
Abstract
The sponge-associated microbial community contributes to the overall health and adaptive capacity of the sponge holobiont. This community is regulated by the environment and the immune system of the host. However, little is known about the effect of environmental stress on the regulation of host immune functions and how this may, in turn, affect sponge–microbe interactions. In this study, we compared the bacterial diversity and immune repertoire of the demosponge, Neopetrosia compacta, and the calcareous sponge, Leucetta chagosensis, under varying levels of acidification and warming stress based on climate scenarios predicted for 2100. Neopetrosia compacta harbors a diverse microbial community and possesses a rich repertoire of scavenger receptors while L. chagosensis has a less diverse microbiome and an expanded range of pattern recognition receptors and immune response-related genes. Upon exposure to RCP 8.5 conditions, the microbiome composition and host transcriptome of N. compacta remained stable, which correlated with high survival (75%). In contrast, tissue necrosis and low survival (25%) of L. chagosensis was accompanied by microbial community shifts and downregulation of host immune-related pathways. Meta-analysis of microbiome diversity and immunological repertoire across poriferan classes further highlights the importance of host–microbe interactions in predicting the fate of sponges under future ocean conditions.
Similar content being viewed by others


Introduction
Since the industrial revolution, the ocean has taken up a substantial amount of CO2 that has led to reduced pH and CaCO3 saturation state [1]. This global change in ocean chemistry, exacerbated by sea surface warming, can affect many organismal processes, consequently disrupting reef population dynamics and ecosystem functioning [2]. A meta-analysis of climate change-associated studies of abundant benthic groups revealed that sponges are likely winners under future climate scenarios [3].
Marine sponges (Porifera) are a major component of the benthic ecosystem and are responsible for many ecological processes, such as nutrient cycling, ecosystem structuring, reef consolidation, and bio-erosion [4]. Sponges are generally thought to possess exceptional ecological adaptability, stemming from a complex physiology and diverse associated microbiome [5]. However, while most siliceous demosponges exhibit resistance and may even benefit from acidified ocean conditions, calcareous sponges are vulnerable when exposed to lower pH levels [6].
Poriferans forge a close relationship with diverse groups of microorganisms to form a complex structured ecosystem referred to as the holobiont [7]. The microbial community has key roles in nutrient assimilation and metabolism, vitamin synthesis, and defense [8]. Host-associated microbiomes are shaped by various ecological selective forces acting within a holobiont. Resource limitation coupled with interspecific interactions and host immune functions fine-tune microbial populations to maintain holobiont homeostasis [9].
Environmental perturbations can cause changes in the sponge microbiome [5]. For example, elevated temperatures disrupt symbiotic functions in the demosponge, Rhopaloeides odorabile Thompson, Murphy, Bergquist & Evans, 1987, leading to holobiont destabilization and dysbiosis [10]. While disruption of host–microbe interactions usually precedes mass mortalities and is widely observed in holobionts under stress [11], alternative trajectories of microbiome plasticity promote rapid organismal adaptation [12]. For example, the microbiome of Coelocarteria singaporensis (Carter, 1883) at CO2 seeps exhibits the potential for more efficient carbon fixation and nitrogen metabolism in acidified conditions compared to sponges at control sites [13, 14].
Environmental perturbations can also induce changes in the host’s innate immune system, which is involved in sensing microbial cells and activating phagocytosis, cell death, or production of antimicrobial molecules [15,16,17]. For example, elevated temperature induced the coordinated expression of pattern recognition receptors (PRRs), immune-related signaling cascades, and apoptosis regulators in the demosponge, Haliclona (Reniera) tubifera (George & Wilson, 1919) [18].
The immune gene repertoire varies among sponges from different taxa [19, 20] and in species with different microbiome diversity [17, 21]. For example, Stylissa carteri (Dendy, 1889), a low microbial abundance (LMA) sponge, possesses an expanded family of scavenger receptor cysteine rich (SRCR) domain-containing proteins relative to the high microbial abundance (HMA) sponge, Xestospongia testudinaria (Lamarck, 1815) [21]. Moreover, a survey of sponge transcriptomes revealed the absence of certain immune pathway components, such as myeloid differentiation primary response 8 (MyD88), in the calcarean, Sycon ciliatum (Fabricus, 1780) [19]. Distinct combinations of immune molecules may influence microbiome control in the sponge holobiont. Thus, elucidating the links between immune system functions and microbiome structuring may provide a better understanding of inter-species differences in the tolerance of sponges to environmental stressors.
To determine how the sponge-associated microbiome and the host immune gene repertoire are affected by varying stress conditions, we sequenced the 16S rRNA genes and host transcriptomes of the sponges Neopetrosia compacta (Ridley & Dendy, 1886) (class Demospongiae, order Haplosclerida, family Petrosiidae) and Leucetta chagosensis Dendy, 1913 (class Calcarea, order Clathrinida, family Leucettidae) subjected to different combinations of warming and acidification stress according to climate scenarios predicted for 2100. We also compared our results to the microbiomes of 189 other sponge species and the immune gene repertoire of 16 sponge species to elucidate common trends. We hypothesized that changes in sponge microbiome structure will correlate with changes in the expression of certain immune response genes. Our findings suggest that microbiome diversity and stability of host immune functions influence the survivorship of sponge holobionts under perturbed conditions. This highlights the importance of host–microbe interactions in predicting the fate of marine sponges in the face of a rapidly changing ocean.
Materials and methods
Sponge sampling and culture
Six specimens each of N. compacta and L. chagosensis were collected from the Bolinao-Anda Reef Complex in Pangasinan, northwestern Philippines (16.296° N, 120.014° E) in September 2018 with permission from the Philippines Department of Agriculture (Gratuitous Permit No. 0169-19). Sponge identities were confirmed by their morphology [22] and 28S rRNA gene analyses (Fig. S1). Donor sponges were cut into 12 (≈1 cm3) fragments using a sterile razor. Each sponge clone was tagged to enable donor tracking, placed individually into polypropylene mesh baskets, and allowed to heal in situ for 30 days. Healed fragments were brought to the Bolinao Marine Laboratory and allowed to acclimatize for 7 days in aquaria receiving flow-through seawater under ambient conditions of pH 8.0 and 28 °C.
