
Abstract
Major depressive disorder (MDD) and opioid use disorder (OUD) are common, potentially fatal, polygenic disorders that are moderately heritable and often co-occur. We examined the unique and shared associations of polygenic risk scores (PRS) for these disorders with µ-opioid receptor (MOR) concentration and endogenous opioid response during a stressful stimulus. Participants were 144 healthy European-ancestry (EA) subjects (88 females) who underwent MOR quantification scans with [11C]carfentanil and PET and provided DNA for genotyping. MOR non-displaceable binding potential (BPND) was measured in 5 regions of interest (ROIs) related to mood and addiction. We examined associations of PRS both at baseline and following opioid release calculated as the ratio of baseline and stress-challenge scans, first in the entire sample and then separately by sex. MOR availability at baseline was positively associated with MDD PRS in the amygdala and ventral pallidum. MDD and OUD PRS were significantly associated with stress-induced opioid system activation in multiple ROIs, accounting for up to 14.5% and 5.4%, respectively, of the variance in regional activation. The associations were most robust among females, where combined they accounted for up to 25.0% of the variance among the ROIs. We conclude that there is a pathophysiologic link between polygenic risk for MDD and OUD and opioid system activity, as evidenced by PRS with unique and overlapping regional associations with this neurotransmitter system. This link could help to explain the high rate of comorbidity of MDD and OUD and suggests that opioid-modulating interventions could be useful in treating MDD and OUD, both individually and jointly.
Similar content being viewed by others
Introduction
Major depressive disorder (MDD) and opioid use disorder (OUD) are common, highly comorbid conditions that adversely affect public health, both individually and jointly [1, 2]. In 2019, 19.3 million US adults experienced a major depressive episode and 47,000 died by suicide [3], while nearly 1.6 million suffered from OUD and nearly 50,000 died from an opioid overdose [3]. Although the etiology of these disorders is not completely understood, there is evidence of shared genetic liability of MDD with opioid use and OUD [4]. Endogenous opioid system dysregulation, which has been associated with the pathophysiology of MDD [5, 6], MDD-associated suicide [7, 8], and the development and maintenance of drug dependence [9], is a potential shared neurobiological mechanism contributing to both disorders.
MDD and OUD are polygenic traits whose estimated heritability in genetic epidemiologic studies is 31–42% [10] and 23–54% [11], respectively. The availability of summary statistics from large genome-wide association studies (GWAS) of MDD [10] and OUD [12] make it possible to calculate polygenic risk scores (PRS) in other samples. Given the high polygenicity of many complex traits, including psychiatric phenotypes, polygenic risk metrics account for a fuller range of genetic effects than single nucleotide polymorphisms (SNPs). Further, polygenic risk may be shared across disorders with overlapping characteristics, pathophysiology, or comorbid presentations [13] and contribute to the comorbidity.
The µ-opioid receptor protein (MOR) is widely distributed in both emotion- and addiction-related circuits [14] and is known to regulate the hedonic value of natural and drug rewards [15], mood, and stress responses [16]. The receptor is required for the analgesic, rewarding, tolerance- and withdrawal-inducing effects of opioid drugs [17, 18] and can impact reward-related signaling through several other basic mechanisms, including modulation of the mesolimbic dopamine system [9]. An A118G base change encoding an Asn40Asp exchange in OPRM1, which encodes MOR, has been associated with changes in MOR non-displaceable binding potential (BPND) measured using positron emission tomography (PET) [19,20,21]. Notably, the strength of genetic associations with molecular targets is typically greater than with behavioral traits (e.g., depressive symptom severity, opioid dose) or non-specific functional measures (e.g., fMRI BOLD signal or metabolism), possibly because of the closer association of gene products with neural signaling mechanisms than with indirect measures [22].
