinical results have suggested that CDK9 inhibitors may synergize with BCL-2 inhibitors, PARP inhibitors, EGFR inhibitors, BET inhibitors, and 5-fluorouracil [29, 119, 140,141,142]. Moreover, it was reported that the CDK7/9 inhibitor PHA-767491 overcomes bone marrow stroma-mediated drug resistance and sensitizes AML cells to BH3-mimetics [143]. (3) Identifying sensitive patient groups. Three selective CDK4 and CDK6 inhibitors have recently received FDA approval for the treatment of hormone receptor (HR)-positive, advanced-stage breast cancer in combination with antihormonal agents [144, 145]. This success demonstrates the importance of defined robust predictive biomarkers to select patients who may benefit from these therapies. Acute inhibition of CDK9 results in transient transcriptional suppression and preferential depletion of proteins with short half-lives, such as MCL-1 and MYC. MCL-1 and MYC-driven cancers, such as leukemia, prostate cancer and breast cancer, may therefore be satisfactory candidates for CDK9 inhibitors [17, 146, 147]. However, current clinical trial data are limited and, even yielded contradictory effects. The selection of correct target patients is crucial and needs to be improved through the study of larger samples and clinical experiments. Moreover, biomarker strategies should be established for patient selection because CDK9 mutations are rare in cancer. Further investigations on biomarkers of CDK9 overactivation may help identify specific target cancer patients and thus improve clinical outcomes.
In summary, CDK9’s role has increasingly been the focus of international attention, especially in cancer. We eagerly wait for the entry of CDK9 inhibitors into the clinical armamentarium against cancer and optimal therapies mode, which may improve the lives of cancer patients in the future.
References
Franco LC, Morales F, Boffo S, Giordano A. CDK9: A key player in cancer and other diseases. J Cell Biochem. 2018;119:1273–84.
Bacon CW, D’Orso I. CDK9: a signaling hub for transcriptional control. Transcription. 2019;10:57–75.
Poon E, Liang T, Jamin Y, Walz S, Kwok C, Hakkert A, et al. Orally bioavailable CDK9/2 inhibitor shows mechanism-based therapeutic potential in MYCN-driven neuroblastoma. J Clin Invest. 2020;130:5875–92.
Štětková M, Growková K, Fojtík P, Valčíková B, Palušová V, Verlande A, et al. CDK9 activity is critical for maintaining MDM4 overexpression in tumor cells. Cell Death Dis. 2020;11:754.
Wang X, Yu C, Wang C, Ma Y, Wang T, Li Y, et al. Novel cyclin-dependent kinase 9 (CDK9) inhibitor with suppression of cancer stemness activity against non-small-cell lung cancer. Eur J Med Chem. 2019;181:111535.
Del ReM, Bertolini I, Crucitta S, Fontanelli L, Rofi E, De Angelis C, et al. Overexpression of TK1 and CDK9 in plasma-derived exosomes is associated with clinical resistance to CDK4/6 inhibitors in metastatic breast cancer patients. Breast Cancer Res Treat. 2019;178:57–62.
Wang R, Cao XJ, Kulej K, Liu W, Ma T, MacDonald M, et al. Uncovering BRD4 hyperphosphorylation associated with cellular transformation in NUT midline carcinoma. Proc Natl Acad Sci USA. 2017;114:E5352–e5361.
Lee DJ, Zeidner JF. Cyclin-dependent kinase (CDK) 9 and 4/6 inhibitors in acute myeloid leukemia (AML): a promising therapeutic approach. Expert Opin Investig Drugs. 2019;28:989–1001.
Rahaman MH, Kumarasiri M, Mekonnen LB, Yu M, Diab S, Albrecht H, et al. Targeting CDK9: a promising therapeutic opportunity in prostate cancer. Endocr Relat Cancer. 2016;23:T211–t226.
Rahaman MH, Lam F, Zhong L, Teo T, Adams J, Yu M, et al. Targeting CDK9 for treatment of colorectal cancer. Mol Oncol. 2019;13:2178–93.
Cassandri M, Fioravanti R, Pomella S, Valente S, Rotili D, Del Baldo G, et al. CDK9 as a valuable target in cancer: from natural compounds inhibitors to current treatment in pediatric soft tissue sarcomas. Front Pharmacol. 2020;11:1230.
Bazarbachi A. CDK9 inhibition for ATL therapy. Blood. 2017;130:1074–5.
Ma H, Seebacher NA, Hornicek FJ, Duan Z. Cyclin-dependent kinase 9 (CDK9) is a novel prognostic marker and therapeutic target in osteosarcoma. EBioMedicine. 2019;39:182–93.
Zhang H, Pandey S, Travers M, Sun H, Morton G, Madzo J, et al. Targeting CDK9 reactivates epigenetically silenced genes in cancer. Cell. 2018;175:1244–58.e1226.
