
Abstract
Ischemic preconditioning induced by brief periods of coronary occlusion and reperfusion protects the heart from a subsequent prolonged ischemic insult. In this study we investigated whether a short-term nonischemic stimulation of hypertrophy renders the heart resistant to subsequent ischemic injury. Male mice were subjected to transient transverse aortic constriction (TAC) for 3 days followed aortic debanding on D4 (T3D4), as well as ligation of the left coronary artery to induce myocardial infarction (MI). The TAC preconditioning mice showed markedly improved contractile function and significantly reduced myocardial fibrotic area and apoptosis following MI. We revealed that TAC preconditioning significantly reduced MI-induced oxidative stress, evidenced by increased NADPH/NADP ratio and GSH/GSSG ratio, as well as decreased mitochondrial ROS production. Furthermore, TAC preconditioning significantly increased the expression and activity of SIRT3 protein following MI. Cardiac-specific overexpression of SIRT3 gene through in vivo AAV-SIRT3 transfection partially mimicked the protective effects of TAC preconditioning, whereas genetic ablation of SIRT3 in mice blocked the protective effects of TAC preconditioning. Moreover, expression of an IDH2 mutant mimicking deacetylation (IDH2 K413R) in cardiomyocytes promoted myocardial IDH2 activation, quenched mitochondrial reactive oxygen species (ROS), and alleviated post-MI injury, whereas expression of an acetylation mimic (IDH2 K413Q) in cardiomyocytes inactivated IDH2, exacerbated mitochondrial ROS overload, and aggravated post-MI injury. In conclusion, this study identifies TAC preconditioning as a novel strategy for induction of an endogenous self-defensive and cardioprotective mechanism against cardiac injury. Therapeutic strategies targeting IDH2 are promising treatment approaches for cardiac ischemic injury.
Similar content being viewed by others


Introduction
Myocardial infarction (MI) remains a leading cause of morbidity and mortality worldwide and is commonly caused by thrombotic occlusion of a coronary artery, resulting in cardiomyocyte death, reparative fibrotic healing, left ventricular remodeling, and ultimately heart failure [1]. Despite progress in reperfusion therapy, nearly 10% of MI subjects die during their index hospitalization, and 25% of survivors progress to develop chronic heart failure [2]. One explanation for these poor outcomes is that reperfusion therapies, such as thrombolytic agent treatment and primary percutaneous coronary intervention, do not reverse cardiomyocyte death or ameliorate the deterioration of myocardial function. Myocardial ischemia-induced DNA damage and mitochondrial reactive oxygen species (ROS) overload are the primary contributors to cardiomyocyte death and mitochondrial impairment [3]. It has been reported that therapeutic strategies targeting mitochondria ameliorate post-MI damage [4, 5]. However, the possible mechanisms for attenuation of mitochondrial damage post-MI remain elusive.
The mitochondrial matrix protein isocitrate dehydrogenase 2 (IDH2) is a major source of NADPH and catalyzes the oxidative decarboxylation of isocitrate to α-ketoglutarate [6]. Moreover, IDH2 has been shown to play a pivotal role in maintaining mitochondrial redox balance by producing mitochondrial NADPH, which is an essential cofactor required for the conversion of oxidized glutathione (GSSG) to reduced glutathione (GSH) [7, 8]. Recently, IDH2 was shown to be a functional target of SIRT3 and to mediate pro-oncogenic properties in cells overexpressing SIRT3 [7, 8]. Furthermore, global induction of mutant IDH2 expression in adult mice leads to dilated cardiomyopathy, white-matter lesions, and muscular dystrophy [9]. These findings motivated us to investigate whether IDH2 can serve as a cardioprotectant against post-MI damage.
Local ischemic preconditioning, a phenomenon in which brief periods of ischemia followed by reperfusion protect cardiomyocytes from subsequent prolonged ischemic insult, has received considerable attention since it was first reported by Murry et al. in 1986 [10]. Given the obvious clinical implications, intense research has been conducted to dissect the potential mechanisms underlying the cardioprotection conferred by preconditioning. In addition to ischemia, preconditioning with mechanical stretch, heat stress, metabolic challenge, and pharmacological agents can induce protective effects [11,12,13]. Recently, Wei et al. reported that retraction of short-term pressure overload increases the resistance of the heart to the development of pathological hypertrophy in a process termed hypertrophic preconditioning [14]. However, whether such cardioprotection extends beyond pathological hypertrophy to myocyte viability and remodeling remains unexplored. If it does, the cardioprotective effects of hypertrophic preconditioning can be applied to different pathological cardiac states such as long-term pressure overload and ischemic insult. Thus, this study was performed to determine whether myocardial hypertrophic preconditioning can attenuate post-MI injury and slow its progression to heart failure. In particular, the role of IDH2 in hypertrophic preconditioning was investigated.
Materials and methods
Animals
Male C57BL/6N mice were supplied by the Shanghai Laboratory Animal Center. SIRT3-knockout (KO) mice were obtained from Dr Wei-li Shen from Shanghai Jiaotong University (Shanghai, China). All the protocols used conformed to the recommendations in the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (8th Edition, NRC 2011), and they were approved by the Institutional Review Board of Zhongshan Hospital at Fudan University.
Grouping and treatment
Six studies were conducted, as described below.
-
(1)
The effect of hypertrophic preconditioning induced by 3 days of transverse aortic constriction (TAC) followed by 4 days of debanding on post-MI injury was examined. Four groups were used: (1) the Sham group, in which wild-type (WT) mice were subjected to a sham operation; (2) the MI group, in which WT mice were subjected to MI; (3) the T3D4 group, in which WT mice were subjected to TAC for 3 days, aortic debanding and a sham operation 4 days later; and (4) the T3D4 + MI group, in which WT mice were subjected to TAC for 3 days, aortic debanding and ligation of the left coronary artery 4 days later.
-
(2)
The effects of SIRT3 deficiency on the cardioprotection induced by hypertrophic preconditioning were assessed in eight groups: (1) the Sham group, in which WT mice were subjected to a sham operation; (2) the MI group, in which WT mice were subjected to MI; (3) the T3D4 group, in which WT mice were subjected to 3 days of TAC, debanding of the aorta and a sham operation 4 days later; (4) the T3D4 + MI group, in which WT mice were subjected to 3 days of TAC, debanding of the aorta and ligation of the left coronary artery 4 days later; (5) the SIRT3-KO + Sham group, in which SIRT3-KO mice were subjected to a sham operation; (6) the SIRT3-KO + MI group, in which SIRT3-KO mice were subjected to MI; (7) the SIRT3-KO + T3D4 group, in which SIRT3-KO mice were subjected to 3 days of TAC, debanding of the aorta and ligation of the left coronary artery 4 days later; and (8) the SIRT3-KO + T3D4 + MI group, in which SIRT3-KO mice were subjected to 3 days of TAC, debanding of the aorta and ligation of the left coronary artery 4 days later.
-
(3)
The effects of SIRT3 deficiency on post-MI injury were assessed in four groups: (1) the WT + Sham group, in which WT mice were subjected to a sham operation; (2) the WT + MI group, in which WT mice were subjected to MI; (3) the SIRT3-KO + Sham group, in which SIRT3-KO mice were subjected to a sham operation; and (4) the SIRT3-KO + MI group: in which SIRT3-KO mice were subjected to MI.
