
Abstract
As we age, we experience changes in our nighttime sleep and daytime wakefulness. Individuals afflicted with Alzheimer’s disease (AD) can develop sleep problems even before memory and other cognitive deficits are reported. As the disease progresses and cognitive changes ensue, sleep disturbances become even more debilitating. Thus, it is imperative to gain a better understanding of the relationship between sleep and AD pathogenesis. We postulate a bidirectional relationship between sleep and the neuropathological hallmarks of AD; in particular, the accumulation of amyloid-β (Aβ) and tau. Our research group has shown that extracellular levels of both Aβ and tau fluctuate during the normal sleep−wake cycle. Disturbed sleep and increased wakefulness acutely lead to increased Aβ production and decreased Aβ clearance, whereas Aβ aggregation and deposition is enhanced by chronic increased wakefulness in animal models. Once Aβ accumulates, there is evidence in both mice and humans that this results in disturbed sleep. New findings from our group reveal that acute sleep deprivation increases levels of tau in mouse brain interstitial fluid (ISF) and human cerebrospinal fluid (CSF) and chronic sleep deprivation accelerates the spread of tau protein aggregates in neural networks. Finally, recent evidence also suggests that accumulation of tau aggregates in the brain correlates with decreased nonrapid eye movement (NREM) sleep slow wave activity. In this review, we first provide a brief overview of the AD and sleep literature and then highlight recent advances in the understanding of the relationship between sleep and AD pathogenesis. Importantly, the effects of the bidirectional relationship between the sleep−wake cycle and tau have not been previously discussed in other reviews on this topic. Lastly, we provide possible directions for future studies on the role of sleep in AD.
Similar content being viewed by others


Introduction
The purpose of sleep is one of the fundamental unsolved mysteries of biology, although we do know that the quantity, quality, and timing of sleep influence a vast array of biological functions [1,2,3,4,5]. During normal healthy aging, changes in sleep architecture and increases in sleep disturbances are a physiological hallmark. From early adulthood (ages 16–25) to midlife (ages 36–50), the percentage of nonrapid eye movement (NREM) slow wave sleep (SWS) (i.e. “deep sleep”, discussed below) decreases significantly. This is compensated for by an increased percentage of lighter sleep but not significant increases in time spent awake (sleep fragmentation), or decreases in time spent in rapid eye movement (REM) sleep [6]. Many elderly people who are 65 years or older have increased sleep fragmentation, increased early-morning awakening, increased sleep latency, decreased sleep quality, and difficulty with sleep maintenance [7,8,9]. The age-related decline of quantity and quality of NREM SWS is related to increased sleep fragmentation and difficulty with sleep maintenance during sleeping time [10]. According to several studies, sleep impacts various aspects of memory processing and cognitive performance [11, 12]. Therefore, sleep disruption has been postulated as a potential risk factor for cognitive decline [13].
Currently, around 47 million people worldwide are living with dementia. Moreover, the number of individuals with dementia is expected to increase to about 90 million by 2030 [14]. Of all dementias, AD is the most common contributor and accounts for up to 75% of all dementia cases [15]. In the United States, deaths due to AD have increased by 71% between 2000 and 2013, ranking this disease as the sixth leading cause of death [16]. AD is a progressive neurodegenerative disorder affecting wide areas of the cerebral cortex and hippocampus [17]. Even though symptoms of AD patients can differ greatly from one person to another, AD generally results in a progressive decline in memory and cognition. The two histopathological markers of AD are extracellular amyloid-β (Aβ) plaques composed predominantly of the Aβ peptide and neurofibrillary tangles (NFTs) as well as neuropil threads in neurons comprised of abnormally aggregated, hyperphosphorylated tau [18]. These pathological hallmarks are accompanied by progressive impairment of memory, language, and cognition, as well as a prominent innate immune response, synaptic loss, and brain atrophy [19,20,21,22,23]. Importantly, sleep disturbances are also prevalent, and are a highly disruptive behavioral manifestation of AD [24, 25].
Recent studies report that sleep disturbances negatively affect cognitive function and functional impairment in patients with AD [26,27,28,29]. Approximately 25–66% of AD patients exhibit sleep disturbances [28, 29]. Aβ pathology begins to accumulate during the preclinical stage of AD, about 15–20 years before cognitive symptoms manifest. Changes in sleep also occur during the preclinical stage of AD [25]. In one study, cognitively normal individuals identified as having amyloid pathology had decreased sleep efficiency compared to those that were amyloid negative [30]. Changes in sleep efficiency and quality seem to precede the onset of cognitive decline in AD patients and progress in parallel with both cognitive dysfunction and the progression of AD pathology [25].
Several studies have shown a role for the sleep−wake cycle in the progression of AD. Extracellular Aβ in brain ISF as well as CSF fluctuates diurnally: soluble Aβ levels are higher during wakefulness and lower during sleep [31, 32]. Acute sleep deprivation in humans increases soluble Aβ in CSF by 25–30% via increased overnight Aβ production relative to sleeping controls [33]. In APP transgenic mice, chronic sleep deprivation accelerates Aβ deposition, whereas in orexin knockout mice, which sleep 10% more than control littermates, there is decreased Aβ deposition [31, 34]. One of the primary functions of orexin, a hypothalamic neurotransmitter, is to promote and stabilize wakefulness and reduce REM sleep via orexinergic neurons in the hypothalamus. The fibers of these neurons project to many different nuclei that regulate the sleep−wake cycle, such as the locus coeruleus (LC) and septal nuclei [35, 36]. Several studies have shown that there is a strong association between orexinergic neurotransmission dysfunction, sleep impairment, and decline in memory function and cognition in moderate–severe AD patients [37,38,39]. In addition, Aβ deposition disrupts the sleep−wake cycle in APP transgenic mice resulting in increased wakefulness [40]. A new study from our group has found that ISF tau also fluctuates diurnally. Furthermore, sleep deprivation increases levels of tau in mouse brain ISF and human CSF, and chronic sleep deprivation accelerates the spreading of tau protein aggregates in specific brain networks in a tau seeding/spreading model [41].
To date, there is mounting evidence that there is a bidirectional relationship between sleep disturbance and AD pathology. This review will highlight recent studies on the bi-directional relationship between sleep and AD from the molecular level to the clinical level. Importantly, the effects of the bi-directional relationship between the sleep−wake cycle and tau have not been previously discussed in other reviews on this topic. We now cite and discuss papers on this topic and discuss findings that are not yet present in our other reviews on this topic. Furthermore, we will briefly discuss the potential role of apolipoprotein E (apoE) and microglia as well as other factors relevant to AD and sleep. These include Aβ accumulation and tau-mediated neurodegeneration, and how APOE, especially the APOE ε4 allele, impacts Aβ and tau [42,43,44,45]. A recent study showed that disrupted sleep was more frequent in males than in females and in carriers of the APOE ε4 allele as compared to noncarriers [46]. Based on a few studies, acute and chronic sleep deprivation in humans results in the induction of both cellular and humoral immunological responses [47]. Chronic sleep deprivation in rodents has been associated with microglial activation and astrocytic phagocytosis in the brain [48]. This low-grade systemic inflammation induced by sleep loss could facilitate neuroinflammation, which could aggravate AD-related brain pathology [49]. We will also highlight questions to address in future studies to test ideas implicated by a bi-directional relationship between sleep and AD.
Basic concepts of sleep
Sleep architecture in humans
Sleep is composed of two very different physiological states: REM sleep and NREM sleep. NREM sleep is further divided into three stages, with stage N1 being the lightest and stages N2 and N3 progressively getting into deeper sleep. In the normal young adult, polysomnographic recordings including electroencephalogram (EEG) show five cyclical sleep stages [50]. During wakefulness with eyes open, the EEG signals show mainly beta activity (frequencies higher than 13 hertz (Hz)), but during wakefulness with eyes closed, alpha activity dominates (8–13 Hz). During stage NREM N1, a transition period from wakefulness to sleep that occupies about 5% of total sleep time, the alpha activity slows to a theta rhythm (4–8 Hz). Two identifiable characteristic phasic features develop during NREM stage N2; sleep spindles (bursts of rhythmic sigma (12–14 Hz) waves) and K-complexes (waves of distinct negative high-voltage peak followed by a distinct positive high-voltage deflection) on a theta wave background. Sleep spindles may be linked to sleep-dependent memory consolidation and cognition [51]. K-complexes have been suggested to react to external stimuli during sleep and to protect sleep as an essential part of the synchronization of NREM sleep [52]. N2 is a period of “light” sleep that occupies about 50% of the sleep period. During stage N3, which occupies about 20% of sleeping time, high-amplitude, low frequency delta waves (0.5–4 Hz) dominate leading to its description as slow-wave sleep (SWS) or “deep” sleep. Lastly, REM sleep, which accounts for about 25% of total sleep time, is characterized as desynchronized sleep due to the physiological similarities to waking states including rapid and low-voltage desynchronized brain waves [50, 53]. EEG desynchrony in REM sleep is largely controlled by acetylcholine (ACh) tone, which is radically different from EEG desynchrony during wake periods [54]. REM sleep is also characterized by muscle atonia (REM sleep atonia). During REM sleep, skeletal muscles are essentially paralyzed to minimize the movements [53]. Sleep is typically organized into 90-min cycles of NREM/REM in humans. However, most stage N3 sleep occurs during the first half of the night, and most REM sleep in the last half of the night [55]. Sleep/wake cycles are tightly regulated in the brain by many brain regions, neurotransmitters, and flip-flop switches [56]. Sleep regulating areas are found throughout the brain, including the brainstem, midbrain, thalamus, hypothalamus, and basal forebrain [56].