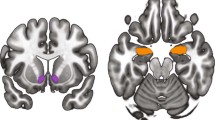
We examined the association of genome-wide PRS for MDD and OUD with between- and within-subject changes in endogenous opioid neurotransmission, measured as MOR BPND during baseline and stress-challenge conditions [23]. We focused on 5 prototypical brain regions involved in the regulation of emotion and mood and the emotional, incentive, and compulsive mechanisms of OUD: namely, the subgenual anterior cingulate, ventral pallidum, amygdala, nucleus accumbens, and dorsal striatum (see Supplementary Fig. S1). Following analyses in the overall sample of study participants and given sex differences in risk for MDD and OUD and sex-specific variation in their expression, clinical course, treatment response, and MOR-mediated neurotransmission [24, 25], we analyzed the data separately by sex. All analyses controlled for the effects of the A118G SNP. Finally, we tested the specificity of the findings using a PRS derived from a GWAS of height [26], for which we expected no association.
We hypothesized that: (1) individually, the MDD and OUD PRS would account for unique variance in stress-induced changes in MOR-mediated neurotransmission; (2) the associations with MDD and OUD PRS would be overlapping, given their high rate of comorbidity and genetic correlation; and (3) the MDD and OUD PRS would show differential effects in males and females.
Materials and methods
Samples
Data from 191 participants from 5 separate [11C] carfentanil PET brain imaging studies were aggregated to form the current sample (see Supplementary Fig. S2). All procedures used in these studies were approved by the Institutional Review Board and the Radioactive Drug Research Committee of the University of Michigan. Written informed consent was obtained from all subjects prior to the performance of the study procedures.
The protocol and sample characteristics for each of these studies are described in detail elsewhere [20, 27,28,29,30]. In brief, at intake, each participant was screened for DSM-IV Axis I disorders using a structured interview administered by a psychiatrist or psychiatric nurse. All participants were right-handed. Substance use disorders, including OUD, and recent use of antipsychotic medication were exclusionary, as was recreational drug use. Prior to scanning, subjects underwent urine testing for the following drugs: amphetamine/methamphetamine, barbiturates, benzodiazepines, cocaine, alcohol, methadone, opioids, phencyclidine, cannabinoids, tricyclic antidepressants, and acetaminophen.
Phenotyping
A subsample of 179 participants met our inclusion and exclusion criteria and had genome-wide genotype and [11C]carfentanil PET data available for analysis. The participants’ genetic ancestry was estimated from the measured genotypes as a combination of European, African, Native American, East Asian, and South Asian populations using 1000 Genomes Project phase 3 samples as the reference [31] (see Supplementary Figs S3 and S4). Because the PRS were derived from European-ancestry individuals and these scores do not translate between populations in a straightforward way, we included only subjects whose proportion of European ancestry was >0.8 (n = 144; 56 males, 88 females; age 18–58 years). These participants (see Supplementary Table S1) included 74 healthy controls; 37 participants with a primary diagnosis of chronic non-specific back pain (CNBP); and 33 participants with a current primary Axis I or II disorder. Six CNBP patients also had comorbid Axis I disorders. None of the individuals who underwent the pain-stress challenge had a diagnosis of MDD. Age and baseline affective ratings and psychophysiological responses during the pain challenge by sex and genotype are shown in Supplementary Table S2. The mean (SD) age among females was 33.1 years (10.6) years and among males 32.0 (10.8) years (p = 0.52).
Neuroimaging
Each participant underwent a resting 90-min [11C]carfentanil scan for the measurement of MOR BPND (see Supplementary Methods for details), a 90-min pain-stress scan that consisted of a 45-min baseline session without intervention followed by a 45-min session that contained a pain-stress challenge, or both (i.e., two separate scans in randomized order). Typically, and to achieve full quantification, we utilized 2 scans–baseline and challenge–in random order, using a subtraction method. Of the total sample, 109 individuals underwent the pain-stress challenge, during which they were exposed to moderate levels of muscular pain through the computer-controlled infusion of small amounts of 5% hypertonic saline into the relaxed masseter muscle [32]. Given that genomic studies require large samples to yield adequate statistical power, we augmented the available sample by including individuals who had both baseline and challenge data obtained in a single session. In these individuals, we calculated BPND early in the scan (control) vs. late in the scan (challenge). The only difference in the data that come from early vs. late in a single scan session and those that come from two separate scan sessions is the decay of 11C, a physical constant. Otherwise, subtraction vs. ratio methods yield nearly identical results, as would be expected, with correlations ranging from 0.94 to 0.97 among the ROIs.