-
(4)
To investigate whether direct SIRT3 activation protects the heart from post-MI injury, a SIRT3 adeno-associated virus (AAV-SIRT3, 1011 v.g./100 µL per mouse) or a control virus (AAV-EGFP, 1011 v.g./100 µL per mouse) was injected into the tail vein of each mouse in the following four groups: (1) the AAV-EGFP + Sham group, in which WT mice were injected with AAV-EGFP and subjected to a sham operation; (2) the AAV-EGFP + MI group, in which WT mice were injected with AAV-EGFP and subjected to MI; (3) the AAV-SIRT3 + Sham group, in which WT mice were injected with AAV-SIRT3 and subjected to a sham operation; and (4) the AAV-SIRT3 + MI group, in which WT mice were injected with AAV-SIRT3 and subjected to MI.
-
(5)
To investigate the effects of IDH2 mutation on post-MI injury with an IDH2 lysine 413 deacetylation mimic (IDH2 K413R) and an IDH2 lysine 413 acetylation mimic (IDH2 K413Q), six groups were established: (1) the IDH2 WT + Sham group, in which WT mice expressing WT IDH2 were subjected to a sham operation; (2) the IDH2 WT + MI group, in which WT mice expressing WT IDH2 were subjected to MI; (3) the IDH2 K413Q + Sham group, in which WT mice expressing IDH2 K413Q were subjected to a sham operation; (4) the IDH2 K413Q + MI group, in which WT mice expressing IDH2 K413Q were subjected to MI; (5) the IDH2 K413R + Sham group, in which WT mice expressing IDH2 K413R were subjected to a sham operation; and (6) the IDH2 K413R + MI group, in which WT mice expressing IDH2 K413R were subjected to MI.
-
(6)
To investigate the effects of IDH2 mutation with IDH2 K413R and IDH2 K413Q on hypertrophic preconditioning-induced cardioprotective effects, twelve groups were established: (1) the IDH2 WT + MI group, in which WT mice expressing WT IDH2 were subjected to MI; (2) the IDH2 WT + T3D4 + MI group, in which WT mice expressing WT IDH2 were subjected to 3 days of TAC, debanding of the aorta and ligation of the left coronary artery 4 days later; (3) the IDH2 K413R + MI group, in which WT mice expressing IDH2 K413R were subjected to MI; (4) the IDH2 WT + T3D4 + MI group, in which WT mice expressing IDH2 K413R were subjected to 3 days of TAC, debanding of the aorta and ligation of the left coronary artery 4 days later; (5) the IDH2 K413Q + MI group, in which WT mice expressing IDH2 K413Q were subjected to MI; (6) the IDH2 K413Q + T3D4 + MI group, in which WT mice expressing IDH2 K413Q were subjected to 3 days of TAC, debanding of the aorta and ligation of the left coronary artery 4 days later; (7) the SIRT3-KO + IDH2 WT + MI group, in which SIRT3-KO mice expressing WT IDH2 were subjected to MI; (8) the SIRT3-KO + IDH2 WT + T3D4 + MI group, in which SIRT3-KO mice expressing WT IDH2 were subjected to 3 days of TAC, debanding of the aorta and ligation of the left coronary artery 4 days later; (9) the SIRT3-KO + IDH2 K413R + MI group, in which SIRT3-KO mice expressing IDH2 K413R were subjected to MI; (10) the SIRT3-KO + IDH2 WT + T3D4 + MI group, in which SIRT3-KO mice expressing IDH2 K413R were subjected to 3 days of TAC, debanding of the aorta and ligation of the left coronary artery 4 days later; (11) the SIRT3-KO + IDH2 K413Q + MI group, in which SIRT3-KO mice expressing IDH2 K413Q were subjected to MI; and (12) the SIRT3-KO + IDH2 K413Q + T3D4 + MI group, in which SIRT3-KO mice expressing IDH2 K413Q were subjected to 3 days of TAC, debanding of the aorta and ligation of the left coronary artery 4 days later.
The viral constructs AAV9-CMV-IDH2(K413R)-P2A-EGFP-3FLAG and AAV9-CMV-IDH2(K413Q)-P2A-EGFP-3FLAG and a WT construct (AAV9- CMV-IDH2-P2A-EGFP-3FLAG) were administered to the animals through tail vein injection (1011 v.g./100 µL per mouse). Myocardial IDH2 activity was analyzed 1 month after virus injection. The viral constructs AAV9-CMV-SIRT3-P2A-EGFP-3FLAG and AAV9-CMV-P2A-EGFP-3FLAG were also administered to the animals through tail vein injection (1011 v.g./100 µL per mouse). The expression of SIRT3 in the myocardium was determined at 1 month post injection. The surgeon was blinded to the treatment allocation.
TAC-induced hypertrophic preconditioning
C57BL/6 male mice (8 weeks, 22–25 g) were subjected to minimally invasive TAC, debanding or sham operation, as described elsewhere [15]. The mice were anesthetized with a single intraperitoneal injection of 1% pentobarbital sodium (Sigma-Aldrich, St. Louis, MO, USA, 50 mg/kg). A horizontal skin incision ~1 cm in length was made at the level of the suprasternal notch. Once the trachea was located, a 5-mm longitudinal cut was made down the sternum, the thymus was retracted and the aortic arch was located with the help of a retractor. A wire with a snare on the end was passed under the aorta between the origin of the right innominate and left common carotid arteries. A 6-0 silk suture was snared with the wire and pulled back around the aorta. A bent 27-gauge needle was then placed next to the aortic arch, and the suture was snugly tied around the needle and the aorta. After ligation, the needle was quickly removed. The skin was closed, and mice were allowed to recover on a warming pad until they were fully awake. The sham procedure was identical except that the aorta was not ligated. The debanding operation was performed by carefully removing the ligature after 3 days of TAC, and then MI surgery was performed 4 days later. After recovery from general anesthesia, each animal was subcutaneously injected with meloxicam (5 mg/kg, Sigma-Aldrich, St. Louis, MO, USA) to mitigate pain.
In vivo adeno-associated virus delivery
The virus AAV9-CMV-IDH2(K413R)-P2A-EGFP-3FLAG was chosen for transfection of IDH2 K413R. The viral construct AAV9-CMV-IDH2(K413Q)-P2A-EGFP-3FLAG was chosen for transfection of IDH2 K413Q. A WT construct (AAV9-CMV-IDH2-P2A-EGFP-3FLAG) without IDH2 mutation was used as a control. The virus was purchased from OBio (Shanghai, China) and administered to the animals through tail vein injection (1011 v.g./100 µL). Myocardial IDH2 activity was analyzed 1 month after virus injection.
MI protocol
The surgical procedures were performed as described in previous study [16]. Briefly, mice were anesthetized with 2% isoflurane, and the heart was manually exposed without intubation via a tiny thoracic incision. A ligation was made around the left anterior descending coronary artery 2–3 mm from its origin using a 6-0 silk suture, and then the chest was closed. Mice that fully recovered from the surgical procedure were returned to the standard animal housing conditions.