Stress response experiments
Stress response experiments were conducted in independently-aerated 10 L aquaria with flow-through seawater. Temperatures were regulated using 300 W submersible heaters (EHEIM GmbH & Co. KG, Baden Wurttemberg, Germany), levels of injected CO2 manipulated using a mass flow controller, and illumination was provided by daylight LED lamps following a 12:12 light:dark photoperiod. Conditions were designed to simulate the present day and predicted 2100 Representative Concentration Pathway (RCP) 6.0 and 8.5 scenarios [23]. Treatment conditions included (i) pH 8.0, 28 °C (Present Day), (ii) pH 7.6, 28 °C (Acidification), (iii) pH 8.0, 32 °C (Warming), (iv) pH 7.8, 30 °C (RCP 6.0), and (v) pH 7.6, 32 °C (RCP 8.5). Each treatment was represented by four independent replicate aquaria containing three fragments of each sponge species, with each fragment originating from a different sponge donor (n = 12 fragments per treatment per species). Temperature and pH levels were changed gradually (temperature: +1 °C/day, pH: −0.5/day) until the desired conditions were reached (Fig. S2). Treatment conditions were maintained for 2 days then the experiment was terminated because tissue necrosis had begun to manifest in some fragments. Surviving sponges were washed with ultraviolet-filtered seawater, any necrotic tissues were excised, and the remaining healthy tissues were flash-frozen in liquid nitrogen for transport, then stored at −80 °C.
Light and temperature in the tanks were monitored using submersible loggers (HOBO pendant, Onset Computer Corp., Bourne, MA, USA), pH was measured using a SevenGo Duo Pro pH meter (Mettler Toledo, Columbus, OH, USA), and dissolved oxygen and salinity were measured using a multiparameter meter (Pro 2030, YSI Inc., Yellow Springs, OH, USA). Seawater carbonate chemistry parameters (Table S1) were calculated using the CO2SYS package [24].
16S rRNA gene sequencing and analysis
Total genomic DNA was extracted from sponge tissues (three biological replicates for each treatment per species) using the DNeasy PowerSoil Pro Kit (Mo Bio, Carlsbad, CA, USA) following the manufacturer’s protocol. Replicate samples for each treatment were selected from different donor sponges. DNA extracts were sent to Macrogen, South Korea for sequencing. Bacterial 16S rRNA V3–V4 hypervariable region was amplified from the extracted DNA using barcoded primers Bakt_341F (5’-CCTACGGGNGGCWGCAG-3’) and Bakt_805R (5’-GACTACHVGGGTATCTAATCC-3’) [25]. Paired-end sequencing (300 bp) was performed on the MiSeq platform (Illumina, Inc., San Diego, CA, USA) following the dual-index sequencing strategy.
16S rRNA analysis was conducted using QIIME2 version 2019.7 [26]. Chimeric sequences and singletons were removed and amplicon errors were corrected using the DADA2 package [27]. Denoised forward and reverse reads were then assembled into contigs. Taxonomic assignment of contigs was carried out using a Naïve Bayes classifier trained on SILVA version 132 [28]. The classifier was set to include V3–V4 regions of 16S rRNA genes at 99% sequence similarity. Sequence reads from chloroplasts and mitochondria were removed from the final set of Amplicon Sequence Variants (ASVs). Raw sequence reads are available on the NCBI Short Read Archive under BioProject PRJNA689294.
Microbial community composition analysis and functional prediction
Rarefied ASV libraries were produced through random down-sampling to the smallest library size (51,306). Alpha diversity indices were computed using Phyloseq [29]. Community distance matrices based on Bray–Curtis dissimilarity index were estimated using vegan [30] and visualized by non-metric multidimensional scaling. Permutational analysis of variance (PERMANOVA) and analysis of similarity (ANOSIM) tests were performed in vegan [30] to evaluate changes in microbiome structure and composition across treatments. Differentially abundant ASVs (log fold change ≥ |2|, Benjamini–Hochberg (BH)-adjusted p value ≤ 0.1) were identified using Phyloseq-DESeq [31] through pairwise comparisons between Present Day samples versus samples subjected to the other treatments. Only ASVs with >10 counts in at least 2 libraries were included in the DESeq analysis.
Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt2) [32] and Tax4Fun2 [33] were used to predict the functional profiles of the sponge microbiomes. Differentially abundant KEGG orthologs (KOs) (BH-adjusted p value ≤ 0.05) were identified using Phyloseq-DESeq [31] through pairwise comparisons between Present Day samples versus samples subjected to the other treatments. The combined set of differentially abundant KOs was searched against the KEGG database using the KEGG mapper-search pathway mapping tool [34] to infer cellular functions that are influenced by the different treatment conditions. Details of functional prediction and analysis are described in Supplementary Methods.
Transcriptome sequencing, assembly, and annotation
Total RNA was extracted from 50 to 100 mg of N. compacta and L. chagosensis tissues using TRIzol (Invitrogen, Waltham, MA, USA) following the manufacturer’s protocol. Contaminating DNA was removed using the TURBO DNA-free Kit (Invitrogen). RNA concentration was determined using a NanoDrop spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, USA). The integrity of RNA extracts was evaluated using gel electrophoresis on 1% agarose in 1× TBE and the Agilent 2200 TapeStation System (Agilent Technologies, Santa Clara, CA, USA). Libraries were prepared from three samples per treatment, except for the L. chagosensis Warming and RCP 6.0 treatments, for which we were only able to obtain high-quality RNA for two samples each. Replicate samples for each treatment were selected from different donor sponges. Barcoded libraries were prepared at Macrogen, South Korea using the Truseq RNA Library Preparation Kit (Illumina, Inc.). mRNA-enriched libraries were sequenced on the Novaseq 6000 platform (Illumina, Inc.) to generate 100 bp paired-end reads.
Raw sequence reads were visualized with FastQC v0.11.8 (Babraham Bioinformatics) and trimmed using Trimmomatic v0.32 [35]. Poor-quality bases (quality score < 3) at the start and end of the reads, as well the first 15 bases from the start of the reads, were removed. Reads were also trimmed if the average per-base quality within a 4-base sliding window fell below 20. Trimmed reads < 36 bases long were excluded from further analyses. De novo transcriptome assembly was carried out using Trinity [36]. Transcripts with 90% sequence similarity were clustered and the longest representative contigs (> 300 bp) were retained. Reads were mapped back to the assemblies and isoforms with zero isoform percentage (IsoPct) were removed to eliminate putative misassembled transcripts. Isoforms with the highest combined IsoPct or longest length were retained for each transcript to generate a reference transcriptome for each species. Raw sequence reads were deposited in the NCBI Short Read Archive database under BioProject PRJNA689294. The reference transcriptomes used in this study are available at DDBJ/EMBL/GenBank under the accession numbers GIYW00000000 (N. compacta) and GIYV00000000 (L. chagosensis).
Peptides were predicted using the Transdecoder package in Trinity and annotated by alignment against the UniProtKB/Swiss-Prot database (April 2020). The top Blastp hit for each peptide was used as input into OmicsBox (BioBam, Valencia, Spain) [37] to predict gene ontology (GO) annotations. Protein domains were identified by mapping the peptide sequences against Pfam 32.0 database [38] using HMMER v3.3 [39].