Sleep changes during normal aging
As discussed in the introduction, with aging, sleep architecture changes with a reduction of NREM SWS, an increase in lighter sleep, changes in REM sleep stage appearance, and increased sleep fragmentation [50]. The decrease in SWS is accompanied by a decrease in both the number and amplitude of delta waves. According to Van Cauter et al., from early adulthood to midlife (age 16–25 to 36–50 years), the percentage of NREM SWS decreases significantly, which is replaced by an increased percentage of lighter sleep without significant increases in sleep fragmentation or decreases in REM sleep [6]. Ohayon et al. also reported that in children and adolescents, the percentage of NREM SWS was significantly negatively correlated with age, and percentages of stage N2 and REM sleep significantly changed with age [10]. Based on their sleep data from a large meta-analysis of PSG-verified sleep, in adulthood (from young to middle (ages 40–60 years) to older (ages >60 years)), as age increases, there are several significant decreases in total sleep time, sleep efficiency, percentage of NREM SWS, percentage of REM sleep, and REM latency, whereas there are significant increases in sleep latency, percentage of stage N1, percentage of stage N2, and wake after sleep onset (WASO) [10]. However, stage N2 and REM sleep remain unchanged until very old age [57, 58], although the two identifiable EEG features of stage N2 (sleep spindles and K-complexes) are less well formed with age [59]. While the duration of stage N2 does not dramatically change until old age, measures of sleep spindles during N2 sleep do dramatically change at the same time as NREM SWS changes begin to occur during early adulthood [60]. These significant changes in sleep with aging have been documented in almost all people older than 60 years and are worse in elderly people with the APOE ε4 allele [61]. As sleep in elderly individuals becomes “lighter and more fragmented”, many factors can induce sleep changes, such as medication, nocturia, bodily pain, and other physical or mental health conditions [50]. Mander et al. reported that decreased frontal brain SWA during sleep predicts memory decline in older adults [62]. They also reported that brain volume loss in medial prefrontal cortex (mPFC) gray matter, an area known to be important for NREM regulation, predicted the loss of SWA in the elderly. The loss of sleep spindles and SWA are a predictive risk factor in age-related decline of memory retention and cognition.
Sleep changes in Alzheimer’s disease (AD)
Sleep architecture changes are exaggerated in patients with mild cognitive impairment (MCI), which is often a prodromal phase of AD, and in dementia due to AD [63, 64]. The occurrence rate of sleep disturbances in MCI patients has been reported to range from 8.8 to 45.5% [65, 66]. Furthermore, sleep disordered breathing (SDB) and sleep behavior disorders occur more frequently in MCI patients than in normal subjects [13]. Excessive daytime sleepiness (EDS), sundowning, and insomnia are among the most common sleep disturbances in dementia due to AD and originate from changes in sleep architecture and the circadian rhythm [67]. EDS, defined as sleepiness that interferes with daily activities, is common in patients with dementia and may have a significant impact on quality of life of AD patients. Sundowning is defined as agitated behavior occurring primarily after the sun goes down; for example, pacing, yelling out, getting violent, or wandering around at night. Insomnia is defined as complaints of difficulty falling asleep, difficulty staying asleep, or early-morning awakenings that have a significant impact on daytime function [68]. Insomnia as a disorder affects 30−50% of older adults [69]. AD patients have been reported to have decreased sleep efficiency, NREM SWS, and total sleep time, as well as decreased REM sleep and increased latency to REM sleep [67, 70, 71]. The occurrence rate of sleep disturbances in AD patients has been documented to range from 25 to 60% [29, 72], and AD patients have more frequent sleep problems than cognitively normal old adults (18.3−27.6%) [73]. Sleep disturbances are associated with AD pathology during the preclinical phase of AD. A study in cognitively normal, late middle-aged adults has shown a decrease in sleep efficiency, measured by actigraphy, and an increase in napping in subjects that are amyloid positive compared to those that are amyloid negative. It suggests a role for Aβ accumulation prior to cognitive decline during preclinical AD (Fig. 1) [30]. A study performed by Spira et al. supported the idea that the deposition of Aβ is linked with shorter sleep [74]. They hypothesized that individuals having more fragmented sleep, shorter sleep duration, and lower sleep quality would have greater amyloid burden than individuals having otherwise uninterrupted sleep. They reported that there is a significant relationship between sleep disturbances (shorter sleep duration and poor sleep quality) and Aβ aggregation, as measured by 11C-PIB positron emission tomography (PET) scanning. They showed that participants reporting >7 h sleep have the least amyloid burden, those reporting >6 to ≤7 h have an intermediate level of burden, and participants reporting ≤6 h sleep duration have the greatest burden. They also found that disturbed sleep, characterized by shorter sleep duration and poor sleep quality, in community-dwelling older adults could be detectable by a sleep questionnaire without a direct measure of sleep using either actigraphy or sleep studies [74]. In addition, several clinical follow-up studies have reported that cognitively normal elderly people with high sleep fragmentation had a 1.5-fold increased risk of AD development, and self-reported sleep decline was associated with a twofold increased risk of developing AD [75, 76]. Recently, Lucey et al. in our group reported that tau accumulation is linked with decreased NREM SWA in low frequencies and it is associated with a greater effect on this activity than Aβ deposition [77]. This finding was noted in cognitively normal and very mildly cognitively impaired individuals.

Prevalence of amyloid deposition by sleep efficiency group. a Prevalence of Cognitively- normal, Clinical Dementia Rating (CDR) 0 participants who have preclinical amyloid pathology exhibit diminished sleep efficiency. Less than 75% and more than 89% represent poor and good sleep efficiency, respectively. The proportion in each group with abnormal Aβ 42 level (≤500 pg/mL) decreases with better sleep efficiency. b Nap days per week was skewed toward zero. Vertical axes represent absolute frequency. Participants who have preclinical amyloid pathology exhibit increased napping. Adapted by permission from JAMA: Neurol [30]
The exact reason for changes in sleep architecture in AD is still unknown, but possibly originates from AD pathology-induced neuronal and synaptic damage in crucial sleep regulating areas and pathways in the brain [78]. Sleep regulating centers are located throughout the basal forebrain (BF), hypothalamus, thalamus, midbrain, pons, and brainstem in the brain, each of which are also areas that can be affected by AD pathology [78]. For example, loss of cholinergic neurons in the BF or noradrenergic neurons in the locus coeruleus (LC) in AD could not only negatively affect cognition, but also sleep−wake regulation [79,80,81]. As discussed above, brain volume loss in mPFC gray matter (an area known to be important for NREM regulation) predicted the loss of SWA. Also, decreased REM sleep duration in people with MCI was strongly associated with gray matter loss in regions which degenerate early in AD, such as the cholinergic BF, the precuneus, the posterior cingulate, and the postcentral gyrus [82, 83]. REM sleep EEG desynchrony is largely controlled by ACh tone, which is radically different from wake EEG desynchrony. This is particularly important for AD, as AD is associated with cholinergic degeneration and loss of REM sleep EEG desynchrony, possibly due to tau accumulation [63, 78]. As we discussed in the section on the basics of sleep, REM sleep is largely controlled by ACh tone following stimulation of cholinergic neurons in precise regions of the brainstem [84]. Using a mouse model of tauopathy, decreased REM and NREM sleep correlated with increased tau pathology in the sublaterodorsal area and parafacial zone in the brainstem [85]. Montplaisir and colleagues have reported that subjects with amnestic MCI (a-MCI) have a significantly slower REM sleep EEG measured from frontal lateral regions than both nonamnestic MCI (na-MCI) and cognitively healthy control subjects [86]. They also reported that the EEG slowing is a good marker of neurodegeneration in REM sleep behavior disorder (RBD) patients, a condition characterized by loss of muscle atonia during REM sleep [87]. RBD is strongly associated with synucleinopathies, such as Parkinson’s disease and Lewy body dementia (LBD) [88, 89]. More than 80% of RBD patients eventually develop a neurodegenerative disease, usually a synucleinopathy [89]. Montplaisir and colleagues also reported that MCI is commonly observed in RBD patients [90]. They suggested that RBD may be caused by neurodegeneration in the brainstem areas controlling REM sleep paralysis [89]. Additionally, as we discussed in the introduction briefly, there is a strong association between orexinergic system dysfunction, sleep impairment, and decline in memory function and cognition in moderate–severe AD patients [37,38,39]. Several studies observed a significant increase in CSF orexin concentration in moderate–severe AD patients [37, 38, 91]. The increased orexin level caused by orexinergic system dysfunction may cause further sleep impairment and secondarily lead to further exacerbation of AD pathology [91].