PET scans were acquired, reconstructed, and processed as described in detail previously [30]. Details of the specific scanning procedures and analytic methods used in this sample are provided in Supplementary Methods.
Genotyping, imputation, and quality control
Genotyping was performed using the Infinium PsychArray 24v1.2 BeadChip (lllumina, San Diego, CA), with quality control performed using SNP clustering in Illumina Genome Studio (https://www.illumina.com/techniques/microarrays/array-data-analysis-experimentaldesign/genomestudio.html). Details on quality control and imputation are provided in Supplementary Methods.
Principal component analysis (PCA) and genetic ancestry
Participants’ genetic ancestry was calculated using a linear regression model that predicted the ancestry proportions based on the top 10 principal components (PCs) from the PCA of the combined set of samples from this study and 1000 Genomes Project (Fig. 1). Additional details on the genetic ancestry methods are provided in Supplementary Methods (including Supplementary Figs. S3 and S4).
Polygenic risk scores
PRS were calculated using the EUR reference panel in PRScs [33] and genome-wide association summary statistics from the largest available GWAS meta-analyses of MDD [10] and OUD [12]. We ran the Markov chain Monte Carlo procedure in PRScs for 1,100 iterations, burning in the first 100 iterations. Other PRScs parameters were set to the default values.
Statistical analyses
Regressions of MOR BPND onto PRS
Linear regressions of BPND on MDD and OUD PRS included sex (for the overall analysis), age, the A118G SNP genotype, and the first 10 ancestry PCs as covariates. The BPND measurements and PRS were quantile normalized to the normal distribution to avoid outliers and disproportionately influential observations. Stress-induced changes in MOR-mediated neurotransmission were calculated by taking the ratio of (1) the initial 45 min of the scan, prior to the introduction of the pain challenge to (2) the last 45 min of the pain scan. As reductions in BPND are thought to reflect increased acute endogenous MOR activity, lower ratio values are indicative of higher endogenous opioid system activation.
Following analyses that included the entire cohort, we examined effects in males and females separately. We estimated models for each PRS predicting BPND for the 5 ROIs: subgenual anterior cingulate, ventral pallidum, amygdala, nucleus accumbens, and dorsal striatum. We report the regression estimates for MDD and OUD PRS, along with the respective standard errors, t-statistics, p-values, and incremental R2 (explained variation relative to the model with only the covariates). The effects of the two PRS were also modeled in the same equation to identify the additive variance in the MOR system functional measures that were accounted for by the genetic risk metrics. Within each of three analyses (overall, male, and female), we calculated the False Discovery Rate (FDR) to correct for multiple testing using the Benjamini-Hochberg procedure [34] and an FDR threshold of q < 0.05 for significance.
Because the participants were recruited from 5 different studies, we included sensitivity analyses that included study as a dummy variable in the models.
The analyses were conducted using R 4.0.2, with the linear regression analysis using built-in function lm. The incremental R2 is calculated as \(R^2 = \left( {R_{{{{{\rm{f}}}}}}^2 - R_0^2} \right)/\left( {1 - R_0^2} \right)\), where \(R_f^2\) and \(R_0^2\) are the values of the explained variation for the linear models with and without the main predictor and with the full set of covariates. Note that incremental R2 takes values between 0 and 1, as does the standard R2. The false discovery rate (q value) was calculated using build-in R function p.adjust.

Illustration of multiple regression analyses of PRS and stress-induced endogenous opioid release controlling for age and 10 ancestry principal components. Mask generated by combining all 5 regions of interest.
The association of the PRS for height [26] with receptor availability during the pain challenge was modeled in the same way as the PRS for OUD and MDD.
Results
Table 1 shows the associations of MDD and OUD PRS with baseline levels of MOR BPND and Table 2 shows the associations of these PRS with the measure of stress-induced endogenous opioid release. Both tables include estimates of the unique variance in the MOR system functional measures accounted for by each PRS and the additive effects of the two PRS.
Associations of baseline MOR BPND with MDD and OUD PRS
As shown in Table 1, in the full sample, the MDD PRS was significantly associated with baseline MOR BPND only in the ventral pallidum (R2 = 4.9%) and the amygdala (R2 = 5.5%). There were no significant effects when the analyses were conducted separately in males and females. The OUD PRS was not significantly associated with MOR BPND in any of the ROIs. The combined effects of the OUD and MDD PRS was not substantially greater than the effects of the MDD PRS alone.