Chou J, Quigley DA, Robinson TM, Feng FY, Ashworth A. Transcription-associated cyclin-dependent kinases as targets and biomarkers for cancer therapy. Cancer Discov. 2020;10:351–70.
Cidado J, Boiko S, Proia T, Ferguson D, Criscione SW, San Martin M, et al. AZD4573 is a highly selective CDK9 inhibitor that suppresses MCL-1 and induces apoptosis in hematologic cancer cells. Clin Cancer Res. 2020;26:922–34.
Boffo S, Damato A, Alfano L, Giordano A. CDK9 inhibitors in acute myeloid leukemia. J Exp Clin Cancer Res. 2018;37:36.
Gressel S, Schwalb B, Decker TM, Qin W, Leonhardt H, Eick D, et al. CDK9-dependent RNA polymerase II pausing controls transcription initiation. Elife. 2017;6:e29736.
Yu F, Shi G, Cheng S, Chen J, Wu SY, Wang Z, et al. SUMO suppresses and MYC amplifies transcription globally by regulating CDK9 sumoylation. Cell Res. 2018;28:670–85.
Ma X, Yang T, Luo Y, Wu L, Jiang Y, Song Z, et al. TRIM28 promotes HIV-1 latency by SUMOylating CDK9 and inhibiting P-TEFb. Elife. 2019;8:e42426.
Etchegaray JP, Zhong L, Li C, Henriques T, Ablondi E, Nakadai T, et al. The histone deacetylase SIRT6 restrains transcription elongation via promoter-proximal pausing. Mol Cell. 2019;75:683–699.e687.
Brauns-Schubert P, Schubert F, Wissler M, Weiss M, Schlicher L, Bessler S, et al. CDK9-mediated phosphorylation controls the interaction of TIP60 with the transcriptional machinery. EMBO Rep. 2018;19:244–56.
Mbonye U, Wang B, Gokulrangan G, Shi W, Yang S, Karn J. Cyclin-dependent kinase 7 (CDK7)-mediated phosphorylation of the CDK9 activation loop promotes P-TEFb assembly with Tat and proviral HIV reactivation. J Biol Chem. 2018;293:10009–25.
Zhao Z, Tang KW, Muylaert I, Samuelsson T, Elias P. CDK9 and SPT5 proteins are specifically required for expression of herpes simplex virus 1 replication-dependent late genes. J Biol Chem. 2017;292:15489–500.
Wang J, Dean DC, Hornicek FJ, Shi H, Duan Z. Cyclin-dependent kinase 9 (CDK9) is a novel prognostic marker and therapeutic target in ovarian cancer. FASEB J. 2019;33:5990–6000.
Li X, Seebacher NA, Xiao T, Hornicek FJ, Duan Z. Targeting regulation of cyclin dependent kinase 9 as a novel therapeutic strategy in synovial sarcoma. J Orthop Res. 2019;37:510–21.
Kretz AL, Schaum M, Richter J, Kitzig EF, Engler CC, Leithäuser F, et al. CDK9 is a prognostic marker and therapeutic target in pancreatic cancer. Tumour Biol. 2017;39:1010428317694304.
He S, Fang X, Xia X, Hou T, Zhang T. Targeting CDK9: a novel biomarker in the treatment of endometrial cancer. Oncol Rep. 2020;44:1929–38.
McLaughlin RP, He J, van der Noord VE, Redel J, Foekens JA, Martens JWM, et al. A kinase inhibitor screen identifies a dual cdc7/CDK9 inhibitor to sensitise triple-negative breast cancer to EGFR-targeted therapy. Breast Cancer Res. 2019;21:77.
Parvathareddy SK, Siraj AK, Masoodi T, Annaiyappanaidu P, Al-Badawi IA, Al-Dayel F, et al. Cyclin-dependent kinase 9 (CDK9) predicts recurrence in Middle Eastern epithelial ovarian cancer. J Ovarian Res. 2021;14:69.
Boiko S, Proia T, San Martin M, Gregory GP, Wu MM, Aryal N, et al. Targeting Bfl-1 via acute CDK9 inhibition overcomes intrinsic BH3-mimetic resistance in lymphomas. Blood. 2021;137:2947–57.
Carter BZ, Mak PY, Tao W, Warmoes M, Lorenzi PL, Mak D, et al. Targeting MCL-1 dysregulates cell metabolism and leukemia-stroma interactions and resensitizes acute myeloid leukemia to BCL-2 inhibition. Haematologica. 2020. https://doi.org/10.3324/haematol.2020.260331.
Luedtke DA, Su Y, Ma J, Li X, Buck SA, Edwards H, et al. Inhibition of CDK9 by voruciclib synergistically enhances cell death induced by the Bcl-2 selective inhibitor venetoclax in preclinical models of acute myeloid leukemia. Signal Transduct Target Ther. 2020;5:17.