Even though this review does not cover the relationship between circadian rhythm dysfunction and AD, many studies have reported a decline of behavioral circadian rhythms in aged mice and humans [92, 93]. Furthermore, a common symptom seen in AD patients includes dysfunction of circadian rhythms, such as increased nocturnal activity, decreased diurnal activity, and core body temperature phase delay and amplitude decrease [93,94,95,96]. Sleep–wake cycle and circadian rhythms are often considered to be interchangeable [63]. Neuronal and synaptic damage by AD pathology in circadian regulating areas (e.g. hypothalamic suprachiasmatic nucleus (SCN)) increase the dysfunction of cellular circadian rhythms, which lies at the root of parts of the sleep disturbances seen in AD [97,98,99].
Basic concepts of Alzheimer’s disease (AD)
Brief history regarding clinical, pathological, and genetic features of Aβ and Tau
Amyloid-β (Aβ)
In 1906, Alois Alzheimer, a German psychiatrist, first reported the presence of a “peculiar substance” in the brain of a patient. However, the identity of the substance remained a mystery until the early 1980s [100]. In 1984, Glenner and Wong identified fragments of the Aβ peptide from meningeal vessels of patients with Down syndrome with amyloid pathology and successfully sequenced the N-terminal 24 amino acids of Aβ [101]. In 1985, Masters et al. isolated amyloid plaque cores from post-mortem AD brains, as well as characterized and sequenced Aβ using a variety of different techniques [102]. In 1987, the first genetic link to AD was identified by two research groups who discovered that the gene from which Aβ is derived, known as the amyloid β precursor protein (APP), is located on human chromosome 21 [103, 104]. All individuals with trisomy 21 or partial trisomy 21 who have three copies of APP develop Aβ deposition by their fourth decade of life [105]. In 1991, Goate and colleagues first identified a disease-causing mutation of dominantly inherited AD in the APP gene (V717I) in families with early-onset AD. Aβ is produced by the sequential proteolytic cleavage of APP by β‑secretase and γ‑secretase, leading to Aβ peptide formation with peptides of various length (between 38 and 43 amino acids) [106, 107]. Mutations in γ-secretase component genes Presenilin-1 (PSEN1), Presenilin-2 (PSEN2), as well as the APP gene lead to dominantly inherited AD [108,109,110,111]. Many of the mutations in these genes (APP, PSEN1 and PSEN2) lead to an increase in longer forms of Aβ (42 and 43) and decrease in shorter forms (Aβ38 and 40) resulting in early-onset Aβ aggregation [112, 113]. Most mutations in the APP gene modify APP processing and increase the ratio of Aβ42 to Aβ40 in the plasma of patients [114, 115]. Moreover, mutations in PSEN1 and PSEN2 also result in an increased Aβ42/Aβ40 ratio [111]. Two major isoforms of Aβ are 40 amino acids (Aβ40) and the less soluble 42 amino-acid form (Aβ42). Aβ42 differs from Aβ40 in that it has two extra hydrophobic amino acids at the C-terminus and is more aggregation prone than shorter Aβ isoforms [106, 116]. Due to the hydrophobic nature of the peptides, soluble monomeric Aβ can self-aggregate to form extracellular oligomers, protofibrils, fibrils, and plaques, the first hallmarks of AD pathology [106].
Based on studies of proteolytic processing, Aβ is a normal product of APP metabolism and is produced at high levels in neurons, as well as in other cell types [17]. The physiological function of APP is still unknown, but studies have indicated that it may be involved in cell health, growth, and synaptic plasticity [22, 117]. A recent paper demonstrates that a secreted form of APP can serve as a ligand for a specific type of GABA receptor [118]. AD is associated with the accumulation of insoluble Aβ plaques in the extracellular space of the brain, as well as in the walls of cerebral blood vessels [17]. Multiple studies suggest that Aβ accumulation and the conformational change it undergoes results in aggregates with high β-sheet structure that is critical in AD pathogenesis [119, 120]. Aβ deposition in the human brain generally begins in areas of the neocortex that overlap with a brain network known as the “default mode network” prior to its progression through multiple other areas of the neocortex [17, 121]. Knowledge of the specific genetic mutation and associated Aβ42/Aβ40 ratio can help predict the average age of onset of AD, as confirmed in the Dominantly Inherited Alzheimer Network (DIAN) studies [122] and a meta-analysis [123].
In late-onset sporadic AD, Aβ accumulation is believed to be due to defective brain clearance of Aβ or increased Aβ seeding propensity [124]. It may also be possible that some individuals have increased Aβ production [124]. Aβ accumulation is influenced by the strongest genetic risk factor for late-onset sporadic AD pathology, apolipoprotein E (APOE), specifically the APOE ε4 allele. APOE ε4 is consistently linked to earlier and more abundant Aβ aggregation and is a predictor of Aβ accumulation in normal older people. APOE ε4 increases the risk of developing AD by ~4-fold with one allele and by ~12 fold with two alleles relative to the APOE ε3/ε3 genotype. One allele of ε2 decreases risk by ~0.6 relative to the APOE ε3/ε3 genotype [42,43,44]. However, these apoE isoforms also have Aβ-independent functions that may affect AD risk and rate of progression [43, 44]. For example, our group reported that APOE ε4 also strongly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy (P301S) via modulation of the brain’s innate immune response [45]. This suggests that apoE could significantly affect tau-mediated neurodegeneration independent of Aβ pathology. As Aβ is considered a key trigger in the AD pathological cascade, we will discuss how Aβ pathology appears to affect AD pathology progression as well as sleep disturbances and vice versa.
Tau
In 1906, Alois Alzheimer also first reported and described NFTs [125]. The primary component of NFTs was not identified as tau protein until 1985 [126]. Historically, tau protein was isolated from porcine brain extracts as a heat-stable and highly soluble protein that is a key component for microtubule (MT) assembly in axons [127]. Tau is a microtubule-associated protein that interacts with tubulin to stabilize microtubules [128, 129]. The function of tau is regulated by the degree of phosphorylation. The hyperphosphorylation and intracellular aggregation of tau protein is considered the second hallmark of AD, and develops in the neocortex subsequent to Aβ aggregation and deposition [23]. Mutations in the microtubule-associated protein tau (MAPT) gene cause frontotemporal dementia with Parkinsonism linked to chromosome 17 (FTLD-17) and these mutations provide genetic evidence that dysfunction in tau can trigger neurodegeneration without Aβ [130,131,132]. Tauopathies, characterized by abnormal tau aggregation in the brain, consist of several different neurodegenerative diseases. The distinction between primary and secondary tauopathies can be characterized by abnormal tau aggregates in different brain regions involving different networks and cell types, as well as specific tau isoforms and conformations [133,134,135]. Primary tauopathies include frontotemporal lobar degeneration (FTLD) associated with tau mutations, aging-related tau astrogliopathy (ARTAG), argyrophilic grain disease (AgD), corticobasal degeneration (CBD), primary age-related tauopathy (PART), progressive supranuclear palsy (PSP), chronic traumatic encephalopathy (CTE), and Pick’s disease (PiD). Secondary tauopathies occur when tau coexists and is likely antecedent to another brain pathology (i.e. Aβ). AD is a secondary tauopathy [135,136,137].
Tau can form insoluble filaments that aggregate as NFTs in AD and other tauopathies [138]. Many studies have shown that NFTs and regions of tau accumulation correlate with the clinical symptoms of dementia in AD [139,140,141]. Unlike Aβ, tau pathology in the brain strongly correlates with cognitive impairment and neurodegeneration in AD. NFTs target selective populations of neurons and specific layers of the cortex as well as specific nuclei in the brainstem [139,140,141]. During the second decade of life during normal aging, hyperphosphorylated, aggregated tau accumulates in the brainstem LC and other subcortical nuclei known to be key sites in sleep−wake regulation [142,143,144]. However, this does not correlate with or predict neurodegeneration or later neurodegenerative disease as it occurs in everyone. By the age of ~50–60, NFTs start to occur in the medial temporal lobe during normal aging; however, these NFTs are also not correlated with neurodegeneration or neurodegenerative disease, provided they do not extend beyond this area, which usually only occurs if Aβ deposition also develops. During the late preclinical stages of AD, tau pathology, such as NFTs and neuritic plaque-associated tau spreads from the medial temporal lobes including the hippocampus and entorhinal cortex to neocortical regions [17].
Many research teams have generated cellular and animal models, using both vertebrate and invertebrate species, to understand the role of tau in AD and other tauopathies and to develop diagnostic and therapeutic approaches [133, 145, 146]. In the adult human brain, six isoforms of tau are expressed and generated due to alternative splicing of the MAPT gene located on chromosome 17q21 [147]. Alternative splicing of exons 2 and 3 produce tau containing zero (0N), one (1N), or two (2N) inserts at the N-terminus. In addition, the absence or presence of exon 10 leads to tau isoforms that contain either three (3R) or four (4R) C-terminal microtubule-binding domains, with 3R tau isoforms binding microtubules less strongly than 4R tau [148]. From all possible alternative splicing, six tau isoforms can be produced: 3R0N, 3R1N, 3R2N, 4R0N, 4R1N, and 4R2N. In the normal human brain, the expression levels of 3R and 4R tau occur at a 1:1 ratio. However, several tauopathies are characterized by the changes in the ratio of 3R:4R tau isoforms that accumulate [130,131,132]. For example, patients with argyrophilic grain disease (AgD), progressive supranuclear palsy (PSP), and corticobasal degeneration (CBD) have higher 4R tau than 3R tau. However, patients with Pick’s disease (PiD) have higher 3R than 4R tau and patients with AD and chronic traumatic encephalopathy (CTE) have both high 3R and 4R tau [136].