Associations of endogenous opioid system activation with MDD and OUD PRS
In the full sample, the MDD PRS was significantly associated with stress-induced endogenous opioid system activation in all 5 ROIs: the subgenual anterior cingulate (R2 = 12.0%), ventral pallidum (R2 = 11.4%), amygdala (R2 = 14.5%), nucleus accumbens (R2 = 6.0%), and dorsal striatum (12.8%) (Table 2). Among females, the MDD PRS was significantly associated with opioid release in the ventral pallidum (R2 = 18.5%), amygdala (R2 = 13.4%), nucleus accumbens (R2 = 7.1%), and the dorsal striatum (R2 = 10.6%). The association in the subgenual anterior cingulate was not significant (q = 0.075). Among males, there were no significant associations of the MDD PRS with endogenous opioid release.
In the full sample, the OUD PRS was significantly positively associated with stress-induced endogenous opioid activation in four of the ROIs: subgenual anterior cingulate (R2 = 4.7%), ventral pallidum (R2 = 5.4%), amygdala (R2 = 4.8%), and dorsal striatum (R2 = 5.4%). Clear sex differences were also evident here. Among females, associations with the OUD PRS accounted for significant and substantial variance in 4 ROIs: ventral pallidum (R2 = 16.6%), amygdala (R2 = 9.5%), nucleus accumbens (R2 = 10.7%), and dorsal striatum (R2 = 13.0%). Among females, the association in the subgenual anterior cingulate was not significant (q = 0.075).
The variance in opioid activation accounted for when we include both PRS in the model (see Table 2, far right column) exceeds the variance accounted for by each of the PRS individually. Thus, in addition to independent associations of the PRS with opioid activation, there is evidence of shared associations between the PRS. For example, among females, the variance accounted for individually in the ventral pallidum was 18.5% and 16.6% for the MDD and OUD PRS, respectively, and 25.0% for the two PRS when modeled together. Thus, of the total variance accounted for when the two individual estimates are summed (35.1%), approximately 10.1% is shared by the two PRS.
Sensitivity analyses that included study as a dummy variable showed results similar to those presented above, though some of the associations reported above were rendered non-significant. The findings are shown in Supplementary Tables S3 and S4.
As shown in Supplementary Table S5, a similar analysis of the association of a PRS for height with endogenous opioid release during the pain condition yielded no significant associations, supporting the specificity of the findings for the MDD and OUD PRS.
Discussion
We found a significant association between the MDD PRS and baseline MOR BPND in two regions: the ventral pallidum and amygdala, where it accounted for ~5% of the variance in MOR in vivo availability. This association’s directionality is the same as that reported in post-mortem studies of depression-linked suicide [7]. There were no significant associations between the OUD PRS and MOR BPND at baseline. We observed significant associations between both MDD and OUD PRS and the endogenous opioid system responses to an experimental stressor. In the overall sample, the association with MDD PRS accounted for up to 14.5% and the OUD PRS for up to 5.3% of the variance in the opioid response in ROIs implicated in reward, motivation, and affect, processes involved in the development of both mood and substance use disorders. For example, opioid mechanisms in the nucleus accumbens and ventral pallidum are thought to play a critical role in encoding hedonic and incentive value [35] and governing drug-seeking behavior and, in the case of the dorsal striatum, compulsive, habitual behaviors [36, 37]. Further, subgenual anterior cingulate MOR activity has been associated with the processing of emotional and social stimuli [38, 39] and implicated in the pathophysiology of MDD, including the response to the administration of antidepressants and MDD-associated suicide [5,6,7,8] and addictions [40, 41]. The findings reported here are in line with previous reports that greater MOR system drive, reflected by greater stress-induced endogenous opioid release, contributes to risk-taking traits linked to affective dysregulation, MDD [6], and problematic drug use [42].