Bagashev A, Fan S, Mukerjee R, Claudio PP, Chabrashvili T, Leng RP, et al. Cdk9 phosphorylates Pirh2 protein and prevents degradation of p53 protein. Cell Cycle. 2013;12:1569–77.
Wu J, Liang Y, Tan Y, Tang Y, Song H, Wang Z, et al. CDK9 inhibitors reactivate p53 by downregulating iASPP. Cell Signal. 2020;67:109508.
Yao JY, Xu S, Sun YN, Xu Y, Guo QL, Wei LB. Novel CDK9 inhibitor oroxylin A promotes wild-type P53 stability and prevents hepatocellular carcinoma progression by disrupting both MDM2 and SIRT1 signaling. Acta Pharmacol Sin. 2021. https://doi.org/10.1038/s41401-021-00708-2.
Shan B, Zhuo Y, Chin D, Morris CA, Morris GF, Lasky JA. Cyclin-dependent kinase 9 is required for tumor necrosis factor-alpha-stimulated matrix metalloproteinase-9 expression in human lung adenocarcinoma cells. J Biol Chem. 2005;280:1103–11.
Zhang J, Liu S, Ye Q, Pan J. Transcriptional inhibition by CDK7/9 inhibitor SNS-032 abrogates oncogene addiction and reduces liver metastasis in uveal melanoma. Mol Cancer. 2019;18:140.
Ali MA, Choy H, Habib AA, Saha D. SNS-032 prevents tumor cell-induced angiogenesis by inhibiting vascular endothelial growth factor. Neoplasia. 2007;9:370–81.
Kryštof V, Rárová L, Liebl J, Zahler S, Jorda R, Voller J, et al. The selective P-TEFb inhibitor CAN508 targets angiogenesis. Eur J Med Chem. 2011;46:4289–94.
Su YT, Chen R, Wang H, Song H, Zhang Q, Chen LY, et al. Novel targeting of transcription and metabolism in glioblastoma. Clin Cancer Res. 2018;24:1124–37.
Huang WL, Abudureheman T, Xia J, Chu L, Zhou H, Zheng WW, et al. CDK9 inhibitor induces the apoptosis of B-cell acute lymphocytic leukemia by inhibiting c-Myc-mediated glycolytic metabolism. Front Cell Dev Biol. 2021;9:641271.
Sharifzad F, Ghavami S, Verdi J, Mardpour S, Mollapour Sisakht M, Azizi Z, et al. Glioblastoma cancer stem cell biology: potential theranostic targets. Drug Resist Updat. 2019;42:35–45.
Andersson ER, Lendahl U. Therapeutic modulation of Notch signalling–are we there yet? Nat Rev Drug Discov. 2014;13:357–78.
Xie Q, Wu Q, Kim L, Miller TE, Liau BB, Mack SC, et al. RBPJ maintains brain tumor-initiating cells through CDK9-mediated transcriptional elongation. J Clin Invest. 2016;126:2757–72.
Qin JJ. Is CDK9 a promising target for both primary and metastatic osteosarcoma? EBioMedicine. 2019;40:27–8.
Beauchamp EM, Abedin SM, Radecki SG, Fischietti M, Arslan AD, Blyth GT, et al. Identification and targeting of novel CDK9 complexes in acute myeloid leukemia. Blood. 2019;133:1171–85.
Giraud S, Hurlstone A, Avril S, Coqueret O. Implication of BRG1 and cdk9 in the STAT3-mediated activation of the p21waf1 gene. Oncogene. 2004;23:7391–8.
Yang L, Zhou F, Zhuang Y, Liu Y, Xu L, Zhao H, et al. Acetyl-bufalin shows potent efficacy against non-small-cell lung cancer by targeting the CDK9/STAT3 signalling pathway. Br J Cancer. 2021;124:645–57.
Liu X, Song J, Zhang Y, Wang H, Sun H, Feng X, et al. ASF1B promotes cervical cancer progression through stabilization of CDK9. Cell Death Dis. 2020;11:705.
Zhang H, Park SH, Pantazides BG, Karpiuk O, Warren MD, Hardy CW, et al. SIRT2 directs the replication stress response through CDK9 deacetylation. Proc Natl Acad Sci USA. 2013;110:13546–51.
Kosciuczuk EM, Mehrotra S, Saleiro D, Kroczynska B, Majchrzak-Kita B, Lisowski P, et al. Sirtuin 2-mediated deacetylation of cyclin-dependent kinase 9 promotes STAT1 signaling in type I interferon responses. J Biol Chem. 2019;294:827–37.
Segatto M, Fittipaldi R, Pin F, Sartori R, Dae KK, Zare H, et al. Epigenetic targeting of bromodomain protein BRD4 counteracts cancer cachexia and prolongs survival. Nat Commun. 2017;8:1707.
Donati B, Lorenzini E, Ciarrocchi A. BRD4 and Cancer: going beyond transcriptional regulation. Mol Cancer. 2018;17:164.