Expression analysis
Transcript abundance was estimated by mapping reads to the reference transcriptomes using RNA-Seq by Expectation Maximization [40] with bowtie alignment [41]. Differentially expressed transcripts were identified using the edgeR [42] package in R. We used the generalized linear model functionality and applied the likelihood ratio testing method, which is recommended for datasets with few replicates [42]. Expected counts were converted to counts per million (CPM) and only genes with > 10 CPM in at least two libraries were included in edgeR analysis. Genes were considered differentially expressed if upregulation or downregulation was > 4-fold relative to the controls with a BH-adjusted p value < 1 × 10−5. Pairwise comparisons were conducted between Present Day samples and samples subjected to the other treatments. Functional enrichment analysis for differentially expressed transcripts was done using the topGO package [43] in R. Only GO terms with a p value < 0.05 were considered significantly enriched. Protein–protein interactions for sponge homologs of genes involved in the human innate immune response were retrieved from the STRING v.11 database [44]. Interaction networks were visualized using Cytoscape v.3.7.2 [45]. Relative expression of sponge gene homologs in each treatment relative to the Present Day control was computed as the average sum of transcripts per million (TPM).
Meta-analysis of sponge microbiome diversity and predicted functions
Selected datasets from the Sponge Microbiome Project [46] were retrieved from the Qiita database under Study ID: 10793 (March 2020). Sequences from healthy adult individuals viz. Demospongiae (n = 1441), Calcarea (n = 20), Homoscleromorpha (n = 41), and Hexactinellida (n = 2) were included in the analysis. Alpha diversity indices were computed with Phyloseq [29]. The predicted functions of 154 sponge microbiomes, as described in a previous study [47], were shared by Miguel Lurgi (CNRS-Paul Sabatier University, France). The relative abundance of KEGG level 2 functions were clustered and visualized using pheatmap in R to observe general trends. Details of this analysis are described in Supplementary Methods.
Identification of the immunological repertoire of sponge hosts
Predicted peptide sequences of representative demosponges (Amphimedon queenslandica Hooper & van Soest, 2006 [48], H. tubifera [49], Petrosia (Petrosia) ficiformis (Poiret, 1789) [19], X. testudinaria, S. carteri [21], Aplysina aerophoba (Nardo, 1833), Dysidea avara (Schmidt, 1862) [17]), calcareans (S. ciliatum, Leucosolenia complicata (Montagu, 1814) [50], Pericharax orientalis van Soest & De Voogd, 2015, Clathrina sp. [51]), homoscleromorphs (Oscarella carmela Muricy & Pearse, 2004, Corticum candelabrum Schmidt, 1862), and a hexactinellid (Aphrocallistes vastus (Schulze, 1886)) were annotated against the Pfam 32.0 [38] database. Information on the source of the sponge sequences is listed in Table S2. All sponge species cited in the study are listed in Table S3.
NACHT domain-containing genes, with bona fide or tripartite NLR gene architecture [52], were identified from the predicted peptides. Amino acid sequences corresponding to the NACHT domain (PF05729) were used for phylogenetic comparisons. Multiple sequence alignment was performed using Clustal Omega [53] and the aligned sequences were manually trimmed (Data S1). The best-fit substitution model (LG+G+F) was identified based on Bayesian Information Criterion using ProtTest v3.4.2 [54]. Bayesian inference analysis was performed in MrBayes v.3.2 [55] with two independent MCMC runs and four chains per run. The analysis was sampled every 100 trees until the average standard deviation of split frequencies was < 0.01. The first 25% of trees were discarded as burn-in.
Statistical analyses and visualization
Expression levels (in TPM) of host PRRs were tested for normality using Shapiro–Wilk test and homogeneity of variance through Levene’s test. Statistical differences were calculated using either Welch’s t test (for normal distribution) or Wilcoxon test (for non-normal distribution) with p values < 0.05 considered statistically significant. All visualizations were done using ggplot2 [56] in R.
Results and discussion
Sponges exhibit differential survival under ocean warming and acidification
Fragments of N. compacta (Fig. 1a) remained healthy under different combinations of pH and temperature stress (Fig. S3). In contrast, L. chagosensis (Fig. 1b) showed visible tissue necrosis under the Warming, RCP 6.0, and RCP 8.5 conditions but not in the Acidification only treatment (Fig. S3). While up to 75% (9 out of 12) of N. compacta fragments survived the most extreme condition at RCP 8.5, only 25% (3 out of 12) of the L. chagosensis fragments survived in the RCP 8.5 treatment after just 2 days of sustained exposure (Fig. 1c and Table S4). These observations are comparable to the findings of other studies that reported the high survivorship of demosponges and the predicted susceptibility of calcareous sponges subjected to these climate change-associated stressors [3, 6, 18].

Representative images of a Neopetrosia compacta and b Leucetta chagosensis in their natural habitats. c Survival probability of N. compacta (top) and L. chagosensis (bottom) throughout the duration of the experiment visualized using Kaplan–Meier survival analysis (n = 12 fragments per species per treatment).
Survivorship correlates with changes in the sponge microbiome
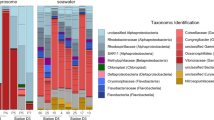
Rarefaction curves indicate the completeness of ASVs detected in N. compacta (Fig. S4A) and L. chagosensis (Fig. S4B) across the different treatments. N. compacta is associated with a diverse microbiome (species richness = 188.00 ± 27.78; Shannon index = 3.68 ± 0.02) with enrichment of the Chloroflexi-related SAR202 clade (22.2%) and phototrophic groups, including Nostocales (8.2%) and Synechococcales (0.1%). On the other hand, L. chagosensis harbored a less diverse microbiome (species richness = 150.33 ± 53.58; Shannon index = 2.04 ± 0.66) composed primarily of Oceanospirillales (56.2%) and Deltaproteobacteria SAR324 clade (27.3%) (Fig. 2a, b and Figs. S5 and S6A).

a Relative abundance of major (≥ 1%) bacterial Orders in the Neopetrosia compacta and Leucetta chagosensis microbiomes. b Non-metric multidimensional scaling (NMDS) clustering of sponge-associated microbial communities. Alpha diversity and microbial community structure of c N. compacta and d L. chagosensis under variable stress conditions. Graphs show NMDS clustering of samples. Box plots of Simpson index (1/D) are shown above each graph. Colors represent different treatments. e Plot of differentially abundant Amplicon Sequence Variants (ASVs) in L. chagosensis relative to the Present Day samples. f Bubble plot of differentially enriched KEGG pathways in L. chagosensis relative to the Present Day samples. Bubble size indicates relative abundance.