Immunoblotting
Samples were taken from the ischemic zone at 3 days after MI. The expression of myocardial IDH2 (Abcam, 1:1000), acetylated IDH2 (at lysine 413) (GeneTel Laboratories LLC, 1:1000), SIRT3 (CST, 1:1000), COX4 (Abcam, 1:1000), β-actin, (Abcam, 1:1000), and GAPDH (CST, 1:1000) was determined by immunoblotting [17]. The protein band densities were quantified with ImageJ 1.37 (National Institutes of Health, Bethesda, MD, USA).
Mitochondrial SIRT3 and IDH2 activity assessment
Myocardial mitochondria taken from the ischemic zone were isolated at 3 days after MI. SIRT3 activity was determined by utilizing a SIRT3 fluorescent assay kit (BPS Bioscience, San Diego, CA, USA), and IDH2 activity was quantified by using an IDH2 activity assay kit (Sigma-Aldrich, St. Louis, MO, USA) according to the manufacturer’s protocol.
Doppler echocardiography
Each mouse was anesthetized with 1% isoflurane at 4 weeks after MI. M-mode images of the left ventricle were obtained at the level of the papillary muscle tips using a Vevo 770 imaging system (VisualSonics, Toronto, Canada). The left ventricular internal diastolic diameter (LVIDd) and left ventricular internal systolic diameter (LVIDs) were recorded. Left ventricular fractional shortening (LVFS) was calculated according to the following formula: LVFS = [(LVIDd − LVIDs)/LVIDd] × 100. The left ventricular ejection fraction (LVEF) was calculated by using the spherical formula.
Myocardial fibrosis
Hearts were harvested after evaluation of cardiac function at 4 weeks after MI. The tissues were cut into sections 4–5-µm thick after 4% paraformaldehyde fixation and stained with Masson’s trichrome. The severity of myocardial fibrosis was evaluated as the percentage of the Masson’s trichrome-stained area to the total left ventricular area.
Detection of Caspase-3 activity in heart tissue
Myocardial Caspase-3 activity was used to assay apoptosis levels with a Caspase Fluorometric Assay Kit (BioVision, Mountain View, CA, USA) at 3 days after MI. Briefly, 100 μg of total protein from tissues was loaded per assay with AFC-conjugated substrates specific for caspase-3 (IEDT, LEHD, and ATAD) at final concentrations of 50 μM. The samples were read by a fluorimeter equipped with a 400-nm excitation filter and a 505-nm emission filter. The activity of Caspase-3 was calculated against the mean value of Caspase-3 activity from the corresponding control.
Mitochondrial morphology assessment
After MI for 3 days, hearts demonstrating infarctions were fixed with 2% glutaraldehyde for 2 h, fixed in 1% OsO4 for 2 h, and embedded in resin. Ultrathin sections were stained with uranyl acetate and lead citrate and observed under an electron microscope (Philips CM-120; Philips Electronic Instruments). Random fields of view were acquired by an electron microscope technician.
Mitochondrial function assessment
Myocardial ATP levels were determined using a commercial ATP assay kit (Beyotime, Shanghai, China) following the manufacturer’s protocol. After MI for 3 days, the tissue was homogenized and centrifuged (12,000 × g, 5 min), and the supernatant was harvested and mixed with a working dilution of the reagent in a 96-well plate. The relative light units (RLUs) were assessed with a microplate reader.
Measurement of superoxide production
Myocardial superoxide production was measured by detecting lucigenin-enhanced chemiluminescence after MI for 3 days. The RLUs emitted were recorded and integrated over 30-s intervals for 5 min. Superoxide production was normalized to the heart weight.
Mitochondrial NADPH/NADP, GSH and GSSG assays
Myocardial mitochondria were isolated after MI for 3 days. NADP and NADPH levels were determined using an NADP/NADPH Assay Kit (Beyotime, Shanghai, China) according to the manufacturer’s instructions. A GSH and GSSG Assay Kit (Beyotime, Shanghai, China) was used to measure the mitochondrial GSH and GSSG levels according to the manufacturer’s instructions.
Statistical analysis
The data are shown as the means ± SDs. Statistical analysis was performed with unpaired Student’s t test for two-group comparisons. For multigroup comparisons, one-way ANOVA followed by a Bonferroni post hoc test was used. A value of P < 0.05 was considered to indicate statistical significance. All statistical analyses were performed using GraphPad Prism version 7.0 (GraphPad Prism lnc, San Diego, CA, USA).
Results
Hypertrophic preconditioning conferred cardioprotection and alleviated post-MI injury
TAC was performed in male mice for 3 days and then withdrawn for 4 days. The mice were subsequently subjected to regional myocardial ischemia via permanent coronary artery ligation to determine whether transient prohypertrophic stress exerts cardioprotective effects against MI. Preconditioning with TAC for 3 days induced mild myocardial hypertrophy, as evidenced by an increased heart weight/body weight ratio (Fig. S1a), heart weight/tibial length ratio (Fig. S1b), and cardiomyocyte cross sectional area (Fig. S1c), which may be considered compensatory hypertrophy. MI significantly decreased the LVEF and LVFS (Fig. 1a), whereas transient TAC for 3 days reversed the MI-induced decreases in LVEF and LVFS at 4 weeks post MI. Consistent with improved cardiac systolic function, TAC preconditioning significantly decreased the myocardial fibrotic area at 4 weeks post MI (Fig. 1b). Mechanistically, hypertrophic preconditioning markedly hindered MI-elicited mitochondrial ROS production (Fig. 1c–e), as evidenced by increased NADPH/NADP ratios (Fig. 1c), elevated GSH/GSSG ratios (Fig. 1d), and decreased superoxide production (Fig. 1e) in the hearts of mice subjected to hypertrophic preconditioning. Moreover, cardiac MI-elicited apoptotic properties were lost in preconditioned mice, as evidenced by alleviated mitochondrial impairment (Fig. 1f), increased ATP content (Fig. 1g) and decreased caspase-3 levels (Fig. 1h). These results suggest that hypertrophic preconditioning conferred cardioprotection against post-MI injury.

a Representative M-mode echocardiography images and cardiac function in WT mice subjected to MI. b Representative photographs of Masson’s trichrome staining and the extent of fibrosis expressed as the percentage of the Masson’s trichrome-stained area to the total left ventricular area in WT mice at 4 weeks post MI. The blue area indicates fibrosis. Scale bar = 50 μm. c NADPH/NADP ratios in MI-injured myocardial tissues. d GSH/GSSG ratios in MI-injured cardiac tissues. e Quantitative analyses of superoxide production in cardiac tissues. f Representative photographs of mitochondria in MI-injured myocardial tissues obtained under transmission electron microscopy. Scale bar = 2 μm. g Mitochondrial function in WT mice, as evaluated by mitochondrial ATP content analysis at 3 days post MI. h Caspase-3 activity in WT mice at 3 days post MI. n = 6–8 animals per group, *P < 0.05.