Analyses conducted separately by sex revealed that associations involving both MDD and OUD PRS were driven largely by effects in females, where they accounted for up to 18.5% and 16.6%, respectively, of the variance in opioid system activation. These findings are consistent with well-described sex differences in the clinical presentation and prevalence of the disorders [43] and in endogenous opioid system functioning [24, 25]. While the prevalence of virtually all substance use disorders, including OUD, is higher in males than females, the antecedents and clinical manifestations of opioid misuse and OUD appear to be sex specific. For example, the risk factors for prescription opioid use differ by sex [44], with childhood trauma having a greater impact on the likelihood of developing OUD among females than males [45]. Further, among individuals with a history of opioid misuse or OUD, females are more likely to report a history of depression and anxiety [44,45,46,47]. Although the most robust predictors of opioid misuse once opioid use has been initiated—a history of substance abuse and of legal problems—may be predictive of opioid misuse only among males [44], females are more likely to experience severe adverse consequences, to proceed from initiation to chronic use more rapidly, and to exhibit higher addiction severity when initiating treatment [48, 49].
In all ROIs, the sum of the variance accounted for by the two PRS exceeded the variance accounted for when both PRS were included in the model. As can be seen in Table 2, the variance in endogenous opioid system activation shared by the two PRS in the overall sample ranges from 1.2% to 3.2%, while among females it ranges from 6.0% to 10.1%. The overlapping variance accounted for by the two PRS may underlie both the high rate of comorbidity of the two disorders seen in epidemiologic and genetic studies and the greater likelihood of the co-occurrence of these disorders in females [2, 4].
Whereas the study sample was comprised of subjects from 5 smaller studies, which provided greater statistical power for the PRS analyses, we conducted sensitivity analyses that controlled for study. These analyses showed a modest reduction overall in the observed associations with the MDD and OUD PRS and some statistically significant findings were rendered non-significant. However, in most cases, the variance in MOR BPND accounted for by the MDD and OUD PRS remained large, supporting the demonstrated associations between these PRS and MOR availability at baseline and endogenous opioid release during a stressful stimulus.
The genetic overlap accords with a recent mendelian randomization study of prescription opioid use and MDD, in which the genetic liability for prescription opioid use was associated with increased risk of MDD [4]. Stress-induced endogenous opioid release has been linked to both emotion dysregulation and trait impulsiveness [42, 50] and reflects one of the most consistently recognized stress-regulatory mechanisms across species [51]. The associations between MDD and OUD PRS and the opioid response to a pain stressor remained evident after controlling for the A118G SNP in OPRM1. Thus, the predominant genetic effects of the PRS appear to be independent of this widely studied functional polymorphism.
The more robust associations for both PRS among females is likely due, at least in part, to the greater proportion of females (58.7%) in the target sample. Thus, associations with these PRS might be evident in a larger male target sample. The GWAS input sample for the OUD PRS was predominantly (89.8%) male [12], which contrasts with a nearly equal sex distribution (51.2% female) in the MDD GWAS input sample [10]. The future availability of an OUD input sample with a more equal sex distribution might yield a PRS that accounts for more variance than the one used here.
There are study limitations that need to be considered. First, the sample was comprised exclusively of EAs, as the number of non-EA individuals for whom scan data were available was too small to permit analysis. Similar analyses in other population groups would require large, diverse samples of individuals who have undergone MOR imaging and genome-wide genotyping. They would also require the availability of summary statistics from GWAS conducted in large, diverse samples to provide input for the calculation of PRS in the target samples. To obtain a large enough sample for the analyses that we performed, we combined data from multiple studies comprising healthy subjects and those with chronic pain or Axis I or Axis II psychiatric disorders. We did not stratify the analyses by diagnostic group, as they were too small to permit such analyses. Finally, because participants with a history of substance use disorders, including OUD, were excluded, and stress-induced endogenous opioid release data were available in few MDD-diagnosed volunteers, the findings cannot be generalized to either OUD- or MDD-affected populations, where they could be used to address risk directly. Because the exclusion of more than a few affected subjects could have biased the findings, larger studies are needed in subjects with MDD and OUD to draw meaningful conclusions.
Despite these limitations, the aggregation of samples in a secondary analysis of data acquired across studies yielded a large study sample, enabling us to conduct analyses that would otherwise not be possible due to the high cost and limited availability of PET. Notably, molecular measurements such as those utilized here are typically more closely associated with gene function than more global, metabolically-driven measurements [22]. Future studies are needed to examine how these genetic effects on opioid neurotransmission are manifested in individuals with established MDD and OUD, which are common, disabling disorders.