To explore the possible roles of microbiome dynamics in the stress response of the two sponge holobionts, we described the shifts in the taxonomic profiles of their bacterial communities following exposure to simulated conditions. The microbiome of N. compacta showed no significant change in structure when subjected to the various stressors (PERMANOVA: Pr(>F) = 0.993; ANOSIM: significance = 0.927; Fig. 2c and Table S5). The few ASVs that showed a significant change in abundance (log fold change ≥ |2|, BH-adjusted p value ≤ 0.1) include a member of Microbacteriaceae (ASV749), which decreased in Acidification and RCP 6.0 conditions, and ASVs affiliated with the families Endozoicomonadaceae (ASV842), Cellvibrionaceae (ASV2840), Rhodobacteraceae (ASV73, ASV1088, ASV887, ASV2836, ASV1181, and ASV2292), Alteromonadaceae (ASV2836), Nitrincolaceae (ASV1181), and Colwelliaceae (ASV2292), which increased in RCP 6.0 (Fig. S7 and Table S6).
In contrast, the L. chagosensis microbiome exhibited apparent changes with the treatments, although not statistically supported (PERMANOVA: Pr(>F) = 0.954; ANOSIM: significance = 0.925; Fig. 2d and Table S5). A total of 37 ASVs exhibited a significant change in abundance (log fold change ≥ |2|, BH-adjusted p value ≤ 0.1), with 21 decreasing and 16 increasing (Fig. 2e). In the treatments with high sponge mortality (i.e. Warming, RCP 6.0, and RCP 8.5), there was reduced abundance of ASVs classified under SAR324 clade (ASV2477) and Endozoicomonadaceae (ASV2219). On the other hand, members of presumptive opportunistic taxa, such as Vibrionales (ASV90, ASV688), Rhodobacterales (ASV1216, ASV1190, ASV1738, ASV2661, ASV1725), and Rhizobiales (ASV1658) [57], increased in relative abundance under these treatments (Fig. 2e; Fig. S8B, and Table S7).
Taxa that proliferated in L. chagosensis microbiome under stress conditions were predicted to invest more in biofilm formation (Fig. 2f), which may be advantageous for active secretion of virulence factors to invade the host cell [58]. This trait, along with the enrichment of functions related to antimicrobial molecule production, two-component signaling, and bacterial chemotaxis, may support competitive colonization of sponge tissues [59]. Changes in the L. chagosensis microbiome were also predicted to correlate with a shift in metabolic potential, which may further contribute to the decline of the holobiont. However, these observations need to be further verified through metagenomic analysis.
Differential regulation of host immune functions accompanies sponge microbiome shifts
The reference transcriptomes of N. compacta and L. chagosensis are composed of 69,202 (N50 = 1150) and 92,629 (N50 = 1475) transcripts, respectively. The quality of the assemblies is comparable to other poriferan transcriptomes [19, 49] (Table S8 and Fig. S9). We used these transcriptomes to determine the expression patterns of immune-related genes to reveal how the host innate immune system is affected by acidification and warming. A total of 1596 genes (Acidification = 74, Warming = 308, RCP 6.0 = 501, RCP 8.5 = 713) were found to be differentially expressed in L. chagosensis, whereas only 70 genes (Acidification = 3, Warming = 7, RCP 6.0 = 12, RCP 8.5 = 48) were differentially expressed in N. compacta.
GO enrichment analysis of differentially expressed L. chagosensis genes revealed an increase in functions related to the antibacterial humoral response and endosome organization under Acidification treatment, which may help the sponge to avoid pathobiont invasion (Fig. 3a). On the other hand, scavenger receptors (SRCRs), secretin G-protein coupled receptors, and nucleotide-binding domain and leucine-rich repeat-containing genes (NLRs) were repressed under Warming, RCP 6.0, and RCP 8.5 conditions (Fig. 3b). The reduction in the expression of these genes and the sensor proteins that they encode, along with the repression of bactericidal permeability-increasing protein (BPI) and lipopolysaccharide-binding protein (LBP) (Fig. 3c), may result in impaired recognition of microbial cells or molecules, which, in turn, influences the regulation of downstream effectors of the immune response [60].

a Gene Ontology (GO) enrichment analysis for upregulated and downregulated transcripts in Neopetrosia compacta and Leucetta chagosensis under the different treatments. Only immune-related GO terms are presented. Asterisks indicate significant enrichment (p < 0.05). b Expression levels of major pattern recognition receptors are presented as log2 transformed TPM values in N. compacta (left) compared with L. chagosensis (right). NLRX refers to NACHT-containing genes with bona fide NLR architecture. Colors represent different treatments. Asterisks indicate significant change in expression relative to the Present Day samples, as determined through Welch’s t test or Wilcoxon test (p < 0.05). c Protein interaction network of immune-related genes in N. compacta (left) and L. chagosensis (right). Relative expression of genes was computed as the sum of TPM values relative to the Present Day samples. Genes with at least one differentially expressed transcript (log2 fold change ≥ |2|, p < 1 × 10−5) are marked by thick black borders. Genes with no ortholog in the transcriptomes are shown in light gray. The network is based on human protein–protein interactions.
Decreased expression of genes involved in tumor necrosis factor (TNF) signaling suggests that L. chagosensis may no longer be able to deploy synchronized expression of diverse aspects of innate immunity [61]. Although the TNF receptor (TNFR) was upregulated, the TNF ligand, activator disintegrin and metalloproteinase domain-containing protein 10 (ADAM10), and the adapter protein TNF receptor-associated factor 5 (TRAF5) were downregulated (Fig. 3c). Repression of responses regulated through this pathway is further supported by the downregulation of immune-related transcription factors, such as interferon regulatory factor 5 (IRF5) and nuclear factor NF-kappa-B p105 subunit (NFKB1), coupled with the increased levels of the NF-kB inhibitor (IKB) and inhibitor of NF-kB kinase (IKK) [62]. Indeed, the downregulation of macrophage-expressed gene protein 1 (MPEG1), allograft inflammatory factor-1 (AIF1), initiator caspase CASP2/9, and executioner caspase CASP3/6/7 suggest inhibited antimicrobial, inflammatory, and apoptotic mechanisms [63,64,65]. Genes with anti-apoptotic functions, including the apoptosis regulator (BCL2), Bcl-2-like protein 1 (BCL2L1), and X-linked inhibitor of apoptosis (XIAP), were negatively regulated as well. These results generally suggest that L. chagosensis may not be able to restore immune homeostasis under combined Warming and Acidification conditions.