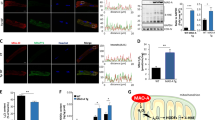
Hypertrophic preconditioning activated SIRT3 signaling and deacetylated IDH2
SIRT3 has been identified as an important mitochondrial deacetylation enzyme. Specifically, SIRT3 directly binds to and deacetylates IDH2, thereby enhancing IDH2 activity and contributing to the regulation of ROS homeostasis [7, 8]. The role of SIRT3 in regulating protein deacetylation modifications and their involvement in diverse physiological and pathophysiological conditions has implicated this deacetylase as a potential therapeutic target. However, the roles of SIRT3 activation and protein deacetylation modifications in ischemia/reperfusion injury, particularly in subjects with hypertrophic preconditioning, have remained elusive. Both the expression and activity of SIRT3 were markedly decreased in the MI group after MI injury but were markedly enhanced in the T3D4 + MI group at 3 days post MI (Fig. 2a, b). Next, we analyzed the acetylation status of the SIRT3 substrate, IDH2, using an antibody that specifically detects IDH2 acetylation at K413 to determine whether increased SIRT3 levels were associated with its increased activity. IDH2 activity has been shown to be increased by deacetylation at K413 by SIRT3 [7, 8]. Consistent with increased SIRT3 levels, we observed increased activity of SIRT3, as revealed by reduced acetylation of IDH2 following the retraction of short-term pressure overload in mice with MI injury (Fig. 2b). IDH2 activity is tightly regulated by acetylation of IDH2 lysine residues. Therefore, we assessed myocardial IDH2 activity. Compared with the sham-operated group, the MI group showed a marked reduction in IDH2 activity, whereas transient TAC promoted IDH2 activation by inducing IDH2 deacetylation (Fig. 2b). Thus, SIRT3 may be involved in the cardioprotective effects of hypertrophic preconditioning.

a Myocardial SIRT3 protein expression and activity in WT mice subjected to MI. b Myocardial IDH2-K413-Ac protein expression and IDH2 activity in WT mice subjected to MI. n = 6 animals per group, *P < 0.05.
SIRT3 was required for the cardioprotective effects of hypertrophic preconditioning
We examined myocardial morphology and function in response to MI injury in the presence or absence of hypertrophic preconditioning in SIRT3-KO mice to explore whether and how SIRT3 regulates the protective effects of hypertrophic preconditioning after myocardial ischemia. SIRT3 deficiency was confirmed by immunoblotting (Fig. S2). Under conditions of MI, genetic ablation of SIRT3 significantly reduced the protective effects of hypertrophic preconditioning, as evidenced by exacerbated cardiac dysfunction (Fig. 3a) and an expanded myocardial fibrotic area (Fig. 3b). Based on these findings, SIRT3 is required for hypertrophic preconditioning-induced cardioprotection against post-MI injury. We further determined whether SIRT3 depletion may attenuate preconditioning-induced antioxidant and antiapoptotic activities. Under conditions of MI, SIRT3-KO completely prevented the effects of preconditioning on oxidative stress (Fig. 3c–e) and cell apoptosis (Fig. 3f–h). SIRT3-KO markedly inhibited the preconditioning-elicited reduction in oxidative stress (Fig. 3c–e), as evidenced by reduced NADPH/NADP ratios (Fig. 3c), decreased GSH/GSSG ratios (Fig. 3d), and increased superoxide production (Fig. 3e) in SIRT3-KO mouse hearts subjected to preconditioning followed by MI. Moreover, the preconditioning-elicited antiapoptotic properties were lost in SIRT3-KO mice, as evidenced by aggravated mitochondrial impairment (Fig. 3f), diminished ATP content (Fig. 3g), and increased caspase-3 levels (Fig. 3h). These results suggest a pivotal role for myocardial hypertrophic preconditioning in the control of mitochondrial ROS production and apoptosis through SIRT3.

a Representative M-mode echocardiography images and cardiac function in SIRT3-KO mice subjected to MI. b Representative photographs of Masson’s trichrome staining and the extent of fibrosis expressed as the percentage of the Masson’s trichrome-stained area to the total left ventricular area in WT mice and SIRT3-KO mice at 4 weeks post MI. The blue area indicates fibrosis. Scale bar = 50 μm. c NADPH/NADP ratios in MI-injured myocardial tissues. d GSH/GSSG ratios in MI-injured cardiac tissues. e Quantitative analysis of superoxide production in cardiac tissues. f Representative photographs of mitochondria in SIRT3-KO mice obtained under transmission electron microscopy at 3 days post MI. Scale bar = 2 μm. g Mitochondrial function in WT mice and SIRT3-KO mice, as evaluated by mitochondrial ATP content analysis at 3 days post MI. h Caspase-3 activity in WT mice and SIRT3-KO mice at 3 days post MI. n = 6–8 animals per group, *P < 0.05.
Loss of SIRT3 increased IDH2 acetylation and exacerbated post-MI injury in mice
To investigate the potential effects of SIRT3 on IDH2 acetylation, cardiac samples from WT and SIRT3-KO mice were harvested, and the expression of total IDH2 and IDH2-K413-Ac was detected by immunoblotting. Even though the total IDH2 expression did not significantly differ among all groups, SIRT3 deficiency significantly enhanced acetylation of IDH2 (Fig. 4a). Moreover, we observed decreased IDH2 activity (Fig. 4a) in the mitochondria of SIRT3-KO mice, indicating that loss of SIRT3 reduces IDH2 activity in vivo. At 4 weeks post MI, we found that SIRT3-KO mice exhibited worse cardiac function (Fig. 4b) and fibrosis (Fig. 4c) than the WT mice subjected to MI. Moreover, we observed that genetic ablation of SIRT3 in the myocardium markedly promoted mitochondrial ROS production (Fig. 4d–f), as evidenced by the decreased NADPH/NADP ratios (Fig. 4d), decreased GSH/GSSG ratios (Fig. 4e), and increased superoxide production (Fig. 4f) in SIRT3-KO mouse hearts compared with WT mouse hearts following MI injury. Furthermore, MI-elicited apoptotic properties were aggravated in SIRT3-ablated mice, as evidenced by exacerbated mitochondrial impairment (Fig. 4g), diminished ATP content (Fig. 4h), and increased caspase-3 levels (Fig. 4i). These data suggest that SIRT3 plays a pivotal role in the maintenance of cardiac survival and function by quenching mitochondrial ROS and deacetylating IDH2.

a Myocardial IDH2-K413-Ac protein expression and IDH2 activity in WT mice and SIRT3-KO mice subjected to MI, as assessed at 3 days post MI. b Representative M-mode echocardiography images and cardiac function in WT mice and SIRT3-KO mice subjected to MI (acquired at 4 weeks post MI). c Representative photographs of Masson’s trichrome staining and the extent of fibrosis expressed as the percentage of the Masson’s trichrome-stained area to the total left ventricular area in WT mice and SIRT3-KO mice at 4 weeks post MI. The blue area indicates fibrosis. Scale bar = 50 μm. d NADPH/NADP ratios in MI-injured myocardial tissues. e GSH/GSSG ratios in MI-injured cardiac tissues. f Quantitative analysis of superoxide production in cardiac tissues. g Representative photographs of mitochondria in WT mice and SIRT3-KO mice obtained under transmission electron microscopy. Scale bar = 2 μm. h Mitochondrial function in WT mice and SIRT3-KO mice, as evaluated by mitochondrial ATP content analysis at 3 days post MI. i Caspase-3 activity in WT mice and SIRT3-KO mice at 3 days post MI. Mito mitochondria. n = 6–8 animals per group, *P < 0.05.