References
Martins SS, Fenton MC, Keyes KM, Blanco C, Zhu H, Storr CL. Mood and anxiety disorders and their association with non-medical prescription opioid use and prescription opioid-use disorder: longitudinal evidence from the National Epidemiologic Study on Alcohol and Related Conditions. Psychol Med. 2012;42:1261–72.
Fink DS, Hu R, Cerdá M, Keyes KM, Marshall BDL, Galea S, et al. Patterns of major depression and nonmedical use of prescription opioids in the United States. Drug Alcohol Depend. 2015;153:258–64.
SAMHSA. Results from the 2019 national survey on drug use and health. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2020.
Rosoff DB, Smith GD, Lohoff FW. Prescription opioid use and risk for major depressive disorder and anxiety and stress-related disorders: A multivariable Mendelian randomization analysis. JAMA Psychiatry. 2021;78:151–60.
Zubieta J-K, Ketter TA, Bueller JA, Xu Y, Kilbourn MR, Young EA, et al. Regulation of human affective responses by anterior cingulate and limbic mu-opioid neurotransmission. Arch Gen Psychiatry. 2003;60:1145–53.
Peciña M, Karp JF, Mathew S, Todtenkopf MS, Ehrich EW, Zubieta J-K. Endogenous opioid system dysregulation in depression: implications for new therapeutic approaches. Mol Psychiatry. 2019;24:576–87.
Gabilondo AM, Meana JJ, García-Sevilla JA. Increased density of mu-opioid receptors in the postmortem brain of suicide victims. Brain Res. 1995;682:245–50.
Escribá PV, Ozaita A, García-Sevilla JA. Increased mRNA expression of alpha2A-adrenoceptors, serotonin receptors and mu-opioid receptors in the brains of suicide victims. Neuropsychopharmacology. 2004;29:1512–21.
Darcq E, Kieffer BL. Opioid receptors: drivers to addiction? Nat Rev Neurosci. 2018;19:499–514.
Howard DM, Adams MJ, Clarke T-K, Hafferty JD, Gibson J, Shirali M, et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat Neurosci. 2019;22:343–52.
Crist RC, Reiner BC, Berrettini WH. A review of opioid addiction genetics. Curr Opin Psychol 2019;27:31–35.
Zhou H, Rentsch CT, Cheng Z, Kember RL, Nunez YZ, Sherva RM, et al. Association of OPRM1 functional coding variant with opioid use disorder: a genome-wide association study. JAMA Psychiatry. 2020. https://doi.org/10.1001/jamapsychiatry.2020.1206
Anderson JS, Shade J, DiBlasi E, Shabalin AA, Docherty AR. Polygenic risk scoring and prediction of mental health outcomes. Curr Opin Psychol. 2019;27:77–81.
Erbs E, Faget L, Scherrer G, Matifas A, Filliol D, Vonesch JL, et al. A mu-delta opioid receptor brain atlas reveals neuronal co-occurrence in subcortical networks. Brain Struct Funct. 2015;220:677–702.
Charbogne P, Kieffer BL, Befort K. 15 years of genetic approaches in vivo for addiction research: Opioid receptor and peptide gene knockout in mouse models of drug abuse. Neuropharmacology. 2014;76 Pt B:204–17.
Hsu DT, Sanford BJ, Meyers KK, Love TM, Hazlett KE, Walker SJ, et al. It still hurts: altered endogenous opioid activity in the brain during social rejection and acceptance in major depressive disorder. Mol Psychiatry. 2015;20:193–200.
Sora I, Takahashi N, Funada M, Ujike H, Revay RS, Donovan DM, et al. Opiate receptor knockout mice define mu receptor roles in endogenous nociceptive responses and morphine-induced analgesia. Proc Natl Acad Sci USA. 1997;94:1544–9.
Matthes HWD, Maldonado R, Simonin F, Valverde O, Slowe S, Kitchen I, et al. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature. 1996;383:819–23.