In contrast to the calcareous sponge, N. compacta exhibited activation of the complement system and cytokine-induced processes under RCP 8.5 conditions (Fig. 3a). An increased level of TNF, along with the upregulation of interleukin-1 receptor-associated kinase-4 (IRAK4), TRAF5, and NFKB1, indicates that the TNF-NFkB and Myd88-dependent signaling pathways were activated [66, 67] (Fig. 3c). The increased expression of AIF1, CASP2/9, and CASP3/7, along with the BCL2 and XIAP, also suggest active inflammatory and apoptotic functions [63, 65]. These indicate that N. compacta may be able to sustain its ability for symbiont recognition and pathogen clearance under the conditions that were tested.
Sponge classes exhibit disparate microbiome diversity and immunological repertoires
Evaluation of microbiome diversity in representatives from all sponge classes showed that demosponges and homoscleromorphs typically harbor microbial communities with a wide range of taxonomic diversity, whereas hexactinellids and calcareans are generally associated with less diverse microbiomes (Fig. 4a). Low microbial abundance has been reported among members of Calcarea and Hexactinellida through microscopy [68], metagenomics [46, 69], or culture-based techniques [68]. However, comparison of predicted microbiome functions across diverse sponge species revealed no distinct functional differentiation among sponge microbiomes in relation to either host type or phylogeny (Fig. S10). This suggests that other factors may contribute to shaping the sponge microbiome and its functional potential [47]. Further metagenome sequencing studies will be needed to more accurately capture the functional profiles of sponge-associated microbiomes.

a Taxonomic diversity (Shannon index) of bacterial communities of healthy sponge adults retrieved from the Sponge Microbiome Project. Yellow and blue circles represent diversity of the bacterial communities of N. compacta and L. chagosensis, respectively. Demo Demospongiae, Calc Calcarea, Homo Homoscleromorpha, Hexa Hexactinellida. b Phylogenetic relationships of sponges included in immune repertoire comparisons. The species tree was inferred from all genes by OrthoFinder [92]. Sponge class is indicated by colored circles at the nodes while host type is indicated by symbols next to the species name. c Abundance of peptides containing selected Pfam domains across species. Bubble size indicates the percentage of peptides containing a specific domain relative to the total number of predicted peptides in each species. d Abundance and diversity of SRCR-containing peptides across species. The percentage of SRCR-containing peptides (left) and the count of SRCR peptides associated with other PFAM domains (right) are presented for each species. e Diversification of NLRs in sponges. The phylogenetic tree was derived from Bayesian analysis. Numbers on selected branches represent Bayesian posterior probabilities. The outer color strips indicate sponge class, host type, and peptide architectures. Species abbreviations: Aphrocallistes vastus (Avas), Aplysina aerophoba (Aaer), Dysidea avara (Dava), Stylissa carteri (Scar), Amphimedon queenslandica (Aque), Xestospongia testudinaria (Xtes), Haliclona tubifera (Htub), Petrosia ficiformis (Pfic), Neopetrosia compacta (Ncom), Leucosolenia complicata (Lcom), Sycon ciliatum (Scil), Clathrina sp. (Csp), Pericharax orientalis (Pori), Leucetta chagosensis (Lcha), Oscarella carmela (Ocar), Corticium candelabrum (Ccan).
The complement of immune receptors in the sponge host may influence the complexity of its associated microbiome [70]. Comparison of immune-related protein domains in representative sponge species revealed both lineage-specific and host-type-associated abundance patterns (Fig. 4b, c). Regardless of host type, SRCR domain-containing proteins were more abundant in heteroscleromorph demosponges, S. carteri, A. queenslandica, X. testudinaria, H. tubifera, P. ficiformis, and N. compacta. SRCRs are PRRs that recognize a wide array of bacterial ligands [71]. In calcareans, the hexactinellid, A. vastus, and the LMA homoscleromorph, O. carmela, SRCRs are associated with diverse combinations of immune or cell-adhesion domains, whereas in demosponges and the HMA homoscleromorph, C. candelabrum, SRCRs consist mostly of multiple SRCR domains (Fig. 4d). A greater number of genes with the C-type lectin domain (Fig. 4c), which is a soluble or transmembrane PRR known to trigger intracellular signaling to induce a wide range of effector immune responses [72], was also found in calcarean, hexactinellid, and homoscleromorph sponges.
NLRs are a group of intracellular receptors that detect foreign microbes that are able to evade extracellular defenses [73]. These genes likely play a critical role in mediating host–symbiont interactions and in differentiating pathogenic from symbiotic microbes [74]. Bona fide NLRs (NLRX) are characterized by both a central NACHT domain and C-terminal leucine-rich repeats [75]. LMA demosponges and calcareans generally possess an extensive family of NLRs (Fig. 4e). Among the LMA demosponges, 50 NLR genes were identified in D. avara, 25 in S. carteri, six in A. queenslandica, and one in H. tubifera. Twenty-eight NLR genes were found in the calcareans, L. complicata and L. chagosensis, 19 in S. ciliatum, four in P. orientalis, and two in Clathrina sp. Among the 28 NLR genes in L. chagosensis, 11 have a tripartite architecture with either a CARD (NLRC, n = 7) or DEATH domain (NLRD, n = 4) at the N-terminal. Other L. chagosensis NACHT-containing genes that are phylogenetically related to NLRs possess either a CARD (CARD-NACHT, n = 11) or DEATH domain (DEATH-NACHT, n = 3). The co-expansion of tripartite NLRC, CARD-NACHT, and other CARD-containing genes in L. chagosensis, as well as in other calcareans (Fig. 4c), is indicative of enhanced signaling potential to launch immune effector mechanisms [76]. Lineage-specific and host type-associated NLR gene expansion (Fig. 4e and Fig. S11), coupled with the rich complement of other surface receptors, may have evolved to facilitate the maintenance of low microbial diversity in LMA demosponges and calcareans [47] through efficient selection or phagocytic clearance of interacting microorganisms.
Sponge holobionts in the future ocean
Understanding the persistence of sponge holobionts in perturbed conditions requires the elucidation of the animal host, the microbiome, and their interactions. Our study revealed that microbiome diversity and the host immune response may influence the ability of sponge holobionts to persist under future ocean conditions.
The demosponge, N. compacta, exhibited greater tolerance to stress compared with the calcarean, L. chagosensis. The stress tolerance of N. compacta was supported by a stable microbiome with abundant phototrophic members (Fig. S6A, C). Other photosymbiotic sponges, Carteriospongia foliascens (Pallas, 1766) and Cymbastela coralliophila Hooper & Bergquist, 1992, have also been shown to have a higher resistance to future ocean conditions due to enhanced productivity of their cyanobacteria symbionts under elevated inorganic carbon concentration [77, 78]. The increase in relative abundance of photoheterotrophic Rhodobacteraceae (Fig. S7) and the stable population of other photosymbionts in N. compacta (Fig. S8A) may have ameliorated the effects of acidification and warming stress.