In contrast to the human brain, the adult mouse brain only expresses 4R tau [149]. There are many studies that assess the pathological effects and functional consequences of human tau mutations that lead to forms of FTD, with the most commonly studied mutations being glycine residue 272 to valine (G272V), asparagine residue 279 to lysine (N279K), proline residue 301 to leucine or serine (P301L or P301S), valine residue 337 to methionine (V337M), and arginine residue 406 to tryptophan (R406W) [23, 146, 150]. All of these mutations promote aggregation and hyperphosphorylation of tau, which leads to accumulation and neurodegeneration in almost all of these mutant mouse models. The identification of pathogenic mutations in the APP and MAPT genes in AD and FTDP-17, respectively, has provided the opportunity to also study the interaction between Aβ and tau. It is clear that Aβ aggregation and accumulation somehow promotes the spreading or development of tau aggregation in the brain during the process of AD [151, 152]. However, how this occurs is not clear and is a major unsolved area in AD research.
Aβ and Tau
In 1992, Hardy and Higgins proposed the amyloid cascade hypothesis. They postulated, “the deposition of amyloid β protein (AβP), the main component of the plaques, is the causative agent of Alzheimer’s pathology and that neurofibrillary tangles, cell loss, vascular damage, and dementia follow as a direct result of this deposition” [153]. The amyloid cascade hypothesis has been a predominant hypothesis for the pathogenesis of AD for over 25 years and is supported by a multitude of genetic studies that directly implicate Aβ accumulation as the trigger that initiates the AD process [151]. Additional support has come from the generation of a variety of APP or APP/PS1 transgenic mouse models that exhibit some of the main pathological and behavioral features of AD, such as formation of Aβ plaques, synaptic loss, synaptic plasticity alterations, and memory impairment [154]. There is also experimental evidence that tau accumulation and spreading, as well as neurodegeneration secondary to tau, is downstream of Aβ. For example, Roberson et al. showed that a reduction in endogenous tau levels in APP transgenic mice prevents memory impairment, reduces susceptibility to experimentally induced excitotoxic seizures, and decreases early mortality, without altering Aβ levels or plaques [155]. Recently, He et al. reported that Aβ plaques enhance human AD tau seeding and spreading in the mouse brain [156]. They recapitulated the formation of three major types of AD-relevant tau pathologies: (1) tau aggregates in dystrophic neurites surrounding Aβ plaques (NP tau), (2) AD-like NFTs, and (3) neuropil threads (NTs) in Aβ plaque-bearing mouse models without overexpressing tau via injecting human AD-brain-derived pathological tau (AD-tau). However, the “amyloid cascade hypothesis” of AD pathology has recently been challenged (see Ricciarelli and Fedele [157] and Kametani and Hasegawa [158] for in-depth reviews). Although accumulation of Aβ amyloid fibrils are observed in a number of genetically modified AD model mice, NFT formation and nerve cell death are not generally observed [159]. However, this can likely be explained in these mouse models by the fact that mouse tau is less susceptible to aggregation due to differences in genetic sequences. However, as just mentioned, less formation of NFT can be better modeled if human AD-tau fibrils are injected into the mouse brain [156]. Recently, several immunotherapies targeting Aβ in AD model mice were effective in attenuating Aβ deposition in the brain, but to date, the antibodies utilized have not been effective at slowing cognitive decline in humans [158, 160,161,162]. This is perhaps not surprising, as Aβ has been accumulating for ~20 years by the time cognitive decline of AD begins, and cognitive decline correlates more with tau accumulation and not Aβ [151]. Until Aβ accumulation is prevented from occurring in humans and its consequences assessed, it will not be known whether the amyloid hypothesis is valid.
Bidirectional relationship between sleep and AD pathology
Sleep and Aβ
Aβ aggregation begins ~15–20 years before cognitive symptoms in AD [18]. Similarly, sleep changes can begin in the preclinical stage of AD and are a predictive risk factor for neurodegeneration and development of cognitive decline. Several studies have reported that sleep disturbances and increased wakefulness increase the accumulation of Aβ. In imaging studies of cognitively normal adults, poor sleep quality, such as shorter sleep time, decreased sleep efficiency, and increased latency to sleep strongly correlate with the accumulation of Aβ measured by PET scanning [30, 74, 163]. Furthermore, subjects with more sleep problems have a greater Aβ burden measured by PET in the prefrontal and insular regions of the brain [164]. Sleep duration has also been documented to have a strong relationship with Aβ burden. Shorter sleep duration was associated with greater accumulation of Aβ, measured by PET, in the precuneus, angular gyrus, and frontal medial orbital cortex in the brain [165]. In addition, SWS arousals correlate with higher CSF levels of Aβ42 in humans with MCI symptoms, but not in controls [82].
The role of sleep in amyloid pathogenesis has been further assessed with sleep deprivation studies in humans and animal models. Even one night of sleep deprivation in healthy young adults significantly increased morning Aβ42 levels in the CSF [166]. Similarly, a recent study found that sleep deprivation increased overnight CSF levels of Aβ38, Aβ40, and Aβ42 levels by 25−30% due to increased overnight CSF Aβ production compared to controls who had a night of normal sleep [33]. Aβ is generated by neurons and secreted into the brain interstitial fluid (ISF). To investigate ISF Aβ metabolism, our group monitored hippocampal Aβ levels using in vivo microdialysis in both wild-type control mice and human APP transgenic (Tg2576) mice, which overexpress a mutated form of human APP [31]. Aβ levels were significantly increased during the dark period (active time for mice) compared to the light period (greater sleep time) in both control C57BL6 and Tg2576 mice. ISF Aβ levels were positively correlated with the amount of wake time. Conversely, ISF Aβ levels were negatively correlated with the amount of sleep time. In humans, CSF Aβ has a 24 h diurnal fluctuation, similar to the fluctuation seen in mouse ISF Aβ [25]. Because increased Aβ levels significantly correlated with wake time, Kang et al. in our group performed acute sleep deprivation (SD) for 6 h at the beginning of the light phase in Tg2576 mice. SD significantly increased ISF Aβ in Tg2576 mice (Fig. 2) [31]. With chronic SD for 20 h daily over a period of 21 days, both Tg2576 and APP/PS1 amyloid-developing model mice had a significant increase in amyloid plaque pathology compared to mice without chronic SD. Conversely, treatment with an orexin receptor antagonist once daily for 8 weeks to increase sleep time in APP/PS1 mice significantly decreased amyloid pathology. While the data from the Kang et al. study [31] suggest that wakefulness is associated with increased Aβ production/release from synapses compared to during sleep, Xie et al. reported that sleep also facilitates clearance of Aβ from the ISF via the glymphatic system [167]. The glymphatic system includes convective fluxes from the para-arterial CSF through ISF and toward the para-venous space that clears neuronal by-products through the systemic circulation [168]. Using two-photon imaging of the brain of living, adult mice, they found that the CSF influx was significantly higher in the brain of sleeping or anesthetized compared to awake mice. They also found that the volume of the interstitial space was >60% greater when mice were sleeping or anesthetized. Therefore, the decrease of Aβ clearance under SD may also contribute to an increase in Aβ levels [167]. As we discussed previously, orexin promotes and stabilizes wakefulness and reduces REM sleep via orexinergic neurons in the hypothalamus. To further understand the role of orexin in AD pathology [31], Roh et al. in our group tested whether orexin release or secondary changes in the sleep-and-wake-cycle via orexin modulation affected Aβ pathology [34]. It was shown that APP/PS1 mice lacking orexin (APP/PS1/OR−/−) had a marked decrease in the amount of Aβ pathology in the brain and an increase in sleep time compared with APP/PS1 mice expressing orexin. Stereotaxic injection of orexin into the hippocampus of APP/PS1 mice did not alter the total amount of sleep/wakefulness or the amount of Aβ pathology. However, sleep deprivation or increasing wakefulness by rescue of orexin expression in orexinergic neurons in APP/PS1/ OR−/− mice increased the amount of Aβ pathology in the brain. Based on these results, it was concluded that wakefulness and sleep deprivation concurrently affect Aβ production and deposition, rather than local orexinergic signaling in the hippocampus [34]. All of the above studies suggest that sleep is a potent modulator of Aβ both in the short term for monomeric Aβ, and in the long term for Aβ deposition and amyloid pathology progression [31].