SIRT3 deacetylated IDH2 and alleviated post-MI damage
Cardiac-specific overexpression of the SIRT3 gene through in vivo AAV-SIRT3 transfection was performed 1 month before MI injury to obtain more direct evidence supporting the cardioprotective effects of SIRT3 against post-MI injury. Myocardial SIRT3 overexpression was demonstrated by increased SIRT3 protein levels and enzyme activity (Fig. 5a). Even though total IDH2 levels did not markedly differ among all groups, SIRT3 overexpression reduced acetylation of IDH2 (Fig. 5b). Moreover, we observed markedly increased IDH2 activity (Fig. 5b) in mitochondria from SIRT3-overexpressing mice subjected to MI, indicating that SIRT3 enhances overall IDH2 activity in mice in vivo. At 4 weeks post MI, we found that the mice that received AAV-SIRT3 exhibited better cardiac function than the control mice that received AAV-EGFP (Fig. 5c). Importantly, treatment with AAV-SIRT3 significantly decreased the myocardial fibrotic area (Fig. 5d). Mitochondrial ROS generation and apoptosis were analyzed to determine whether SIRT3 improved cardiac survival and function by regulating cellular ROS homeostasis. Compared with the AAV-EGFP group, the AAV-SIRT3 group showed considerably less MI-induced oxidative stress (Fig. 5e–g) and apoptosis (Fig. 5h–j). AAV-SIRT3 markedly inhibited MI-elicited overproduction of superoxide (Fig. 5e–g), as evidenced by increased NADPH/NADP ratios (Fig. 5e), increased GSH/GSSG ratios (Fig. 5f), and decreased superoxide production (Fig. 5g) in SIRT3-overexpressing mouse hearts following MI injury. Moreover, cardiac MI-elicited apoptotic properties were lost in SIRT3-overexpressing mice, as evidenced by alleviation of mitochondrial impairment (Fig. 5h), increased ATP content (Fig. 5i), and reductions in caspase-3 levels (Fig. 5j). These results suggest a pivotal role for SIRT3 activation in the control of mitochondrial ROS production.

a Myocardial SIRT3 protein expression and activity in WT mice infected with AAV-EGFP (control virus) or AAV-SIRT3. b Myocardial IDH2-K413-Ac protein expression and IDH2 activity in WT mice infected with AAV-EGFP (control virus) or AAV-SIRT3, as assessed at 3 days post MI. c Representative M-mode echocardiography images and cardiac function in WT mice infected with AAV-EGFP (control virus) or AAV-SIRT3 (acquired at 4 weeks post MI). d Representative photographs of Masson’s trichrome staining and the extent of fibrosis expressed as the percentage of the Masson’s trichrome-stained area to the total left ventricular area in WT mice infected with AAV-EGFP (control virus) or AAV-SIRT3 at 4 weeks post MI. The blue area indicates fibrosis. Scale bar = 50 μm. e NADPH/NADP ratios in MI-injured myocardial tissues. f GSH/GSSG ratios in MI-injured cardiac tissues. g Quantitative analysis of superoxide production in cardiac tissues. h Representative photographs of mitochondria in WT mice infected with AAV-EGFP (control virus) or AAV-SIRT3 obtained under transmission electron microscopy. Scale bar = 2 μm. i Mitochondrial function in WT mice infected with AAV-EGFP (control virus) or AAV-SIRT3, as evaluated by mitochondrial ATP content analysis at 3 days post MI. j Caspase-3 activity in WT mice infected with AAV-EGFP (control virus) or AAV-SIRT3 at 3 days post MI. n = 6–8 animals per group, *P < 0.05.
An IDH2 lysine 413 deacetylation mimic conferred cardioprotective effects against post-MI injury by attenuating oxidative stress-induced apoptotic signaling
IDH2 has been shown to regulate cellular ROS homeostasis by producing mitochondrial NADPH, which is an essential cofactor required for the reduction of GSSG to GSH [18, 19]. The IDH2-K413-Ac status has been shown to direct enzymatic activity in cancer cells, and replacement of a lysine with a glutamine (K413Q) mimics lysine acetylation, whereas replacement with an arginine (K413R) mimics deacetylation [7, 8]. Thus, mutating IDH2-K413 to arginine would be expected to mimic lysine deacetylation, whereas replacement with a glutamine would be predicted to mimic lysine acetylation. We transfected recombinant adeno-associated virus 9 constructs containing WT IDH2, IDH2 K413R (deacetylation mimic), and IDH2 K413Q (acetylation mimic) into mouse hearts through direct tail vein injection to determine whether mimicked IDH2 deacetylation confers cardioprotective effects against post-MI injury. In comparison with IDH2 WT expression, IDH2 K413R expression markedly increased myocardial IDH2 activity, whereas IDH2 K413Q expression decreased IDH2 activity (Fig. 6a). At 4 weeks post MI, we demonstrated that the IDH2 K413R-expressing mice exhibited improved cardiac function (Fig. 6b) and attenuated fibrosis (Fig. 6c), whereas the IDH2 K413Q-expressing mice exhibited aggravated cardiac function (Fig. 6b) and expanded fibrotic areas (Fig. 6c) in comparison with the mice expressing WT IDH2 following MI. Mitochondrial ROS production regulates cell death during myocardial ischemic injury. Therefore, we assessed the effects of IDH2 mutants on oxidative stress (a pivotal upstream mediator of cell death) [20]. We observed that myocardial IDH2 K413R expression markedly inhibited mitochondrial ROS production, as evidenced by increased NADPH/NADP ratios (Fig. 6d), increased GSH/GSSG ratios (Fig. 6e), and reduced superoxide production (Fig. 6f) following MI injury in the hearts of mice treated with IDH2 K413R. In contrast, myocardial IDH2 K413Q expression markedly promoted mitochondrial ROS production, as evidenced by decreased NADPH/NADP ratios (Fig. 6d), decreased GSH/GSSG ratios (Fig. 6e), and increased superoxide production (Fig. 6f) following MI injury in the hearts of mice treated with IDH2 K413Q. Apoptosis is the major contributor to cell death after oxidative injury [21]. We next investigated the effects of IDH2 mutants on the mitochondrial-mediated apoptotic pathway to determine the mechanisms underlying the protective effects of IDH2 K413R on the myocardium. In mouse hearts, MI aggravated mitochondrial impairment (Fig. 6g), diminished ATP content (Fig. 6h), and promoted caspase-3 activation (Fig. 6i), changes that were strongly alleviated by IDH2 K413R transfection. In contrast, IDH2 K413Q expression further exacerbated post-MI injury, as evidenced by increased mitochondrial impairment (Fig. 6g), diminished ATP content (Fig. 6h), and increased caspase-3 activation (Fig. 6i) in IDH2 K413Q-expressing mice compared with WT IDH2-expressing mice following MI. These results demonstrate that deacetylation of IDH2 exerts cardioprotective effects against post-MI injury that involve improvements in the quenching of mitochondrial ROS.