On the other hand, we propose that the susceptibility of L. chagosensis to stressors is due, in part, to the instability of its microbiome, possibly stemming from low taxonomic diversity (Fig. S5) and lower predicted functional redundancy (Figs. S5 and S6B) [79]. While microbiome flexibility in corals [80] and sponges [81] has been proposed as a mechanism for rapid adaptation [12], unstable phases during community restructuring may result in loss of essential functions and offer an opportunity for pathogen invasion. Changes in the predicted metabolic capabilities and increased pathogenic potential of the L. chagosensis microbiome under stress is similar to observations on the dysbiotic metagenomes of the sponge, R. odorabile [10], and the coral, Porites compressa [82].
The difference in microbiome dynamics in the two sponges may also be influenced by differences in host immune functions. Under the simulated stress conditions, sustained levels of surface receptors and expression of immune effectors in N. compacta may have allowed efficient symbiont recognition and pathogenic clearance, whereas the suppression of immune pathways in L. chagosensis may have disrupted the sponge–symbiont interactions and attenuated the host’s defense mechanisms. Our results mirror reports on adaptive or dysbiotic events in other holobionts challenged by various environmental perturbations. For instance, the coral, Montipora aequituberculata, which had a stable bacterial community under elevated temperatures, exhibited regulation of the complement system and phagocytosis [83], while the dissociation of coral–algal symbiosis in Orbicella faveolata following a prolonged thermal anomaly was accompanied by overall reduced expression of genes implicated in the TNF pathway and apoptosis [16].
The HMA–LMA status of sponges correlates with differences in host physiology [84]. HMA species generally have slower filtration rates and a denser mesohyl, while LMA demosponges and calcareans can more rapidly take up large volumes of seawater through their tissues [84,85,86]. For example, Leucetta can filter about 4.56 L h−1 [86] while Neopetrosia problematica (de Laubenfels, 1930) can only take up 0.53 L h−1 [85]. We speculate that species with higher pumping rates and lower tissue density may be more susceptible to perturbations as they have more frequent encounters with pathobionts and are less protected against the external environment. However, as it has been shown that sponge body size can also affect pumping rates [87], further investigation is warranted to test this hypothesis.
Given their synapomorphic calcitic spicules, lower microbiome diversity, and rapid suppression of PRRs and immune effectors under stress, calcareans are likely to be more susceptible to future ocean conditions. It is worth noting, however, that L. chagosensis was able to survive under reduced pH, which corroborates the reported proliferation of L. complicata at pH 7.7 [88]. The invasive calcaronean sponge, Sycettusa hastifera (Row, 1909), has also been shown to tolerate thermo-acidic stress with little change to its microbiome and spicules [89, 90]. Although reduced pH may be less detrimental to sponges compared to elevated temperature [77], the predicted co-occurrence of acidification and warming in the long run may result in the narrowing of organismal thermal tolerance thresholds [91].
Comparison of the patterns of microbiome diversity and immunological repertoire among poriferans provides broader insights into the adaptive capacity of different sponge groups in perturbed ocean conditions. Although sponges are generally predicted to be winners under future ocean scenarios, species- and lineage-specific holobiont features may define their susceptibility or tolerance to various stress events. Further investigations on the roles of microbiome flexibility, immune functions, and other host-specific traits in the stress response of diverse sponge species with different evolutionary histories, morphologies, and microbiome densities are warranted. Given that sponges are critical members of the reef ecosystem, revealing the mechanisms underlying their adaptive success or failure is pivotal in projecting the reef landscape in the future ocean.
Data availability
Raw sequence data from this study are accessible through the NCBI Sequence Read Archive under BioProject PRJNA689294. The datasets we generated and analyzed are available in Figshare (https://figshare.com/projects/Sponge_holobionts_under_future_ocean_conditions/95796).
References
Le Quéré C, Moriarty R, Andrew RM, Canadell JG, Sitch S, Korsbakken JI, et al. Global carbon budget 2015. Earth Syst Sci Data. 2015;7:349–96.
Hoegh-Guldberg O, Mumby PJ, Hooten AJ, Steneck RS, Greenfield P, Gomez E, et al. Coral reefs under rapid climate change and ocean acidification. Science. 2007;318:1737–42.
Bell JJ, Bennett HM, Rovellini A, Webster NS. Sponges to be winners under near-future climate scenarios. Bioscience. 2018;68:955–68.
Bell JJ. The functional roles of marine sponges. Estuar Coast Shelf Sci. 2008;79:341–53.
Pita L, Rix L, Slaby BM, Franke A, Hentschel U. The sponge holobiont in a changing ocean: from microbes to ecosystems. Microbiome. 2018;6:46.
Smith AM, Berman J, Key MM Jr, Winter DJ. Not all sponges will thrive in a high-CO2 ocean: Review of the mineralogy of calcifying sponges. Palaeogeogr Palaeoclimatol Palaeoecol. 2013;392:463–72.
Webster NS, Thomas T. The sponge hologenome. MBio. 2016;7:e00135–16.
Hentschel U, Piel J, Degnan SM, Taylor MW. Genomic insights into the marine sponge microbiome. Nat Rev Microbiol. 2012;10:641–54.
Thompson JR, Rivera HE, Closek CJ, Medina M. Microbes in the coral holobiont: partners through evolution, development, and ecological interactions. Front Cell Infect Microbiol. 2014;4:176.
Fan L, Liu M, Simister R, Webster NS, Thomas T. Marine microbial symbiosis heats up: the phylogenetic and functional response of a sponge holobiont to thermal stress. ISME J. 2013;7:991–1002.
Egan S, Gardiner M. Microbial dysbiosis: rethinking disease in marine ecosystems. Front Microbiol. 2016;7:991.
Voolstra CR, Ziegler M. Adapting with microbial help: microbiome flexibility facilitates rapid responses to environmental change. Bioessays. 2020;42:e2000004.
Botte ES, Nielsen S, Abdul Wahab MA, Webster J, Robbins S, Thomas T, et al. Changes in the metabolic potential of the sponge microbiome under ocean acidification. Nat Commun. 2019;10:4134.
Morrow KM, Bourne DG, Humphrey C, Botté ES, Laffy P, Zaneveld J, et al. Natural volcanic CO2 seeps reveal future trajectories for host–microbial associations in corals and sponges. ISME J. 2015;9:894–908.
Pollock FJ, Lamb JB, van de Water J, Smith HA, Schaffelke B, Willis BL, et al. Reduced diversity and stability of coral-associated bacterial communities and suppressed immune function precedes disease onset in corals. R Soc Open Sci. 2019;6:190355.