Diurnal fluctuation of ISF Aβ levels in the hippocampus of mice and CSF Aβ levels in human subjects. a ISF human Aβ levels expressed as a percentage of basal ISF Aβ levels over six light−dark periods in Tg2576 mice (n = 8). ISF human Aβ in Tg2576 mice has a 24 h diurnal fluctuation. b Mean ISF Aβ levels were 24.4% higher (***P < 0.0001, n = 8) during dark versus light periods. c Acute sleep deprivation (SD) alters ISF Aβ diurnal rhythm. Mice underwent acute SD (gray dashed line) for 6 h at the beginning of the light period. d Mean ISF Aβ levels during SD were 16.8% higher compared to those during the light period 24 h earlier (*P = 0.05, n = 8). e APPswe/PS1dE9 mice after chronic sleep restriction for 21 days showed significantly greater Aβ plaque deposition in multiple subregions of the cortex compared to age-matched control mice (**P < 0.0008, *P < 0.008, n = 9−11 mice per group). Representative photomicrographs of Aβ plaques are shown in f control and g sleep-restricted olfactory bulb, h control and i sleep-restricted piriform cortex, and j control and k sleep-restricted entorhinal cortex. l CSF Aβ1−40 levels from human subjects expressed as a percentage of basal CSF Aβ1−40 levels over 33 h (n = 10). Mean peak CSF Aβ1−40 levels (black bar) at 1900−2100 hours were 27.6% higher than mean CSF Aβ levels (gray bar) at 0900−1100 hours. Data shown are the means ± SEM. Adapted by permission from AAAS: Science [31]
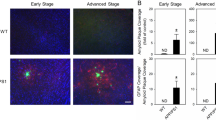
In addition to the sleep/wake cycle regulating Aβ levels, amyloid-precursor protein (APP)/Aβ-depositing mice demonstrate sleep changes. Changes in sleep−wake have been reported in Aβ depositing mice such as APP/PS1, 5xFAD, PDAPP, TgCRND8, and Tg2576 mice [40, 169,170,171,172,173]. These studies have reported that APP/Aβ overproduction model mice show increases in time awake and decreases in both NREM and REM sleep, resembling in some ways sleep changes seen in AD patients. Specifically, APP transgenic mice have increased sleep fragmentation, increased latency to REM sleep, and decreased occurrence of REM sleep bouts. Our group reported that the sleep−wake cycle significantly deteriorated and diurnal fluctuation of ISF Aβ disappeared after amyloid accumulation in the brain of the APPswe/PS1dE9 mouse [40]. To investigate whether Aβ plaque formation is a cause for changes in sleep amount and quality, and diurnal fluctuation of ISF Aβ, Roh et al. vaccinated APP/PS1 mice with subcutaneous injections of synthetic Aβ1–42 starting at 1.5 months. They found that elimination of Aβ deposits in the mouse brain normalized the sleep−wake cycle and diurnal fluctuation of ISF Aβ (Fig. 3) [40]. These studies show the important effects of APP/Aβ on sleep in AD, but do not address the presence of other pathological characteristics of AD such as tau aggregation. How Aβ induces sleep-changes requires further study, but is likely the result of multiple mechanisms. The sleep changes in AD could be due to the effects of Aβ pathology on neuronal populations in multiple cortical regions or in specific nuclei that regulate sleep and wake. For example, in the brain of mouse models of AD or humans, amyloid plaques are found throughout the hypothalamus, which has many sleep regulating regions such as the ventrolateral preoptic area (VLPO), an important sleep-promoting region of the brain. Amyloid pathology in this region is one possible explanation for decreased sleep in AD [78, 174]. Amyloid pathology is also observed in the periaqueductal gray matter (PAG), a dopaminergic wake-active area, in 81% of AD cases [175]. Sleep changes in AD could also be the result of neuronal circuit dysfunction due to amyloid pathology, such as impairment of slow oscillations and coherence in the neocortex, thalamus, and hippocampus [176].

Aβ42-vaccination normalized sleep−wake patterns, diurnal fluctuation of ISF Aβ, and Aβ plaque deposition in the hippocampus and striatum in APPswe/PS1δE9 mice. a, g Sleep−wake cycles in 9-month-old PBS-treated (a) and Aβ42-immunized (g) APPswe/PS1δE9 mice for 2 days shown as minutes awake per hour. d, j Comparison of minutes awake per hour between the dark and the light periods in each group. b, h Diurnal rhythms of ISF Aβ in the hippocampus of 9-month-old PBS-treated (b) and Aβ42-immunized (h) APPswe/PS1δE9 mice for 2 days. e, k Comparison of percent average of absolute values of ISF Aβ in the hippocampus between the dark and the light periods. c, i Diurnal rhythms of ISF Aβ in the striatum of 9-month-old PBS-vaccinated (c) and Aβ42-vaccinated (i) APPswe/PS1δE9 mice for 2 days. f, l Comparison of percent average of absolute values of ISF Aβ in the striatum between the dark and the light periods. m−p Representative brain sections of the hippocampus (m and o) and striatum (n and p) of mice from each group stained with Aβ antibody. q and r Amount of Aβ deposition in the PBS-treated mice and Aβ42-vaccinated mice are shown with amount of Aβ deposition in 6- and 9-month-old APPswe/PS1δE9 mice in the hippocampus (q) and striatum (r), N = 5−6 in each group; two-tailed t test.; *P < 0.05; ***P < 0.001; data shown are the means ± SEM. Adapted by permission from AAAS: Sci Transl Med [40]
As shown above, the relationship between sleep−wake disruption and Aβ deposition is most likely bidirectional. Changes in sleep−wake regulation can accelerate Aβ accumulation, and Aβ deposits conversely lead to sleep disturbance in humans. Consistent with this, a murine model of Aβ amyloidosis showed that significant deterioration of sleep amount and quality closely followed the emergence of Aβ aggregates, and strongly correlated with the progression of Aβ pathology, while the prevention of Aβ accumulation blocked these changes in sleep.
Sleep and Tau
The hyperphosphorylation and intracellular aggregation of tau protein is the second pathological hallmark of AD. Total and phosphorylated tau in human CSF strongly correlates with cognitive decline in preclinical and clinical AD [177, 178]. Also, increases of tau in CSF in preclinical AD is a strong predictor of cognitive decline in AD [179]. Tau phosphorylation and aggregation have been observed in the brainstem, specifically the locus coeruleus (LC), in young adults (70% of 20–30 years old) [142,143,144]. Tau then spreads to other connected regions such as the medial parabrachial nucleus (mPB), the dorsal raphe (DR), and the periaqueductal gray matter (PAG), regions that promote wake arousal, before amyloid is detected [78, 142, 143, 180]. Pre-tangle tau pathology is also found in the hypothalamic tuberomammilary nucleus (TMN), lateral hypothalamus (LH), and the basal forebrain (BF). All of these regions are part of the ascending arousal system and have tau hyperphosphorylation prior to tau or amyloid pathology in the cortex [78]. However, the VLPO in the hypothalamus, which links to and inhibits the ascending arousal system during sleep, does not appear to contain significant NFT pathology, even in AD patients [174]. Subsequently, Lim et al. suggested that neuronal loss due to minor NFT pathology in the VLPO and intermediate nucleus in the hypothalamus might explain sleep fragmentation in normal older adults and AD patients [181]. Sleep dysregulation during normal aging may be affected by abnormal tau phosphorylation in the above brain regions of the ascending arousal system in the absence of Aβ pathology [142, 143, 180]. In AD, the above arousal brain regions, as well as the pedunculopontine tegmental nucleus (PPT) and laterodorsal tegmental nucleus (LDT), have been shown to have robust NFT pathology [182].
Mice with tau pathology have been shown to have sleep disruptions as well. A human tau and APP knock-in mouse (PLB1, carrying a single copy each of mutant human APP and tau transgenes and overexpressing PS1) exhibits increased wake bout duration and decreased REM and NREM sleep bout duration [183, 184]. However, the PLB1 mouse model has the APP knock-in mutation, so the effect of tau pathology alone on sleep is unknown. Holth et al. in our group have reported that a mouse model of tauopathy, the P301S mouse, has decreased REM sleep at 9 months of age and decreased REM and NREM sleep and increased wakefulness at 11 months. The decreased REM and NREM sleep correlated with increased tau pathology in the sublaterodorsal area and parafacial zone in the brainstem [85]. This work suggests that tau pathology alone is sufficient to induce sleep changes in animal models.