a Myocardial IDH2 activity in mouse hearts expressing WT IDH2, IDH2 K413R, or IDH2 K413Q at 3 days post MI. b Representative M-mode echocardiography images and cardiac function in mouse hearts expressing WT IDH2, IDH2 K413R, or IDH2 K413Q at 4 weeks post MI. c Representative photographs of Masson’s trichrome staining and the extent of fibrosis expressed as the percentage of the Masson’s trichrome-stained area to the total left ventricular area in mouse hearts expressing WT IDH2, IDH2 K413R, or IDH2 K413Q at 4 weeks post MI. The blue area indicates fibrosis. Scale bar = 50 μm. d NADPH/NADP ratios in MI-injured myocardial tissues. e GSH/GSSG ratios in MI-injured cardiac tissues. f Quantitative analysis of superoxide production in cardiac tissues. g Representative photographs of mitochondria in mouse hearts expressing WT IDH2, IDH2 K413R, or IDH2 K413Q obtained under transmission electron microscopy. Scale bar = 2 μm. h Mitochondrial function in mouse hearts expressing WT IDH2, IDH2 K413R, or IDH2 K413Q, as evaluated by mitochondrial ATP content analysis at 3 days post MI. i Caspase-3 activity in mouse hearts expressing WT IDH2, IDH2 K413R, or IDH2 K413Q at 3 days post MI. LVIDd left ventricular internal diastolic diameter, LVIDs left ventricular internal systolic diameter; n = 6–8 animals per group, *P < 0.05.
An IDH2 lysine 413 acetylation mimic abrogated hypertrophic preconditioning-induced cardioprotective effects by promoting mitochondrial ROS production
We transfected recombinant adeno-associated virus 9 constructs carrying WT IDH2, IDH2 K413Q (acetylation mimic), and IDH2 K413R (deacetylation mimic) into WT and SIRT3-KO mice through direct tail vein injection to obtain direct evidence that IDH2 acetylation is involved in myocardial hypertrophic preconditioning-induced cardioprotection. Compared with WT IDH2 expression, IDH2 K413Q expression in the myocardium significantly reversed the effects of hypertrophic preconditioning, as evidenced by exacerbated cardiac dysfunction (Fig. 7a) and expanded myocardial fibrotic areas (Fig. 7b). Compared with WT IDH2 expression, IDH2 K413R expression, but not IDH2 K413Q expression, in the myocardium ameliorated MI-elicited cardiac injury, as evidenced by preserved cardiac function (Fig. 7a) and reduced myocardial fibrotic areas (Fig. 7b) in mice lacking SIRT3. However, no additional cardioprotective effects were achieved when we combined myocardial hypertrophic preconditioning and IDH2 K413R expression in SIRT3-KO mice. Based on these findings, the IDH2 lysine 413 acetylation mimic abrogated the hypertrophic preconditioning-induced cardioprotective effect.

a Cardiac function in WT mice and SIRT3-KO mice expressing WT IDH2, IDH2 K413R, or IDH2 K413Q at 4 weeks post MI. b Myocardial fibrotic areas in WT mice and SIRT3-KO mice expressing WT IDH2, IDH2 K413R, or IDH2 K413Q at 4 weeks post MI. The blue area indicates fibrosis. c NADPH/NADP ratios in MI-injured myocardial tissues. d GSH/GSSG ratios in MI-injured cardiac tissues. e Quantitative analyses of superoxide production in cardiac tissues. f Mitochondrial ATP content in WT mice and SIRT3-KO mice expressing WT IDH2, IDH2 K413R, or IDH2 K413Q at 3 days post MI. g Caspase-3 activity in WT mice and SIRT3-KO mice expressing WT IDH2, IDH2 K413R, or IDH2 K413Q at 3 days post MI. n = 6–8 animals per group, *P < 0.05.
IDH2 has been shown to regulate mitochondrial ROS production [8]. Therefore, we next sought to determine whether acetylation of IDH2 is involved in myocardial hypertrophic preconditioning-mediated regulation of mitochondrial ROS generation and apoptosis. We hypothesized that IDH2 acetylation may block preconditioning-induced antioxidant and antiapoptotic activities. Under MI conditions, cardiac IDH2 K413Q expression completely blocked the effects of preconditioning on oxidative stress (Fig. 7c–e) and apoptosis (Fig. 7f, g). IDH2 K413Q expression also markedly inhibited the preconditioning-elicited reductions in superoxide levels, as evidenced by reduced NADPH/NADP ratios (Fig. 7c), decreased GSH/GSSG ratios (Fig. 7d), and elevated superoxide production (Fig. 7e). Moreover, the preconditioning-elicited antiapoptotic properties were lost in IDH2 K413Q-treated mice, as evidenced by aggravated mitochondrial dysfunction (Fig. 7f) and increased caspase-3 levels (Fig. 7g). In contrast, expression of IDH2 K413R markedly hindered MI-elicited mitochondrial ROS production (Fig. 7c–e), as evidenced by increased NADPH/NADP ratios (Fig. 7c), increased GSH/GSSG ratios (Fig. 7d), and decreased superoxide production (Fig. 7e) in the hearts of SIRT3-KO mice treated with IDH2 K413R and then subjected to MI. Moreover, cardiac MI-elicited apoptotic properties were alleviated in IDH2 K413R-treated mice, as evidenced by attenuated mitochondrial impairment (Fig. 7f) and decreased caspase-3 levels (Fig. 7g) in the hearts of SIRT3-KO mice treated with IDH2 K413R following MI. These results suggest a pivotal role for myocardial hypertrophic preconditioning in the control of mitochondrial ROS production and cell death through deacetylation of IDH2.
Discussion
IDH2 catalyzes the oxidative decarboxylation of isocitrate to α-ketoglutarate and is a major source of NADPH [6]. Moreover, IDH2 has been shown to play a pivotal role in maintaining mitochondrial redox balance by regulating ROS production [7, 8]. However, the roles of IDH2 in post-MI damage and specifically in pressure overload-induced preconditioning remain unclear. The current study was performed to clarify these roles.
We make several novel contributions in the present work. First, we provide strong evidence that ischemic myocardial insult induces acetylation of IDH2 without altering total IDH2 levels, thus reducing IDH2 activity. Second, using gain- and loss-of-function approaches, we demonstrate that SIRT3 regulates IDH2 activity by deacetylating IDH2 in the context of MI-induced cell death. Third, using an IDH2 lysine 413 acetylation mimic (IDH2 K314Q) and an IDH2 lysine 413 deacetylation mimic (IDH2 K413R), we demonstrate that the reduction in IDH2 activity caused by IDH2 K314Q mutation aggravates mitochondrial impairment, promotes ROS overload, and exacerbates post-MI damage, whereas the increase in IDH2 activity caused by IDH2 K314R expression preserves intact mitochondria, alleviates ROS overload, and reduces post-MI damage. Fourth, we report, for the first time, that hypertrophic preconditioning attenuates post-MI damage by deacetylating IDH2 though a SIRT3-dependent mechanism (Fig. 8).