Ijaz T, Jamaluddin M, Zhao Y, Zhang Y, Jay J, Finnerty CC, et al. Coordinate activities of BRD4 and CDK9 in the transcriptional elongation complex are required for TGFβ-induced Nox4 expression and myofibroblast transdifferentiation. Cell Death Dis. 2017;8:e2606.
Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell. 2005;19:523–34.
Hajmirza A, Emadali A, Gauthier A, Casasnovas O, Gressin R, Callanan MB. BET family protein BRD4: an emerging actor in NFκB signaling in inflammation and cancer. Biomedicines. 2018;6:16.
Parua PK, Kalan S, Benjamin B, Sansó M, Fisher RP. Distinct Cdk9-phosphatase switches act at the beginning and end of elongation by RNA polymerase II. Nat Commun. 2020;11:4338.
Jones PA, Issa JP, Baylin S. Targeting the cancer epigenome for therapy. Nat Rev Genet. 2016;17:630–41.
Okugawa Y, Grady WM, Goel A. Epigenetic alterations in colorectal cancer: emerging biomarkers. Gastroenterology. 2015;149:1204–25.e1212.
Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27.
Miranda Furtado CL, Dos Santos Luciano MC, Silva Santos RD, Furtado GP, Moraes MO, Pessoa C. Epidrugs: targeting epigenetic marks in cancer treatment. Epigenetics. 2019;14:1164–76.
Nebbioso A, Tambaro FP, Dell’Aversana C, Altucci L. Cancer epigenetics: moving forward. PLoS Genet. 2018;14:e1007362.
Pirngruber J, Shchebet A, Schreiber L, Shema E, Minsky N, Chapman RD, et al. CDK9 directs H2B monoubiquitination and controls replication-dependent histone mRNA 3’-end processing. EMBO Rep. 2009;10:894–900.
Sansó M, Parua PK, Pinto D, Svensson JP, Pagé V, Bitton DA, et al. Cdk9 and H2Bub1 signal to Clr6-CII/Rpd3S to suppress aberrant antisense transcription. Nucleic Acids Res. 2020;48:7154–68.
Winter GE, Mayer A, Buckley DL, Erb MA, Roderick JE, Vittori S, et al. BET bromodomain proteins function as master transcription elongation factors independent of CDK9 recruitment. Mol Cell. 2017;67:5–18.e19.
Pierre R, Kadoch C. Mammalian SWI/SNF complexes in cancer: emerging therapeutic opportunities. Curr Opin Genet Dev. 2017;42:56–67. St.
Van Duyne R, Guendel I, Narayanan A, Gregg E, Shafagati N, Tyagi M, et al. Varying modulation of HIV-1 LTR activity by Baf complexes. J Mol Biol. 2011;411:581–96.
Ali Syeda Z, Langden SSS, Munkhzul C, Lee M, Song SJ. Regulatory mechanism of microRNA expression in cancer. Int J Mol Sci. 2020; 21:1723.
Ganju A, Khan S, Hafeez BB, Behrman SW, Yallapu MM, Chauhan SC, et al. miRNA nanotherapeutics for cancer. Drug Discov Today. 2017;22:424–32.
Abreu FB, Liu X, Tsongalis GJ. miRNA analysis in pancreatic cancer: the Dartmouth experience. Clin Chem Lab Med. 2017;55:755–62.
Deb B, Uddin A, Chakraborty S. miRNAs and ovarian cancer: an overview. J Cell Physiol. 2018;233:3846–54.
Iqbal MA, Arora S, Prakasam G, Calin GA, Syed MA. MicroRNA in lung cancer: role, mechanisms, pathways and therapeutic relevance. Mol Asp Med. 2019;70:3–20.
Iacona JR, Lutz CS. miR-146a-5p: expression, regulation, and functions in cancer. Wiley Interdiscip Rev RNA. 2019;10:e1533.
Bhatia V, Yadav A, Tiwari R, Nigam S, Goel S, Carskadon S, et al. Epigenetic silencing of miRNA-338-5p and miRNA-421 drives SPINK1-positive prostate cancer. Clin Cancer Res. 2019;25:2755–68.
Tarhriz V, Wagner KD, Masoumi Z, Molavi O, Hejazi MS, Ghanbarian H. CDK9 regulates apoptosis of myoblast cells by modulation of microRNA-1 expression. J Cell Biochem. 2018;119:547–54.
Wu T, Qin Z, Tian Y, Wang J, Xu C, Li Z, et al. Recent developments in the biology and medicinal chemistry of CDK9 inhibitors: an update. J Med Chem. 2020;63:13228–57.
Gojo I, Sadowska M, Walker A, Feldman EJ, Iyer SP, Baer MR, et al. Clinical and laboratory studies of the novel cyclin-dependent kinase inhibitor dinaciclib (SCH 727965) in acute leukemias. Cancer Chemother Pharmacol. 2013;72:897–908.