To better understand the effects of neuronal activity on tau release, levels of ISF tau have been studied following modulation by excitatory neuronal activity. Yamada et al. in our group reported that increasing excitatory neuronal activity significantly increased ISF tau levels within hours, as measured by in vivo microdialysis in wild-type mice [185]. Similar findings were noted by Pooler et al. in vitro [186]. These studies suggest the possibility that if there is an increase in neuronal/synaptic activity during wakefulness vs. during sleep, the sleep−wake cycle may regulate extracellular tau levels. Ju et al. in our group found that worse in-home sleep quality in the six nights preceding in-lab sleep disruption was significantly associated with higher CSF tau levels in humans [187]. Recently, Lucey et al. in our group reported that tau pathology is associated with reduced NREM slow wave activity (SWA) in cognitively normal and very mildly cognitively impaired individuals [77]. They found that older people (60 years of age or older) with higher tau accumulation as measured by tau PET, or estimated by the tau/Aβ42 ratio in CSF, have decreased SWS power in the 1–4 Hz range. The participants were given a portable EEG monitor to measure their brain waves during sleep and actigraphy that tracked their body movements all day, at home over a normal week. The 38 participants were measured for Aβ and tau pathology through PET brain scans and 104 people underwent lumbar punctures to provide CSF for the further analysis. Twenty-seven participants did both. The study found that increased tau pathology was associated with decreased delta power in the 1–4 Hz range during SWS. It was previously known that tau pathology was significantly associated with sleep amount and quality in mouse models, and human studies have shown that decreased NREM sleep slow-wave activity (SWA) is associated with Aβ deposition [62, 188]. Importantly, this study suggests that decreased slow-wave sleep also occurs with tau accumulation in people who were either cognitively normal or very mildly impaired, meaning that reduced NREM SWA may be a biomarker for the transition between normal individuals and those soon to become cognitively impaired. Winer et al. have recently reported that poor sleep history in middle age (40–50 years) is a potential early biomarker of tau and Aβ burden developing later in life [189]. They found that participants who had sleep problems in their 40 and 50s had a greater Aβ burden in their brains later in life as measured by amyloid PET scans. Interestingly, participants who experienced sleep disruption in their 50 and 60s had more tau tangles. Impaired NREM sleep spindles-slow oscillations (SOs) coupling (synchronization of slow brain waves throughout the cortex of the sleeping brain) was associated with greater tau accumulation in the medial temporal lobe (MTL). However, NREM sleep spindles-SOs coupling was not associated with Aβ burden [189]. Recently, Kam et al. also evaluated potential relationships between levels of CSF Aβ42, P-tau, and total tau with sleep spindle density during N2 sleep and other biophysical properties of sleep spindles in cognitively normal elderly individuals [190]. They showed that sleep spindle density during N2 sleep was negatively correlated with the level of three AD biomarkers. CSF total tau levels were most significantly associated with the reduction of spindle density and duration [190]. According to this paper, a decrease in sleep spindle activity could be associated with tau pathology. For example, they propose that a decrease in sleep spindles during N2 sleep may represent early dysfunction related to the spread of tau pathology to certain sleep/wake regulating centers suggesting that changes in sleep architecture might be an early measurable indicator of tau pathology [142, 190]. As discussed before, sleep spindles are strongly associated with sleep-dependent memory consolidation and cognition. Therefore, the decline in memory function and cognition in the elderly may be related to the reduction of sleep spindles and NREM sleep spindle-SOs coupling following brain volume loss linked with progression of tau pathology [190]. Crucially, in terms of the sleep/wake cycle influencing tau pathogenesis, Holth et al. in our group recently reported that the sleep−wake cycle regulates brain ISF tau in mice and CSF tau in humans [41]. They tested whether ISF/CSF tau and tau seeding and spreading are affected by the sleep-wake cycle and SD. Mouse ISF tau levels fluctuated with sleep and wake, with increased tau during the night (when mice are more awake) compared to the day (when they sleep more). Sleep deprivation also increased ISF tau levels and the spread of pathological forms of tau in the brain. Mouse ISF tau was increased ~ 90% during dark (wake period) compared to light and ~100% during SD compared to normal sleep control. Specifically, the results showed ~40% increase in ISF tau and ~ 90% increase in lactate in mice in which there was chemogenetically driven wakefulness by clozapine-N-oxide (CNO). To understand the effect of sleep on tau seeding and spreading, they injected recombinant P301S human tau fibrils unilaterally into the hippocampus of 8–9-week-old P301S male mice before the onset of tau pathology. Under chronic SD, 20 h daily for 28 days, tau pathology spread from the hippocampus to the LC and the entorhinal cortex, which was significantly increased with SD. Although Aβ deposition is important in initiating AD, the actual damage in the brain is linked to tau accumulation. The findings from this study suggest that real-life factors such as sleep might affect the rate at which tau pathology spreads through the brain (Fig. 4). As the authors suggested in their paper, “the acute increases in monomeric ISF tau through wakefulness and sleep deprivation may play a normal role in cell signaling, whereas increased release of pathological species may play a role in seeding, spreading, and neurodegeneration” [41]. For future studies, it will be important to identify the different effects of acute and chronic tau increases in ISF, CSF, and in different brain regions on AD progression.

The sleep−wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. a ISF tau exhibits diurnal fluctuation and increases following manual sleep deprivation (SD) but not in the presence of tetrodotoxin (TTX), which attenuates neuronal activity. Manual SD and TTX infusion occurred from 0900 to 1500 hours (shaded), control animals were undisturbed. b Average ISF tau is significantly increased during dark (wake) compared to light (sleep) in control animals, demonstrating diurnal fluctuation (n = 8, paired t test). c Average ISF tau (normalized to baseline) during SD (0900–1500 hours) was significantly increased in sleep-deprived mice compared to controls or mice with SD in the presence of TTX. d CSF tau levels are increased by SD in human subjects (n = 6). e CSF tau levels during SD are significantly increased by 51.5% compared to undisturbed sleep (n = 6). f Total CSF Aβ is significantly correlated with CSF tau in control and SD conditions during the SD time period (n = 6, Pearson’s correlation). g, h Ipsilateral hippocampal AT8 phosphorylated-tau (p-tau) staining in grid control and chronic SD P301S male mice with unilateral hippocampal tau fibril injection. i−l AT8 staining in the LC of SD and control hippocampal-seeded P301S mice (scale bar (h) and (l), 125 μm). m SD does not alter p-tau staining in the ipsilateral hippocampus (n = 14–16). n The ipsilateral LC/hippocampus AT8 ratio is increased in SD mice. o p-Tau is significantly increased in the ipsilateral LC and p trended toward an increase in the contralateral LC of SD compared to control animals. *P < 0.05; **P < 0.01; Data shown are the means ± SEM. Adapted by permission from AAAS: Science [41]
As shown above, many associations between tau pathology and sleep−wake regulation have been reported. The recent studies suggest that there may be a bidirectional relationship between sleep−wake disruption and tau pathology, similar to the bidirectional relationship between sleep−wake disruption and Aβ. Having a well-optimized sleep−wake cycle should be an important treatment target to test in the prevention of AD. Whether improving sleep quality would reduce tau pathology or slow tau pathogenesis and cognition impairment remains to be determined.
Obstructive sleep apnea (OSA) and AD
Obstructive sleep apnea (OSA) is a respiratory disorder characterized by partial or complete cessation or closure of the upper airway during sleep [191]. It causes intermittent hypoxia and sleep disturbance and is estimated to affect 1–4% of middle-aged adults [192] and 24–30% of elderly people [193]. OSA is intrinsically associated with metabolic disorders like diabetes and obesity, intermittent hypoxia and oxidative stress, and cardiovascular comorbidities [194,195,196,197,198]. With these factors, OSA is also considered to be one of the risk factors for dementia and cognitive decline [199,200,201], suggesting that OSA may increase the risk for AD [202]. Osorio et al. reported that individuals with OSA might show signs of MCI and AD at a younger age [201]. Conversely, treating OSA has been shown to delay the age of onset of MCI [201] and to improve cognitive function in AD [203, 204]. Upregulation of oxidative stress during OSA plays an important pathogenic role in hypoxia-induced cerebral dysfunction [205]. Hypoxia is a crucial environmental factor that can trigger neuronal degeneration and axonal dysfunction in both cortex and brainstem. Several studies have shown that cerebral amyloidogenesis and tau phosphorylation are triggered by hypoxia that occurs during OSA [206, 207]. Mice subjected to hypoxic conditions demonstrated upregulation in cerebral amyloid plaque formation and tau phosphorylation, as well as memory impairment [207]. As discussed above, sleep deprivation increases Aβ levels in ISF and CSF through increased release by active neurons; this could also be affected by OSA [33, 166]. Ju et al. in our group have reported that OSA acutely decreases extracellular central nervous system-derived proteins in the CSF of middle-aged people without AD pathology. This suggests that OSA acutely induces slower clearance of soluble proteins from ISF to CSF [208]. Recently, Liguori et al. have reported that subjective cognitive impairment (SCI) patients affected by OSA showed lower CSF Aβ42 concentrations and higher total tau/Aβ42 ratio compared to SCI controls without OSA or OSA patients treated with continuous positive airway pressure (CPAP) (OSA-CPAP) [209]. They also reported that OSA patients showed reduced sleep quality and duration and lower performance on memory, intelligence, and executive tests than SCI controls or OSA-CPAP patients. Furthermore, there was a significant relationship between higher CSF tau protein levels and sleep impairment in the OSA patients [132]. These findings may be due to the fact that a greater percentage of the individual subjects being studied with OSA had concurrent AD pathology. A recent paper published by Ju et al. reported that treated-OSA patients without AD pathology showed increased SWA, and the increased SWA was significantly associated with lower Aβ levels in CSF after treatment [210]. Greater alleviation of OSA was significantly associated with greater decreases in Aβ, suggesting that OSA treatment may affect both Aβ production and clearance.