Hypertrophic preconditioning reduces oxidative stress and inhibits mitochondrial-dependent cell apoptosis by deacetylation of IDH2 through a SIRT3-dependent mechanism.
MI remains a leading cause of morbidity and mortality throughout the world and results in cardiomyocyte death, reparative fibrotic healing, adverse ventricular remodeling, and ultimately heart failure [22]. Hence, development of appropriate therapeutic strategies against post-MI damage is an area of intense interest. In response to myocardial ischemia, mitochondria are primary cellular damage targets, and therapeutic strategies targeting mitochondria ameliorate MI damage [3, 23]. IDH2 is present primarily in mitochondria, serves as a functional target of SIRT3 and mediates pro-oncogenic properties in cells overexpressing SIRT3 [7, 8]. Global induction of mutant IDH2 expression in adult mice leads to dilated cardiomyopathy, white-matter lesions, and muscular dystrophy [9, 24]. However, the effects of IDH2 mutation on post-MI damage have remained elusive. In the present study, we found that myocardial IDH2 activity, but not IDH2 protein expression, is decreased in response to MI, indicating that IDH2 is involved in post-MI damage. IDH2 activity is tightly regulated by acetylation at IDH2 lysine residues [7, 8]. Therefore, we determined the acetylation of IDH2 via immunoblotting using an anti-acetylated lysine antibody. We observed increased acetylated IDH2 expression in MI mice. Furthermore, using IDH2 K314Q and IDH2 K413R, we demonstrated that IDH2 signaling plays a crucial role in maintaining cardiac function, ameliorating myocardial fibrosis, inhibiting cardiomyocyte apoptosis, and alleviating ROS overload. There are several possible mechanisms underlying the cardioprotective effects of IDH2 against post-MI damage. Oxidative stress can be induced by various stresses and is the primary contributor to post-MI cell death and mitochondrial impairment. Mitochondria are not only major sources of ROS but also primary targets of ROS-induced damage in response to MI [4, 5]. Therefore, mitochondrial damage and ROS overproduction can constitute an impairment cycle leading to aggravated myocardial insult. In our study, we found that expression of IDH2 K413Q reduced myocardial IDH2 activity, exacerbated myocardial fibrosis, and impaired cardiac function by aggravating mitochondrial ROS overload, whereas expression of IDH2 K413R enhanced IDH2 activity, reduced myocardial fibrosis, and improved cardiac function by alleviating ROS overload. Collectively, these findings indicate that posttranslational modification of IDH2 plays a crucial role in post-MI remodeling by regulating mitochondrial oxidative stress.
Lysine acetylation is a critical posttranslational modification involved in the regulation of mitochondrial protein activity [25,26,27]. Recently, SIRT3 has been identified as an important mitochondrial deacetylation enzyme along with two other sirtuins, SIRT4 and SIRT5 [28]. SIRT3 can deacetylate a number of mitochondrial proteins and is the most robust mitochondrial deacetylase [29, 30]. SIRT3 is also involved in mitochondrial energy metabolism and regulation of mitochondrial ROS production [31,32,33]. In the current study, SIRT3 was shown to regulate IDH2 enzymatic activity by deacetylating IDH2 (at lysine 413) in the setting of MI, suggesting that IDH2 is a major downstream target of SIRT3 that contributes to the reduction in ROS production [7, 8]. Herein, we demonstrated that MI reduces SIRT3 protein expression and activity, thus regulating the transcriptional activity of IDH2 via acetylation modification. Moreover, SIRT3 overexpression increases IDH2 activity, hinders ROS accumulation, and reverses MI-induced mitochondrial dysfunction via deacetylation of IDH2, whereas SIRT3 deficiency reduces IDH2 activity, aggravates ROS accumulation, and exacerbates MI-induced mitochondrial dysfunction via acetylation of IDH2. In addition, we demonstrated that SIRT3 signaling plays a crucial role in maintaining cardiac function and ameliorating myocardial fibrosis. These results demonstrate that SIRT3-IDH2-modulated redox balance is an indispensable mechanism for MI-induced cardiac damage.
We next determined whether a physiological intervention, myocardial hypertrophic preconditioning, would produce cardioprotection against post-MI injury. We demonstrated, for the first time, that hypertrophic preconditioning induced by transient pressure overload protects hearts from post-MI damage, as evidenced by improved echocardiographic left ventricular function and decreased cardiac fibrosis. To provide mechanistic insight into how hypertrophic preconditioning confers cardioprotection against post-MI damage, we investigated the effects of hypertrophic preconditioning on the SIRT3/IDH2 pathway. The rationale for examining this pathway was threefold. First, MI was shown to increase IDH2 acetylation, resulting in reduced IDH2 enzyme activity. Second, we demonstrated that SIRT3 overexpression caused deacetylation of IDH2 and attenuated post-MI injury. Third, we observed that retraction of 3-day pressure overload increased mitochondrial SIRT3 expression and activity in preconditioned mice. This evidence motivated us to investigate the potential effects of SIRT3/IDH2 signaling in post-MI damage. We found that hypertrophic preconditioning significantly activated SIRT3 signaling and enhanced IDH2 activity. Ablation of endogenous SIRT3 completely blocked the cardioprotective effects of hypertrophic preconditioning, as demonstrated by deteriorated cardiac function, and aggravated cardiac fibrosis. Furthermore, IDH2 activity was enhanced by hypertrophic preconditioning through a SIRT3-dependent deacetylation effect, and expression of IDH2 K413Q in the myocardium reversed the cardioprotective effects of hypertrophic preconditioning against post-MI damage. Taken together, these results imply that deacetylation of IDH2 by SIRT3 plays a crucial role in mediating the cardioprotective effects of hypertrophic preconditioning against post-MI damage.
Conclusion
In summary, we have demonstrated, in a murine model, that IDH2 can serve as a novel therapeutic target for post-MI damage. We provide the first direct evidence that SIRT3 is an upstream regulator of IDH2 that deacetylates IDH2 to disturb the cycle of mitochondrial damage and mitochondrial ROS overload. Finally, we have demonstrated that hypertrophic preconditioning alleviates post-MI remodeling via a SIRT3-dependent IDH2 deacetylation mechanism. We conclude that hypertrophic preconditioning exploits a novel endogenous self-defensive cardioprotective strategy against post-MI injury and therefore represents a potentially attractive therapeutic strategy for the treatment of ischemic heart diseases. Therapeutic strategies targeting IDH2 will be promising approaches for alleviation of post-MI injury.
References
Hill JA, Olson EN. Cardiac plasticity. N Engl J Med. 2008;358:1370–80.
Cung TT, Morel O, Cayla G, Rioufol G, Garcia-Dorado D, Angoulvant D, et al. Cyclosporine before PCI in patients with acute myocardial infarction. N Engl J Med. 2015;373:1021–31.
Cadenas S. ROS and redox signaling in myocardial ischemia-reperfusion injury and cardioprotection. Free Radic Biol Med. 2018;117:76–89.
Pei H, Song X, Peng C, Tan Y, Li Y, Li X, et al. TNF-alpha inhibitor protects against myocardial ischemia/reperfusion injury via Notch1-mediated suppression of oxidative/nitrative stress. Free Radic Biol Med. 2015;82:114–21.