Shapiro GI. Preclinical and clinical development of the cyclin-dependent kinase inhibitor flavopiridol. Clin Cancer Res. 2004;10:4270s–4275s.
Liu G, Gandara DR, Lara PN Jr., Raghavan D, Doroshow JH, Twardowski P, et al. A Phase II trial of flavopiridol (NSC #649890) in patients with previously untreated metastatic androgen-independent prostate cancer. Clin Cancer Res. 2004;10:924–8.
Stadler WM, Vogelzang NJ, Amato R, Sosman J, Taber D, Liebowitz D, et al. Flavopiridol, a novel cyclin-dependent kinase inhibitor, in metastatic renal cancer: a University of Chicago Phase II Consortium study. J Clin Oncol. 2000;18:371–5.
Blum W, Phelps MA, Klisovic RB, Rozewski DM, Ni W, Albanese KA, et al. Phase I clinical and pharmacokinetic study of a novel schedule of flavopiridol in relapsed or refractory acute leukemias. Haematologica. 2010;95:1098–105.
Dispenzieri A, Gertz MA, Lacy MQ, Geyer SM, Fitch TR, Fenton RG, et al. Flavopiridol in patients with relapsed or refractory multiple myeloma: a phase 2 trial with clinical and pharmacodynamic end-points. Haematologica. 2006;91:390–3.
Phelps MA, Lin TS, Johnson AJ, Hurh E, Rozewski DM, Farley KL, et al. Clinical response and pharmacokinetics from a phase 1 study of an active dosing schedule of flavopiridol in relapsed chronic lymphocytic leukemia. Blood. 2009;113:2637–45.
Lin TS, Ruppert AS, Johnson AJ, Fischer B, Heerema NA, Andritsos LA, et al. Phase II study of flavopiridol in relapsed chronic lymphocytic leukemia demonstrating high response rates in genetically high-risk disease. J Clin Oncol. 2009;27:6012–8.
Karp JE, Smith BD, Levis MJ, Gore SD, Greer J, Hattenburg C, et al. Sequential flavopiridol, cytosine arabinoside, and mitoxantrone: a phase II trial in adults with poor-risk acute myelogenous leukemia. Clin Cancer Res. 2007;13:4467–73.
Bible KC, Peethambaram PP, Oberg AL, Maples W, Groteluschen DL, Boente M, et al. A phase 2 trial of flavopiridol (Alvocidib) and cisplatin in platin-resistant ovarian and primary peritoneal carcinoma: MC0261. Gynecol Oncol. 2012;127:55–62.
Shah MA, Kortmansky J, Motwani M, Drobnjak M, Gonen M, Yi S, et al. A phase I clinical trial of the sequential combination of irinotecan followed by flavopiridol. Clin Cancer Res. 2005;11:3836–45.
Rathkopf D, Dickson MA, Feldman DR, Carvajal RD, Shah MA, Wu N, et al. Phase I study of flavopiridol with oxaliplatin and fluorouracil/leucovorin in advanced solid tumors. Clin Cancer Res. 2009;15:7405–11.
Tan AR, Yang X, Berman A, Zhai S, Sparreboom A, Parr AL, et al. Phase I trial of the cyclin-dependent kinase inhibitor flavopiridol in combination with docetaxel in patients with metastatic breast cancer. Clin Cancer Res. 2004;10:5038–47.
Bharate SB, Kumar V, Jain SK, Mintoo MJ, Guru SK, Nuthakki VK, et al. Discovery and preclinical development of IIIM-290, an orally active potent cyclin-dependent kinase inhibitor. J Med Chem. 2018;61:1664–87.
Kumar V, Mintoo MJ, Mondhe DM, Bharate SB, Vishwakarma RA, Bharate SS. Binary and ternary solid dispersions of an anticancer preclinical lead, IIIM-290: In vitro and in vivo studies. Int J Pharm. 2019;570:118683.
Kumar V, Bharate SB, Vishwakarma RA, Bharate SS. Selection of a water-soluble salt form of a preclinical candidate, IIIM-290: multiwell-plate salt screening and characterization. ACS Omega. 2018;3:8365–77.
Mintoo M, Khan S, Wani A, Malik S, Bhurta D, Bharate S, et al. A rohitukine derivative IIIM-290 induces p53 dependent mitochondrial apoptosis in acute lymphoblastic leukemia cells. Mol Carcinog. 2021;60:671–83.
Bettayeb K, Tirado OM, Marionneau-Lambot S, Ferandin Y, Lozach O, Morris JC, et al. Meriolins, a new class of cell death inducing kinase inhibitors with enhanced selectivity for cyclin-dependent kinases. Cancer Res. 2007;67:8325–34.
Echalier A, Bettayeb K, Ferandin Y, Lozach O, Clément M, Valette A, et al. Meriolins (3-(pyrimidin-4-yl)-7-azaindoles): synthesis, kinase inhibitory activity, cellular effects, and structure of a CDK2/cyclin A/meriolin complex. J Med Chem. 2008;51:737–51.