Weerts EM, McCaul ME, Kuwabara H, Yang X, Xu X, Dannals RF, et al. Influence of OPRM1 Asn40Asp variant (A118G) on [11C]carfentanil binding potential: preliminary findings in human subjects. Int J Neuropsychopharmacol. 2013;16:47–53.
Peciña M, Love T, Stohler CS, Goldman D, Zubieta J-K. Effects of the Mu opioid receptor polymorphism (OPRM1 A118G) on pain regulation, placebo effects and associated personality trait measures. Neuropsychopharmacology. 2015;40:957–65.
Ray R, Ruparel K, Newberg A, Wileyto EP, Loughead JW, Divgi C, et al. Human Mu Opioid Receptor (OPRM1 A118G) polymorphism is associated with brain mu-opioid receptor binding potential in smokers. Proc Natl Acad Sci USA. 2011;108:9268–73.
Zhou Z, Zhu G, Hariri AR, Enoch MA, Scott D, Sinha R, et al. Genetic variation in human NPY expression affects stress response and emotion. Nature. 2008;452:997–1001.
Zubieta JK, Smith YR, Bueller JA, Xu Y, Kilbourn MR, Jewett DM, et al. Regional mu opioid receptor regulation of sensory and affective dimensions of pain. Science. 2001;293:311–5.
Zubieta JK, Dannals RF, Frost JJ. Gender and age influences on human brain mu-opioid receptor binding measured by PET. Am J Psychiatry. 1999;156:842–8.
Zubieta J-K, Smith YR, Bueller JA, Xu Y, Kilbourn MR, Jewett DM, et al. mu-opioid receptor-mediated antinociceptive responses differ in men and women. J Neurosci. 2002;22:5100–7.
Wood AR, Esko T, Yang J, Vedantam S, Pers TH, Gustafsson S, et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet. 2014;46:1173–86.
Zubieta J-K. Placebo effects mediated by endogenous opioid activity on mu-opioid receptors. J Neurosci. 2005;25:7754–62.
Peciña M, Bohnert ASB, Sikora M, Avery ET, Langenecker SA, Mickey BJ, et al. Association between placebo-activated neural systems and antidepressant responses: neurochemistry of placebo effects in major depression. JAMA Psychiatry. 2015;72:1087–94.
Zubieta J-K, Heitzeg MM, Smith YR, Bueller JA, Xu K, Xu Y, et al. COMT val158met genotype affects mu-opioid neurotransmitter responses to a pain stressor. Science. 2003;299:1240–3.
Martikainen IK, Pecina M, Love TM, Nuechterlein EB, Cummiford CM, Green CR, et al. Alterations in endogenous opioid functional measures in chronic back pain. J Neurosci. 2013;33:14729–37.
1000 Genomes Project Consortium, Auton AA, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, et al. A global reference for human genetic variation. Nature. 2015;526:68–74.
Stohler CS, Kowalski CJ. Spatial and temporal summation of sensory and affective dimensions of deep somatic pain. Pain. 1999;79:165–73.
Ge T, Chen C-Y, Ni Y, Feng Y-CA, Smoller JW. Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nat Commun. 2019;10:1776.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc. 1995;57:289–300.
Smith KS, Berridge KC. Opioid limbic circuit for reward: interaction between hedonic hotspots of nucleus accumbens and ventral pallidum. J Neurosci. 2007;27:1594–605.
Kalivas PW, Volkow ND. The neural basis of addiction: a pathology of motivation and choice. Am J Psychiatry. 2005;162:1403–13.
Berridge KC, Kringelbach ML. Towards a neuroscience of well-being: implications of insights from pleasure research. In: Happiness studies book series. Springer Netherlands; 2013. p. 81–100.
Loseth GE, Ellingsen D-M, Leknes S. State-dependent μ-opioid modulation of social motivation. Front Behav Neurosci. 2014;8:430.
Hsu DT, Sanford BJ, Meyers KK, Love TM, Hazlett KE, Wang H, et al. Response of the μ-opioid system to social rejection and acceptance. Mol Psychiatry. 2013;18:1211–7.
Koob GF, Volkow ND. Neurobiology of addiction: a neurocircuitry analysis. Lancet Psychiatry. 2016;3:760–73.