Other factors relevant to AD and sleep
Apolipoprotein E (APOE)
Apolipoprotein E (APOE) is mainly produced in the brain by astrocytes and is a major cholesterol carrier that transports lipids between cells [44, 211]. In late-onset AD, which accounts for >99% of cases, the most important genetic risk factor is APOE genotype. APOE appears to influence AD pathogenesis at least in part by affecting Aβ clearance and aggregation [42, 44]. In humans, the APOE gene on chromosome 19 exists as three different polymorphic alleles (ε2, ε3 and ε4). APOE ε3 is the most common (77%) and ε2 the least (8%) common allele in the general population [212]. Roses and colleagues reported that the APOE ε4 allele is the strongest genetic risk factor for late-onset AD. The frequency of the APOE ε4 allele is about 15% in the general population, but is disproportionally represented in up to 50% of the APOE alleles in AD patients [212]. Individuals with one APOE ε4 allele have 3−4 times greater AD risk with an earlier age- of-disease-onset than those without APOE ε4 [44, 213, 214]. Aβ deposition is more abundant in individuals with APOE ε4 than in noncarriers, suggesting that APOE ε4 promotes Aβ aggregation [215,216,217]. Conversely, APOE ɛ2 is associated with decreased risk, suggesting a protective role. The effect of APOE genotype on risk of AD appears to be in part mediated by differential effects of apoE on Aβ accumulation in the brain and its vasculature [217]. Recently, our group reported that APOE ε4 also strongly exacerbates tau-dependent neurodegeneration in a mouse model of tauopathy (P301S) [45]. This suggests that apoE significantly affects tau-mediated neurodegeneration independently of Aβ pathology.
Kadotani et al. reported that APOE ε4 is associated with sleep apnea. A significant portion of individuals with APOE ε4 show SDB [218]. Gottlieb et al. suggested that individuals carrying the APOE ε4 have an increased risk of developing OSA [219]. Moreover, OSA patients carrying the APOE ε4 allele have demonstrated an increased risk of impaired spatial working memory [220]. However, this relationship between APOE ε4 and OSA is controversial. Using a meta-analysis, several studies showed that there was not a strong association between APOE alleles and OSA susceptibility [221, 222]. Interestingly, Drogos et al. reported that sleep quality measured by polysomnography (PSG) and actigraphy in individuals with APOE ε4 alleles was significantly worse than sleep quality of those without APOE ε4 [223]. They suggest that the influence of the APOE ε4 allele on sleep quality may lead to sleep disturbances in individuals at increased risk for dementia. Lim et al. have reported that better sleep consolidation in nearly 700 older persons without dementia substantially attenuated the negative impact of the APOE ε4 allele on incident AD risk [224]. A recent study showed that disrupted sleep was more frequent in males than in females and in carriers of the APOE ε4 allele than in noncarriers [46]. More detailed studies are needed to understand how APOE alleles affect sleep disturbance in AD patients as well as in AD model mice.
Microglia
Microglia are resident immune cells in the central nervous system (CNS) that stem from myeloid progenitor cells [225]. In the healthy state, microglia are highly dynamic, scanning the parenchyma of the brain continuously and responding immediately by rapidly extending and retracting their highly branched processes [226, 227]. Many studies have reported that microglia are activated around amyloid plaques both in humans and in transgenic mouse models that develop amyloid deposition [228,229,230,231,232]. Recent genetic studies have reported that genetic variants in the triggering receptor expressed on myeloid cells 2 (TREM2) gene strongly increase the risk of developing AD, confirming the role of microglia in AD pathogenesis [233]. Previous studies have suggested that loss of TREM2 function exacerbates Aβ plaque-related toxicity, including accumulation of p-tau and neuritic dystrophy around Aβ plaques [234, 235]. Recently, however, our group has reported that TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy (P301S) [236]. This suggests that TREM2 function may protect against amyloid-dependent toxicity early in AD, but subsequent TREM2-dependent microgliosis may lead to exacerbation of injury following the onset of tau pathology [236].
Recent studies have reported that sleep loss affects microglial morphology, phagocytosis, and Aβ clearance [48, 237]. It was found that impairment in the sleep−wake cycle reduces microglial Aβ clearance [237]. In addition, chronic but not acute SD increases microglial phagocytosis of synaptic structures in the mouse frontal cortex without neuroinflammation [48]. Enhanced microglial phagocytosis of synaptic structures linked to chronic sleep disturbance might be exacerbated in AD. Recently, Sprecher et al. reported that poor sleep is associated with CSF biomarkers of amyloid pathology, such as Aβ42, Aβ40, t-tau, p-tau, and chitinase-3-like protein 1 (YKL-40), a biomarker for neuroinflammation/astroglial activation in neurological disease [238]. They showed that poor sleep is associated with lower CSF Aβ42/Aβ40 and higher t-tau/Aβ42, p-tau/Aβ42, and YKL-40/Aβ42. More detailed studies are needed to identify how sleep affects microglial function in AD.
Conclusion
Recent studies in mouse models that develop AD-like pathology and in humans with AD pathology provide evidence for a bidirectional relationship between sleep and AD. Increased wakefulness and decreased NREM sleep acutely increase extracellular Aβ and tau and chronically appear to accelerate Aβ and tau pathology in animal models. Also, sleep disturbance affects the acute, dynamic changes in both Aβ and tau, which may lead to exacerbation of AD pathology. Chronic sleep disturbances can facilitate Aβ plaque formation and tau spreading in animal models, while acute sleep disturbance is more closely linked with transient changes in Aβ and tau levels in the ISF or CSF in animals and humans. Conversely, AD pathology is associated with decreased sleep quality and NREM SWS power (Fig. 5). Currently, the relationships between the sleep−wake cycle and the development of Aβ and tau pathology, as well as other aspects of neurodegeneration, are just beginning to be understood. Further studies are needed to determine whether the dynamics of Aβ and tau are regulated by similar mechanisms of production and clearance and by similar components of sleep.

Model of interactions between sleep and Alzheimer disease (AD). Bidirectional relationships between Aβ and tau and the sleep/wake cycle. The interrelationships and positive feedback loops between sleep, Aβ, tau, AD, and related factors are schematized. OSA indicates obstructive sleep apnea
Future research directions
The relationship between sleep and AD has important implications for the optimal clinical management and potential treatment strategies in patients with AD. As we discussed herein, AD pathology accumulates for 15−20 years before individuals begin to have cognitive dysfunction and functional impairment. Several studies suggest that delaying onset of AD by just 5 years may reduce the treatment costs for AD by 40% [239]. There is currently no proven disease modifying therapy for AD; only symptomatic treatments are available. Several recent clinical trials have failed in the development of possible new treatments against AD [240, 241]. One potential cause for the lack of disease modifying treatments to date is that treatments were started too late in the progression of AD for the targets, such as Aβ. Early changes in sleep, along with biomarkers, may be useful in identifying individuals that are at risk for developing dementia due to AD and enrolling them in preventative clinical trials. Moreover, large-scale longitudinal studies evaluating the efficacy of sleep interventions to influence AD biomarkers and AD risk and progression are needed. As the animal model data show, increased wakefulness and decreased sleep leads to enhanced buildup of AD pathology. Large-scale studies are needed to determine if improving and/or increasing NREM sleep, as well as treating sleep disorders, decreases the accumulation of AD pathology and ultimately cognitive decline due to AD. Other important things to address include the blood brain barrier (BBB), disruption of which may be a possible mechanism by which sleep disruption promotes AD progression [242]. According to several studies, chronic sleep restriction induces the breakdown of the BBB in rodents [243, 244] and selective REM sleep deprivation also leads to impairment of BBB integrity [243]. Impairment of BBB integrity has been observed in AD [245] and can be detected before onset of cognitive decline during aging [242] and even before significant changes in CSF Aβ and tau levels [246]. Further studies are needed to clarify the interaction between BBB integrity and AD and the effect of sleep disruption on the BBB integrity in the preclinical stage, as a candidate biomarker for AD.
We have just reported that tau fluctuates with the sleep/wake cycle and SD increases levels of tau in mouse brain ISF and human CSF and accelerates the spread of tau protein through the brain [41]. It would be of interest to determine if increasing SWS can decrease tau seeding and spreading. More detailed studies are needed to identify the effects of increasing SWS acutely and chronically on tau and other neurodegenerative disease pathways including assessing effects on the innate immune response. Lastly, given the data that tau pathology and CSF tau/Aβ42 are significantly related to quantitative changes in SWA during NREM sleep, follow-up studies are needed to identify whether these quantitative changes in SWA can be utilized as biomarkers that predict prognosis of AD and respond to treatments. Finally, several studies suggest that treatments can improve sleep quality, such as bright light therapy and exercise training in middle-aged and older adults, and OSA therapy with continuous positive airway pressure (CPAP) in patients with OSA [247,248,249,250,251]. Therefore, more detailed clinical studies are needed to identify the effects of increasing the amplitude of the rest-activity cycle with bright light therapy [247, 248] and/or physical activity therapy [249] in individuals with preclinical AD and OSA treatments in patients with OSA and AD [250]. Recently, Perrault et al. [252] and Kompotis et al. [253] reported neuronal mechanisms underlying the beneficial effect of rocking for sleep in humans and mice. Originally, Bayer et al. in the same group reported sleeping on a rocking bed not only accelerated sleep onset, but also enhanced the prevalence of two cardinal sleep signatures (slow oscillations and sleep spindles) measured by scalp EEG [254]. In accordance with previous findings, Perrault et al. also confirmed that a rocking bed with 0.25 Hz entrained a boost in spindle activity resulting in improved memory consolidation in human participants [252]. Similar to humans, Kompotis et al. reported that the rocking with 1 Hz had a remarkable impact on sleep architecture in mice. To improve sleep quality in preclinical AD or in patients with symptomatic AD, more detailed clinical and animal studies utilizing a rocking strategy will be needed. Examination of treatment strategies that aim to increase sleep quality, especially SWS, may be important in determining whether they will be valuable in managing AD symptoms and signs as well as in slowing disease progression.