Wei L, Zhou Y, Qiao C, Ni T, Li Z, You Q, et al. Oroxylin A inhibits glycolysis-dependent proliferation of human breast cancer via promoting SIRT3-mediated SOD2 transcription and HIF1α destabilization. Cell Death Dis. 2015;6:e1714.
Ni T, He Z, Dai Y, Yao J, Guo Q, Wei L. Oroxylin A suppresses the development and growth of colorectal cancer through reprogram of HIF1α-modulated fatty acid metabolism. Cell Death Dis. 2017;8:e2865.
Wei L, Dai Y, Zhou Y, He Z, Yao J, Zhao L, et al. Oroxylin A activates PKM1/HNF4 alpha to induce hepatoma differentiation and block cancer progression. Cell Death Dis. 2017;8:e2944.
Hui H, Chen Y, Yang H, Zhao K, Wang Q, Zhao L, et al. Oroxylin A has therapeutic potential in acute myelogenous leukemia by dual effects targeting PPARγ and RXRα. Int J Cancer. 2014;134:1195–206.
Gupta A, Shah K, Oza MJ, Behl T. Reactivation of p53 gene by MDM2 inhibitors: a novel therapy for cancer treatment. Biomed Pharmacother. 2019;109:484–92.
Xu Z, Wu W, Yan H, Hu Y, He Q, Luo P. Regulation of p53 stability as a therapeutic strategy for cancer. Biochem Pharmacol. 2021;185:114407.
Yang Y, Hu Y, Gu HY, Lu N, Liu W, Qi Q, et al. Oroxylin A induces G2/M phase cell-cycle arrest via inhibiting Cdk7-mediated expression of Cdc2/p34 in human gastric carcinoma BGC-823 cells. J Pharm Pharmacol. 2008;60:1459–63.
Xu ZF, Sun XK, Chen G, Han C, Wang F, Zhang YD. Oroxyloside inhibits human glioma progression by suppressing proliferation, metastasis and inducing apoptosis related pathways. Biomed Pharmacother. 2018;97:1564–74.
Wang J, Ge R, Qiu X, Xu X, Wei L, Li Z, et al. Discovery and synthesis of novel Wogonin derivatives with potent antitumor activity in vitro. Eur J Med Chem. 2017;140:421–34.
Wang J, Li T, Zhao T, Wu T, Liu C, Ding H, et al. Design of wogonin-inspired selective cyclin-dependent kinase 9 (CDK9) inhibitors with potent in vitro and in vivo antitumor activity. Eur J Med Chem. 2019;178:782–801.
Bian J, Ren J, Li Y, Wang J, Xu X, Feng Y, et al. Discovery of Wogonin-based PROTACs against CDK9 and capable of achieving antitumor activity. Bioorg Chem. 2018;81:373–81.
Xu J, Li H, Wang X, Huang J, Li S, Liu C, et al. Discovery of coumarin derivatives as potent and selective cyclin-dependent kinase 9 (CDK9) inhibitors with high antitumour activity. Eur J Med Chem. 2020;200:112424.
Barlaam B, Casella R, Cidado J, Cook C, De Savi C, Dishington A, et al. Discovery of AZD4573, a potent and selective inhibitor of CDK9 that enables short duration of target engagement for the treatment of hematological malignancies. J Med Chem. 2020;63:15564–90.
Krystof V, Cankar P, Frysová I, Slouka J, Kontopidis G, Dzubák P, et al. 4-arylazo-3,5-diamino-1H-pyrazole CDK inhibitors: SAR study, crystal structure in complex with CDK2, selectivity, and cellular effects. J Med Chem. 2006;49:6500–9.
Tong Z, Chatterjee D, Deng D, Veeranki O, Mejia A, Ajani JA, et al. Antitumor effects of cyclin dependent kinase 9 inhibition in esophageal adenocarcinoma. Oncotarget. 2017;8:28696–710.
Yamamoto M, Onogi H, Kii I, Yoshida S, Iida K, Sakai H, et al. CDK9 inhibitor FIT-039 prevents replication of multiple DNA viruses. J Clin Invest. 2014;124:3479–88.
Ajiro M, Sakai H, Onogi H, Yamamoto M, Sumi E, Sawada T, et al. CDK9 inhibitor FIT-039 suppresses viral oncogenes E6 and E7 and has a therapeutic effect on HPV-induced neoplasia. Clin Cancer Res. 2018;24:4518–28.
Sumi E, Nomura T, Asada R, Uozumi R, Tada H, Amino Y, et al. Safety and plasma concentrations of a cyclin-dependent kinase 9 (CDK9) inhibitor, FIT039, administered by a single adhesive skin patch applied on normal skin and cutaneous warts. Clin Drug Investig. 2019;39:55–61.