Volkow ND, Baler RD, Goldstein RZ. Addiction: pulling at the neural threads of social behaviors. Neuron. 2011;69:599–602.
Love TM, Stohler CS, Zubieta J-K. Positron emission tomography measures of endogenous opioid neurotransmission and impulsiveness traits in humans. Arch Gen Psychiatry. 2009;66:1124–34.
Salk RH, Hyde JS, Abramson LY. Gender differences in depression in representative national samples: Meta-analyses of diagnoses and symptoms. Psychol Bull. 2017;143:783–822.
Jamison RN, Butler SF, Budman SH, Edwards RR, Wasan AD. Gender differences in risk factors for aberrant prescription opioid use. J Pain. 2010;11:312–20.
Evans EA, Goff SL, Upchurch DM, Grella CE. Childhood adversity and mental health comorbidity in men and women with opioid use disorders. Addict Behav. 2020;102:106149.
Grella CE, Karno MP, Warda US, Niv N, Moore AA. Gender and comorbidity among individuals with opioid use disorders in the NESARC study. Addict Behav. 2009;34:498–504.
McHugh RK, Devito EE, Dodd D, Carroll KM, Potter JS, Greenfield SF, et al. Gender differences in a clinical trial for prescription opioid dependence. J Subst Abus Treat. 2013;45:38–43.
Back SE, Payne RL, Wahlquist AH, Carter RE, Stroud Z, Haynes L, et al. Comparative profiles of men and women with opioid dependence: results from a national multisite effectiveness trial. Am J Drug Alcohol Abus. 2011;37:313–23.
Hernandez-Avila CA, Rounsaville BJ, Kranzler HR. Opioid-, cannabis- and alcohol-dependent women show more rapid progression to substance abuse treatment. Drug Alcohol Depend. 2004;74:265–72.
Kennedy SE, Koeppe RA, Young EA, Zubieta J-K. Dysregulation of endogenous opioid emotion regulation circuitry in major depression in women. Arch Gen Psychiatry. 2006;63:1199–208.
Valentino RJ, Volkow ND. Untangling the complexity of opioid receptor function. Neuropsychopharmacology. 2018;43:2514–20.
Author contributions
Substantial contributions to the conception or design of the work: HRK, J-KZ, TL. Acquisition, analysis, or interpretation of data for the work: J-KZ, AAS, HRK, HZ, JG, AKB, VK. Drafting the work or revising it critically for important intellectual content: TL, HRK, J-KZ, RLK, EH, JD, ARD, HZ. Final approval of the version to be published: TL, AAS, RLK, ARD, HZ, VK, JG, AKB, EH, JD, J-KZ, HRK. Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved: HRK, J-KZ.
Funding
Supported by grants R01 DA022520, R01 MH086858, R01 DA027494, R01 AT001415, R21 MH069612, P30 DA046345, K01 AA028292 and K23 DA038726 from the U.S. National Institutes of Health; I01 BX003341, I01 CX001734 and the VISN 4 Mental Illness Research, Education and Clinical Center from the U.S. Department of Veterans Affairs; and NARSAD Young Investigator Award # 28686. The views expressed in this article are those of the authors and do not necessarily represent the position or policy of the Department of Veterans Affairs or the United States Government.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
HRK is a scientific advisory board member for Dicerna Pharmaceuticals, Sophrosyne Pharmaceuticals, and Enthion Pharmaceuticals; a consultant for Sobrera Pharmaceuticals; the recipient of research funding and medication supplies for an investigator-initiated study from Alkermes; and a member of the American Society of Clinical Psychopharmacology’s Alcohol Clinical Trials Initiative (ACTIVE Group), which over the past three years was supported by Alkermes, Dicerna, Ethypharm, Lundbeck, Mitsubishi, and Otsuka. HRK and JG hold US patent 10,900,082 titled: “Genotype-guided dosing of opioid agonists,” 26 Jan. 2021. The other authors have no interests to disclose.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Love, T., Shabalin, A.A., Kember, R.L. et al. Unique and joint associations of polygenic risk for major depression and opioid use disorder with endogenous opioid system function. Neuropsychopharmacol. 47, 1784–1790 (2022). https://doi.org/10.1038/s41386-022-01325-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-022-01325-1