Pinzon JH, Kamel B, Burge CA, Harvell CD, Medina M, Weil E, et al. Whole transcriptome analysis reveals changes in expression of immune-related genes during and after bleaching in a reef-building coral. R Soc Open Sci. 2015;2:140214.
Pita L, Hoeppner MP, Ribes M, Hentschel U. Differential expression of immune receptors in two marine sponges upon exposure to microbial-associated molecular patterns. Sci Rep. 2018;8:16081.
Guzman C, Conaco C. Gene expression dynamics accompanying the sponge thermal stress response. PLoS ONE. 2016;11:e0165368.
Riesgo A, Farrar N, Windsor PJ, Giribet G, Leys SP. The analysis of eight transcriptomes from all poriferan classes reveals surprising genetic complexity in sponges. Mol Biol Evol. 2014;31:1102–20.
Germer J, Cerveau N, Jackson DJ. The holo-transcriptome of a calcified early branching metazoan. Front Mar Sci. 2017;4:81.
Ryu T, Seridi L, Moitinho-Silva L, Oates M, Liew YJ, Mavromatis C, et al. Hologenome analysis of two marine sponges with different microbiomes. BMC Genomics. 2016;17:158.
Hooper JNA, Van Soest RWM. Systema Porifera. A guide to the classification of sponges. In: Hooper JNA, Van Soest RWM, editors. Systema Porifera. New York, NY: Springer; 2002. p. 1–7.
Pachauri RK, Allen MR, Barros VR, Broome J, Cramer W, Christ R, et al. Climate change 2014: synthesis report. Contribution of Working Groups I, II and III to the fifth assessment report of the Intergovernmental Panel on Climate Change. Geneva: IPCC; 2014.
Pierrot DE, Lewis E, Wallace DWR. MS Excel program developed for CO2 system calculations. Oak Ridge, TN: Carbon Dioxide Information Analysis Center, Oak Ridge National Laboratory, US Department of Energy, ORNL/CDIAC-IOS; 2006.
Herlemann DP, Labrenz M, Jurgens K, Bertilsson S, Waniek JJ, Andersson AF. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 2011;5:1571–9.
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37:852–7.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590–6.
McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE. 2013;8:e61217.
Dixon P. VEGAN, a package of R functions for community ecology. J Veg Sci. 2003;14:927–30.
McMurdie PJ, Holmes S. Waste not, want not: why rarefying microbiome data is inadmissible. PLoS Comput Biol. 2014;10:e1003531.
Douglas GM, Maffei VJ, Zaneveld JR, Yurgel SN, Brown JR, Taylor CM, et al. PICRUSt2 for prediction of metagenome functions. Nat Biotechnol. 2020;38:685–8.
Asshauer KP, Wemheuer B, Daniel R, Meinicke P. Tax4Fun: predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics. 2015;31:2882–4.
Kanehisa M, Sato Y. KEGG Mapper for inferring cellular functions from protein sequences. Protein Sci. 2020;29:28–35.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 2013;8:1494.
Conesa A, Gotz S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int J Plant Genomics. 2008;2008:619832.
Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42:D222–D30.
Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14:755–63.
Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323.
Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40.
Alexa A, Rahnenführer J. Gene set enrichment analysis with topGO. Bioconductor Improv. 2009;27:1–26.
Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607–D13.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504.
Moitinho-Silva L, Nielsen S, Amir A, Gonzalez A, Ackermann GL, Cerrano C, et al. The sponge microbiome project. Gigascience. 2017;6:1–7.
Lurgi M, Thomas T, Wemheuer B, Webster NS, Montoya JM. Modularity and predicted functions of the global sponge-microbiome network. Nat Commun. 2019;10:992.
Srivastava M, Simakov O, Chapman J, Fahey B, Gauthier ME, Mitros T, et al. The Amphimedon queenslandica genome and the evolution of animal complexity. Nature. 2010;466:720–6.
Guzman C, Conaco C. Comparative transcriptome analysis reveals insights into the streamlined genomes of haplosclerid demosponges. Sci Rep. 2016;6:18774.
Fortunato SA, Adamski M, Ramos OM, Leininger S, Liu J, Ferrier DE, et al. Calcisponges have a ParaHox gene and dynamic expression of dispersed NK homeobox genes. Nature. 2014;514:620–3.
Voigt O, Fradusco B, Gut C, Kevrekidis C, Vargas S, Wörheide G. Carbonic anhydrases: an ancient tool in calcareous sponge biomineralization. Front Genet. 2021;12:624533.
Yuen B, Bayes JM, Degnan SM. The characterization of sponge NLRs provides insight into the origin and evolution of this innate immune gene family in animals. Mol Biol Evol. 2014;31:106–20.
Madeira F, Park YM, Lee J, Buso N, Gur T, Madhusoodanan N, et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019;47:W636–41.
Darriba D, Taboada GL, Doallo R, Posada D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics. 2011;27:1164–5.
Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–4.
Wickham H. ggplot2: elegant graphics for data analysis. New York: Springer; 2016.
McDevitt-Irwin JM, Baum JK, Garren M, Vega Thurber RL. Responses of coral-associated bacterial communities to local and global stressors. Front Mar Sci. 2017;4:262.
Hori K, Matsumoto S. Bacterial adhesion: from mechanism to control. Biochem Eng J. 2010;48:424–34.
Yao J, Allen C. Chemotaxis is required for virulence and competitive fitness of the bacterial wilt pathogen Ralstonia solanacearum. J Bacteriol. 2006;188:3697–708.
Chu H, Mazmanian SK. Innate immune recognition of the microbiota promotes host-microbial symbiosis. Nat Immunol. 2013;14:668–75.
Bazzoni F, Beutler B. The tumor necrosis factor ligand and receptor families. N Engl J Med. 1996;334:1717–25.
Hayden MS, Ghosh S. Regulation of NF-kappaB by TNF family cytokines. Semin Immunol. 2014;26:253–66.
Parrish AB, Freel CD, Kornbluth S. Cellular mechanisms controlling caspase activation and function. Cold Spring Harb Perspect Biol. 2013;5:a008672.
Wiens M, Korzhev M, Krasko A, Thakur NL, Perovic-Ottstadt S, Breter HJ, et al. Innate immune defense of the sponge Suberites domuncula against bacteria involves a MyD88-dependent signaling pathway. Induction of a perforin-like molecule. J Biol Chem. 2005;280:27949–59.
Muller WE, Muller IM. Origin of the metazoan immune system: identification of the molecules and their functions in sponges. Integr Comp Biol. 2003;43:281–92.
Yuen B Deciphering the genomic toolkit underlying animal-bacteria interactions – insights through the demosponge Amphimedon queenslandica. Saint Lucia, QLD: School of Biological Sciences, The University of Queensland; 2016.