Lücking U, Scholz A, Lienau P, Siemeister G, Kosemund D, Bohlmann R, et al. Identification of atuveciclib (BAY 1143572), the first highly selective, clinical PTEFb/CDK9 inhibitor for the treatment of cancer. ChemMedChem. 2017;12:1776–93.
Narita T, Ishida T, Ito A, Masaki A, Kinoshita S, Suzuki S, et al. Cyclin-dependent kinase 9 is a novel specific molecular target in adult T-cell leukemia/lymphoma. Blood. 2017;130:1114–24.
Kinoshita S, Ishida T, Ito A, Narita T, Masaki A, Suzuki S, et al. Cyclin-dependent kinase 9 as a potential specific molecular target in NK-cell leukemia/lymphoma. Haematologica. 2018;103:2059–68.
Brisard D, Eckerdt F, Marsh LA, Blyth GT, Jain S, Cristofanilli M, et al. Antineoplastic effects of selective CDK9 inhibition with atuveciclib on cancer stem-like cells in triple-negative breast cancer. Oncotarget. 2018;9:37305–18.
Tong Z, Mejia A, Veeranki O, Verma A, Correa AM, Dokey R, et al. Targeting CDK9 and MCL-1 by a new CDK9/p-TEFb inhibitor with and without 5-fluorouracil in esophageal adenocarcinoma. Ther Adv Med Oncol. 2019;11:1758835919864850.
Johansson P, Dierichs L, Klein-Hitpass L, Bergmann AK, Möllmann M, Menninger S, et al. Anti-leukemic effect of CDK9 inhibition in T-cell prolymphocytic leukemia. Ther Adv Hematol. 2020;11:2040620720933761.
Bayer AG. An open-label Phase I dose-escalation study to characterize the safety, tolerability, pharmacokinetics, and maximum tolerated dose of BAY 1143572 given in a once-daily or an intermittent dosing schedule in subjects with advanced malignancies. ClinicalTrials. 2017. https://clinicaltrials.bayer.com/study/?id=16519.
Bayer AG. An open-label Phase I dose-escalation study to characterize the safety, tolerability, pharmacokinetics, and maximum tolerated dose of BAY 1143572 given in a once-daily or an intermittent dosing schedule in subjects with advanced acute leukemia. ClinicalTrials. 2018. https://clinicaltrials.bayer.com/study/?id=16520&page=0&SortField=Location_Distance&SortOrder=asc&Keyword=NCT02345382&Status=&Longitude=&Latitude=&ageRange=&conditions=&phases=&gender=&healthyVol=&studyType=&studyResult=&locationCountryInternal=&LocationName=&MileRadius=.
Meng H, Jin Y, Liu H, You L, Yang C, Yang X, et al. SNS-032 inhibits mTORC1/mTORC2 activity in acute myeloid leukemia cells and has synergistic activity with perifosine against Akt. J Hematol Oncol. 2013;6:18.
Walsby E, Lazenby M, Pepper C, Burnett AK. The cyclin-dependent kinase inhibitor SNS-032 has single agent activity in AML cells and is highly synergistic with cytarabine. Leukemia. 2011;25:411–9.
Xie G, Tang H, Wu S, Chen J, Liu J, Liao C. The cyclin-dependent kinase inhibitor SNS-032 induces apoptosis in breast cancer cells via depletion of Mcl-1 and X-linked inhibitor of apoptosis protein and displays antitumor activity in vivo. Int J Oncol. 2014;45:804–12.
Lemke J, von Karstedt S, Abd El Hay M, Conti A, Arce F, Montinaro A, et al. Selective CDK9 inhibition overcomes TRAIL resistance by concomitant suppression of cFlip and Mcl-1. Cell Death Differ. 2014;21:491–502.
Wu Y, Chen C, Sun X, Shi X, Jin B, Ding K, et al. Cyclin-dependent kinase 7/9 inhibitor SNS-032 abrogates FIP1-like-1 platelet-derived growth factor receptor α and bcr-abl oncogene addiction in malignant hematologic cells. Clin Cancer Res. 2012;18:1966–78.
Chen R, Wierda WG, Chubb S, Hawtin RE, Fox JA, Keating MJ, et al. Mechanism of action of SNS-032, a novel cyclin-dependent kinase inhibitor, in chronic lymphocytic leukemia. Blood. 2009;113:4637–45.
Ali MA, Reis A, Ding LH, Story MD, Habib AA, Chattopadhyay A, et al. SNS-032 prevents hypoxia-mediated glioblastoma cell invasion by inhibiting hypoxia inducible factor-1alpha expression. Int J Oncol. 2009;34:1051–60.
Olson CM, Jiang B, Erb MA, Liang Y, Doctor ZM, Zhang Z, et al. Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat Chem Biol. 2018;14:163–70.