Ge H, Zhao M, Lee S, Xu Z. Mitochondrial Src tyrosine kinase plays a role in the cardioprotective effect of ischemic preconditioning by modulating complex I activity and mitochondrial ROS generation. Free Radic Res. 2015;49:1210–7.
Dang L, Su SM. Isocitrate dehydrogenase mutation and (R)-2-hydroxyglutarate: from basic discovery to therapeutics development. Annu Rev Biochem. 2017;86:305–31.
Zou X, Zhu Y, Park SH, Liu G, O’Brien J, Jiang H, et al. SIRT3-mediated dimerization of IDH2 directs cancer cell metabolism and tumor growth. Cancer Res. 2017;77:3990–9.
Yu W, Dittenhafer-Reed KE, Denu JM. SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J Biol Chem. 2012;287:14078–86.
Akbay EA, Moslehi J, Christensen CL, Saha S, Tchaicha JH, Ramkissoon SH, et al. D-2-hydroxyglutarate produced by mutant IDH2 causes cardiomyopathy and neurodegeneration in mice. Genes Dev. 2014;28:479–90.
Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–36.
Hausenloy DJ, Yellon DM. Ischaemic conditioning and reperfusion injury. Nat Rev Cardiol. 2016;13:193–209.
Xia Z, Li H, Irwin MG. Myocardial ischaemia reperfusion injury: the challenge of translating ischaemic and anaesthetic protection from animal models to humans. Br J Anaesth. 2016;117 (Suppl 2):i44–62.
Heusch G. Molecular basis of cardioprotection: signal transduction in ischemic pre-, post-, and remote conditioning. Circ Res. 2015;116:674–99.
Wei X, Wu B, Zhao J, Zeng Z, Xuan W, Cao S, et al. Myocardial hypertrophic preconditioning attenuates cardiomyocyte hypertrophy and slows progression to heart failure through upregulation of S100A8/A9. Circulation. 2015;131:1506–17.
Hu P, Zhang D, Swenson L, Chakrabarti G, Abel ED, Litwin SE. Minimally invasive aortic banding in mice: effects of altered cardiomyocyte insulin signaling during pressure overload. Am J Physiol Heart Circ Physiol. 2003;285:H1261–9.
Gao E, Lei YH, Shang X, Huang ZM, Zuo L, Boucher M, et al. A novel and efficient model of coronary artery ligation and myocardial infarction in the mouse. Circ Res. 2010;107:1445–53.
Ma LL, Li Y, Yin PP, Kong FJ, Guo JJ, Shi HT, et al. Hypertrophied myocardium is vulnerable to ischemia/reperfusion injury and refractory to rapamycin-induced protection due to increased oxidative/nitrative stress. Clin Sci. 2018;132:93–110.
Jo SH, Son MK, Koh HJ, Lee SM, Song IH, Kim YO, et al. Control of mitochondrial redox balance and cellular defense against oxidative damage by mitochondrial NADP+-dependent isocitrate dehydrogenase. J Biol Chem. 2001;276:16168–76.
Noh MR, Kong MJ, Han SJ, Kim JI, Park KM. Isocitrate dehydrogenase 2 deficiency aggravates prolonged high-fat diet intake-induced hypertension. Redox Biol. 2020;34:101548.
Navarro-Yepes J, Burns M, Anandhan A, Khalimonchuk O, Del RL, Quintanilla-Vega B, et al. Oxidative stress, redox signaling, and autophagy: cell death versus survival. Antioxid Redox Signal. 2014;21:66–85.
Shimokawa H, Sunamura S, Satoh K. RhoA/Rho-Kinase in the cardiovascular system. Circ Res. 2016;118:352–66.
Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. Executive summary: heart disease and stroke statistics-2016 update: a report from the American Heart Association. Circulation. 2016;133:447–54.
Ong SB, Samangouei P, Kalkhoran SB, Hausenloy DJ. The mitochondrial permeability transition pore and its role in myocardial ischemia reperfusion injury. J Mol Cell Cardiol. 2015;78:23–34.
Ku HJ, Ahn Y, Lee JH, Park KM, Park JW. IDH2 deficiency promotes mitochondrial dysfunction and cardiac hypertrophy in mice. Free Radic Biol Med. 2015;80:84–92.
Banreti A, Sass M, Graba Y. The emerging role of acetylation in the regulation of autophagy. Autophagy. 2013;9:819–29.
Baek SH, Kim KI. Epigenetic control of autophagy: nuclear events gain more attention. Mol Cell. 2017;65:781–5.
Aon MA, Cortassa S, Juhaszova M, Sollott SJ. Mitochondrial health, the epigenome and healthspan. Clin Sci. 2016;130:1285–305.
Schwer B, Verdin E. Conserved metabolic regulatory functions of sirtuins. Cell Metab. 2008;7:104–12.
Winnik S, Auwerx J, Sinclair DA, Matter CM. Protective effects of sirtuins in cardiovascular diseases: from bench to bedside. Eur Heart J. 2015;36:3404–12.
Kumar S, Lombard DB. Mitochondrial sirtuins and their relationships with metabolic disease and cancer. Antioxid Redox Signal. 2015;22:1060–77.
Dikalova AE, Itani HA, Nazarewicz RR, McMaster WG, Flynn CR, Uzhachenko R, et al. Sirt3 impairment and SOD2 hyperacetylation in vascular oxidative stress and hypertension. Circ Res. 2017;121:564–74.
Zhang X, Ji R, Liao X, Castillero E, Kennel PJ, Brunjes DL, et al. miR-195 regulates metabolism in failing myocardium via alterations in SIRT3 expression and mitochondrial protein acetylation. Circulation. 2018;19:2052–67.
Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, et al. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell. 2010;143:802–12.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 81870290, 81521001, 81600280, and 81800238).
Author information
Authors and Affiliations
Contributions
LLM, SJW, YJM, FJK, JJG, and ZD performed the experiments and analyzed the data. LLM, JJG, and FJK performed the cardiac I/R injury surgery. SJW, AJS, and YZZ revised the manuscript. SJW, YZZ, and JBG designed and supervised the study and wrote the paper. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Ma, Ll., Kong, Fj., Ma, Yj. et al. Hypertrophic preconditioning attenuates post-myocardial infarction injury through deacetylation of isocitrate dehydrogenase 2. Acta Pharmacol Sin 42, 2004–2015 (2021). https://doi.org/10.1038/s41401-021-00699-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-021-00699-0
Keywords
This article is cited by
-
Genome-wide analysis revealed the dysregulation of RNA binding protein-correlated alternative splicing events in myocardial ischemia reperfusion injury
BMC Medical Genomics (2023)
-
SZC-6, a small-molecule activator of SIRT3, attenuates cardiac hypertrophy in mice
Acta Pharmacologica Sinica (2023)
-
Changes of energy metabolism in failing heart and its regulation by SIRT3
Heart Failure Reviews (2023)
-
Pregnancy-induced physiological hypertrophic preconditioning attenuates pathological myocardial hypertrophy by activation of FoxO3a
Cellular and Molecular Life Sciences (2023)