Gauthier ME, Du Pasquier L, Degnan BM. The genome of the sponge Amphimedon queenslandica provides new perspectives into the origin of Toll-like and interleukin 1 receptor pathways. Evol Dev. 2010;12:519–33.
Roue M, Quevrain E, Domart-Coulon I, Bourguet-Kondracki ML. Assessing calcareous sponges and their associated bacteria for the discovery of new bioactive natural products. Nat Prod Rep. 2012;29:739–51.
Steinert G, Busch K, Bayer K, Kodami S, Arbizu PM, Kelly M, et al. Compositional and quantitative insights into bacterial and archaeal communities of South Pacific deep-sea sponges (Demospongiae and Hexactinellida). Front Microbiol. 2020;11:716.
Thomas T, Moitinho-Silva L, Lurgi M, Bjork JR, Easson C, Astudillo-Garcia C, et al. Diversity, structure and convergent evolution of the global sponge microbiome. Nat Commun. 2016;7:11870.
Yap NV, Whelan FJ, Bowdish DM, Golding GB. The evolution of the scavenger receptor cysteine-rich domain of the class a scavenger receptors. Front Immunol. 2015;6:342.
Brown GD, Willment JA, Whitehead L. C-type lectins in immunity and homeostasis. Nat Rev Immunol. 2018;18:374–89.
von Moltke J, Ayres JS, Kofoed EM, Chavarria-Smith J, Vance RE. Recognition of bacteria by inflammasomes. Annu Rev Immunol. 2013;31:73–106.
Robertson SJ, Rubino SJ, Geddes K, Philpott DJ. Examining host-microbial interactions through the lens of NOD: from plants to mammals. Semin Immunol. 2012;24:9–16.
Ting JP, Lovering RC, Alnemri ES, Bertin J, Boss JM, Davis BK, et al. The NLR gene family: a standard nomenclature. Immunity. 2008;28:285–7.
Messier-Solek C, Buckley KM, Rast JP. Highly diversified innate receptor systems and new forms of animal immunity. Semin Immunol. 2010;22:39–47.
Bennett HM, Altenrath C, Woods L, Davy SK, Webster NS, Bell JJ. Interactive effects of temperature and pCO2 on sponges: from the cradle to the grave. Glob Chang Biol. 2017;23:2031–46.
Luter HM, Andersen M, Versteegen E, Laffy P, Uthicke S, Bell JJ, et al. Cross-generational effects of climate change on the microbiome of a photosynthetic sponge. Environ Microbiol. 2020;22:4732–44.
Girvan MS, Campbell CD, Killham K, Prosser JI, Glover LA. Bacterial diversity promotes community stability and functional resilience after perturbation. Environ Microbiol. 2005;7:301–13.
Ziegler M, Grupstra CGB, Barreto MM, Eaton M, BaOmar J, Zubier K, et al. Coral bacterial community structure responds to environmental change in a host-specific manner. Nat Commun. 2019;10:3092.
Ribes M, Calvo E, Movilla J, Logares R, Coma R, Pelejero C. Restructuring of the sponge microbiome favors tolerance to ocean acidification. Environ Microbiol Rep. 2016;8:536–44.
Vega Thurber R, Willner-Hall D, Rodriguez-Mueller B, Desnues C, Edwards RA, Angly F, et al. Metagenomic analysis of stressed coral holobionts. Environ Microbiol. 2009;11:2148–63.
van de Water J, Chaib De Mares M, Dixon GB, Raina JB, Willis BL, Bourne DG, et al. Antimicrobial and stress responses to increased temperature and bacterial pathogen challenge in the holobiont of a reef-building coral. Mol Ecol. 2018;27:1065–80.
Weisz JB, Lindquist N, Martens CS. Do associated microbial abundances impact marine demosponge pumping rates and tissue densities? Oecologia. 2008;155:367–76.
Ludeman DA, Reidenbach MA, Leys SP. The energetic cost of filtration by demosponges and their behavioural response to ambient currents. J Exp Biol. 2017;220:995–1007.
Perea-Blazquez A, Davy SK, Bell JJ. Estimates of particulate organic carbon flowing from the pelagic environment to the benthos through sponge assemblages. PLoS ONE. 2012;7:e29569.
Morganti TM, Ribes M, Yahel G, Coma R. Size is the major determinant of pumping rates in marine sponges. Front Physiol. 2019;10:1474.
Peck LS, Clark MS, Power D, Reis J, Batista FM, Harper EM. Acidification effects on biofouling communities: winners and losers. Glob Chang Biol. 2015;21:1907–13.
Ribeiro B, Padua A, Barno A, Villela H, Duarte G, Rossi A, et al. Assessing skeleton and microbiome responses of a calcareous sponge under thermal and pH stresses. ICES J Mar Sci. 2020:fsaa231.
Lanna E, Klautau M. Life history and reproductive dynamics of the cryptogenic calcareous sponge Sycettusa hastifera (Porifera, Calcarea) living in tropical rocky shores. J Mar Biol Assoc UK. 2018;98:505–14.
Pörtner HO, Langenbuch M, Michaelidis B. Synergistic effects of temperature extremes, hypoxia, and increases in CO2 on marine animals: from Earth history to global change. J Geophys Res. 2005;110:C09S10.
Emms DM, Kelly S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 2019;20:238.
Acknowledgements
We thank Francis Kenith Adolfo, Robert Valenzuela, and Ronald De Guzman for field and hatchery assistance and staff of the Bolinao Marine Laboratory for logistical support. This study was funded by the Department of Science and Technology Philippine Council for Agriculture, Aquatic and Natural Resources Research and Development (QMSR-MRRD-MEC-295-1449) to CC.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Posadas, N., Baquiran, J.I.P., Nada, M.A.L. et al. Microbiome diversity and host immune functions influence survivorship of sponge holobionts under future ocean conditions. ISME J 16, 58–67 (2022). https://doi.org/10.1038/s41396-021-01050-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-021-01050-5
This article is cited by
-
Diversity, composition and potential roles of sedimentary microbial communities in different coastal substrates around subtropical Okinawa Island, Japan
Environmental Microbiome (2024)
-
Gut microbiota modulation enhances the immune capacity of lizards under climate warming
Microbiome (2024)
-
Dynamics, diversity, and roles of bacterial transmission modes during the first asexual life stages of the freshwater sponge Spongilla lacustris
Environmental Microbiome (2024)
-
Microbiome origin and stress-related changes in bacterial abundance of the photosymbiotic sea slug Berghia stephanieae (Á. Valdés, 2005)
Symbiosis (2024)
-
Prokaryotic communities of the French Polynesian sponge Dactylospongia metachromia display a site-specific and stable diversity during an aquaculture trial
Antonie van Leeuwenhoek (2024)