Tong WG, Chen R, Plunkett W, Siegel D, Sinha R, Harvey RD, et al. Phase I and pharmacologic study of SNS-032, a potent and selective Cdk2, 7, and 9 inhibitor, in patients with advanced chronic lymphocytic leukemia and multiple myeloma. J Clin Oncol. 2010;28:3015–22.
Heath EI, Bible K, Martell RE, Adelman DC, Lorusso PM. A phase 1 study of SNS-032 (formerly BMS-387032), a potent inhibitor of cyclin-dependent kinases 2, 7 and 9 administered as a single oral dose and weekly infusion in patients with metastatic refractory solid tumors. Invest N Drugs. 2008;26:59–65.
Richters A, Doyle SK, Freeman DB, Lee C, Leifer BS, Jagannathan S, et al. Modulating androgen receptor-driven transcription in prostate cancer with selective CDK9 inhibitors. Cell Chem Biol. 2021;28:134–147.e114.
Lücking U, Kosemund D, Böhnke N, Lienau P, Siemeister G, Denner K, et al. Changing for the better: discovery of the highly potent and selective CDK9 inhibitor VIP152 suitable for once weekly intravenous dosing for the treatment of cancer. J Med Chem. 2021;64:11651–74.
Byrne M, Frattini MG, Ottmann OG, Mantzaris I, Cordoba R. Phase I study of the PTEFb inhibitor BAY 1251152 in patients with acute myelogenous leukemia. Blood. 2018. https://doi.org/10.1182/blood-2018-99-117257.
Diamond JR, Moreno V, Lim EA, Cordoba R, Boni V. Phase I dose escalation study of the first-in-class selective PTEFb inhibitor BAY 1251152 in patients with advanced cancer: Novel target validation and early evidence of clinical activity. J Clin Oncol. 2018. https://doi.org/10.1200/JCO.2018.36.15_suppl.2507.
Paparidis NF, Durvale MC, Canduri F. The emerging picture of CDK9/P-TEFb: more than 20 years of advances since PITALRE. Mol Biosyst. 2017;13:246–76.
Pirngruber J, Shchebet A, Johnsen SA. Insights into the function of the human P-TEFb component CDK9 in the regulation of chromatin modifications and co-transcriptional mRNA processing. Cell Cycle. 2009;8:3636–42.
Borden KLB. CDK9 and mTOR: trading places. Blood. 2019;133:1167–8.
Phillips DC, Jin S, Gregory GP, Zhang Q, Xue J, Zhao X, et al. A novel CDK9 inhibitor increases the efficacy of venetoclax (ABT-199) in multiple models of hematologic malignancies. Leukemia. 2020;34:1646–57.
Li J, Zhi X, Chen S, Shen X, Chen C, Yuan L, et al. CDK9 inhibitor CDKI-73 is synergetic lethal with PARP inhibitor olaparib in BRCA1 wide-type ovarian cancer. Am J Cancer Res. 2020;10:1140–55.
Emran AA, Tseng HY, Gunatilake D, Cook SJ, Ahmed F, Wang S, et al. A combination of epigenetic BET and CDK9 inhibitors for treatment of human melanoma. J Invest Dermatol. 2021;141:2238–2249.e2212.
E OR, Dhami SPS, Baev DV, Ortutay C, Halpin-McCormick A, Morrell R, et al. Repression of Mcl-1 expression by the CDC7/CDK9 inhibitor PHA-767491 overcomes bone marrow stroma-mediated drug resistance in AML. Sci Rep. 2018;8:15752.
Pandey K, An HJ, Kim SK, Lee SA, Kim S, Lim SM, et al. Molecular mechanisms of resistance to CDK4/6 inhibitors in breast cancer: A review. Int J Cancer. 2019;145:1179–88.
Gao JJ, Cheng J, Bloomquist E, Sanchez J, Wedam SB, Singh H, et al. CDK4/6 inhibitor treatment for patients with hormone receptor-positive, HER2-negative, advanced or metastatic breast cancer: a US Food and Drug Administration pooled analysis. Lancet Oncol. 2020;21:250–60.
Fallah Y, Brundage J, Allegakoen P, Shajahan-Haq AN. MYC-driven pathways in breast cancer subtypes. Biomolecules. 2017;7:53.
Rebello RJ, Pearson RB, Hannan RD, Furic L. Therapeutic approaches targeting MYC-driven prostate cancer. Genes. 2017;8:71.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (82173856), Natural Science Foundation of Zhejiang Province (LY21H300005) and Wenzhou Municipal Science and Technology Bureau (ZY2020025).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. YLS and YMW wrote the original draft equally. YXZ and SJM designed the figures and critically revised the manuscript. LHY, CGZ and XYH revised the manuscript and contributed to writing the final manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Shen, Yl., Wang, Ym., Zhang, Yx. et al. Targeting cyclin-dependent kinase 9 in cancer therapy. Acta Pharmacol Sin 43, 1633–1645 (2022). https://doi.org/10.1038/s41401-021-00796-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-021-00796-0