Funding and disclosure
This review was supported by a grant from NIH P01NS074969 (DMH). DMH reports being a co-founder of C2N Diagnostics, LLC; being on the scientific advisory board of C2N Diagnostics, Denali, and Gentech; and being a consultant for AbbVie and Idorsia. CW declares no competing interests.
References
McEwen BS. Sleep deprivation as a neurobiologic and physiologic stressor: Allostasis and allostatic load. Metabolism. 2006;55:S20–3.
Robles TF, Carroll JE. Restorative biological processes and health. Soc Pers Psychol Compass. 2011;5:518–37.
Chen X, Redline S, Shields AE, Williams DR, Williams MA. Associations of allostatic load with sleep apnea, insomnia, short sleep duration, and other sleep disturbances: findings from the National Health and Nutrition Examination Survey 2005 to 2008. Ann Epidemiol. 2014;24:612–9.
Mullington JM, Haack M, Toth M, Serrador JM, Meier-Ewert HK. Cardiovascular, inflammatory, and metabolic consequences of sleep deprivation. Prog Cardiovasc Dis. 2009;51:294–302.
Tobaldini E, Costantino G, Solbiati M, Cogliati C, Kara T, Nobili L, et al. Sleep, sleep deprivation, autonomic nervous system and cardiovascular diseases. Neurosci Biobehav Rev. 2017;74:321–29.
Van Cauter E, Leproult R, Plat L. Age-related changes in slow wave sleep and REM sleep and relationship with growth hormone and cortisol levels in healthy men. JAMA. 2000;284:861–8.
Cooke JR, Ancoli-Israel S. Normal and abnormal sleep in the elderly. Handb Clin Neurol. 2011;98:653–65.
Ohayon MM. Epidemiology of insomnia: what we know and what we still need to learn. Sleep Med Rev. 2002;6:97–111.
Kang DW, Lee CU, Lim HK. Role of sleep disturbance in the trajectory of Alzheimer’s disease. Clin Psychopharmacol Neurosci. 2017;15:89–99.
Ohayon MM, Carskadon MA, Guilleminault C, Vitiello MV. Meta-analysis of quantitative sleep parameters from childhood to old age in healthy individuals: developing normative sleep values across the human lifespan. Sleep. 2004;27:1255–73.
Nebes RD, Buysse DJ, Halligan EM, Houck PR, Monk TH. Self-reported sleep quality predicts poor cognitive performance in healthy older adults. J Gerontol B Psychol Sci Soc Sci. 2009;64:180–7.
Xu L, Jiang CQ, Lam TH, Liu B, Jin YL, Zhu T, et al. Short or long sleep duration is associated with memory impairment in older Chinese: the Guangzhou Biobank Cohort Study. Sleep. 2011;34:575–80.
Bombois S, Derambure P, Pasquier F, Monaca C. Sleep disorders in aging and dementia. J Nutr Health Aging. 2010;14:212–7.
Van Erum J, Van Dam D, De Deyn PP. Sleep and Alzheimer's disease: a pivotal role for the suprachiasmatic nucleus. Sleep Med Rev. 2018;40:17–27.
Qiu C, Kivipelto M, von Strauss E. Epidemiology of Alzheimer's disease: occurrence, determinants, and strategies toward intervention. Dialog- Clin Neurosci. 2009;11:111–28.
Alzheimer's A. Alzheimer's disease facts and figures. Alzheimers Dement. 2016;12:459–509. 2016
Masters CL, Bateman R, Blennow K, Rowe CC, Sperling RA, Cummings JL. Alzheimer's disease. Nat Rev Dis Prim. 2015;1:15056.
Holtzman DM, Morris JC, Goate AM. Alzheimer's disease: the challenge of the second century. Sci Transl Med. 2011;3:77sr1.
Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT. Inflammation as a central mechanism in Alzheimer's disease. Alzheimers Dement. 2018;4:575–90.
Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med. 2016;8:595–608.
De Strooper B, Karran E. The cellular phase of Alzheimer's Disease. Cell. 2016;164:603–15.
O'Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer's disease. Annu Rev Neurosci. 2011;34:185–204.
Medeiros R, Baglietto-Vargas D, LaFerla FM. The role of tau in Alzheimer's disease and related disorders. CNS Neurosci Ther. 2011;17:514–24.
Moran M, Lynch CA, Walsh C, Coen R, Coakley D, Lawlor BA. Sleep disturbance in mild to moderate Alzheimer's disease. Sleep Med. 2005;6:347–52.
Ju YE, Lucey BP, Holtzman DM. Sleep and Alzheimer disease pathology—a bidirectional relationship. Nat Rev Neurol. 2014;10:115–9.
Wennberg AMV, Wu MN, Rosenberg PB, Spira AP. Sleep disturbance, cognitive decline, and dementia: a review. Semin Neurol. 2017;37:395–406.
Rongve A, Boeve BF, Aarsland D. Frequency and correlates of caregiver-reported sleep disturbances in a sample of persons with early dementia. J Am Geriatr Soc. 2010;58:480–6.
Bianchetti A, Scuratti A, Zanetti O, Binetti G, Frisoni GB, Magni E, et al. Predictors of mortality and institutionalization in Alzheimer disease patients 1 year after discharge from an Alzheimer dementia unit. Dementia. 1995;6:108–12.
Guarnieri B, Adorni F, Musicco M, Appollonio I, Bonanni E, Caffarra P, et al. Prevalence of sleep disturbances in mild cognitive impairment and dementing disorders: a multicenter Italian clinical cross-sectional study on 431 patients. Dement Geriatr Cogn Disord. 2012;33:50–8.
Ju YE, McLeland JS, Toedebusch CD, Xiong C, Fagan AM, Duntley SP, et al. Sleep quality and preclinical Alzheimer disease. JAMA Neurol. 2013;70:587–93.
Kang JE, Lim MM, Bateman RJ, Lee JJ, Smyth LP, Cirrito JR, et al. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science. 2009;326:1005–7.
Huang Y, Potter R, Sigurdson W, Santacruz A, Shih S, Ju YE, et al. Effects of age and amyloid deposition on Abeta dynamics in the human central nervous system. Arch Neurol. 2012;69:51–8.
Lucey BP, Hicks TJ, McLeland JS, Toedebusch CD, Boyd J, Elbert DL, et al. Effect of sleep on overnight cerebrospinal fluid amyloid beta kinetics. Ann Neurol. 2018;83:197–204.
Roh JH, Jiang H, Finn MB, Stewart FR, Mahan TE, Cirrito JR, et al. Potential role of orexin and sleep modulation in the pathogenesis of Alzheimer's disease. J Exp Med. 2014;211:2487–96.
Kilduff TS, Peyron C. The hypocretin/orexin ligand-receptor system: implications for sleep and sleep disorders. Trends Neurosci. 2000;23:359–65.
Peyron C, Tighe DK, van den Pol AN, de Lecea L, Heller HC, Sutcliffe JG, et al. Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci. 1998;18:9996–10015.
Liguori C, Romigi A, Nuccetelli M, Zannino S, Sancesario G, Martorana A, et al. Orexinergic system dysregulation, sleep impairment, and cognitive decline in Alzheimer disease. JAMA Neurol. 2014;71:1498–505.
Liguori C, Placidi F, Albanese M, Nuccetelli M, Izzi F, Marciani MG, et al. CSF beta-amyloid levels are altered in narcolepsy: a link with the inflammatory hypothesis? J Sleep Res. 2014;23:420–4.
Liguori C, Nuccetelli M, Izzi F, Sancesario G, Romigi A, Martorana A, et al. Rapid eye movement sleep disruption and sleep fragmentation are associated with increased orexin-A cerebrospinal-fluid levels in mild cognitive impairment due to Alzheimer's disease. Neurobiol Aging. 2016;40:120–26.
Roh JH, Huang Y, Bero AW, Kasten T, Stewart FR, Bateman RJ, et al. Disruption of the sleep-wake cycle and diurnal fluctuation of beta-amyloid in mice with Alzheimer's disease pathology. Sci Transl Med. 2012;4:150ra22.
Holth JK, Fritschi SK, Wang C, Pedersen NP, Cirrito JR, Mahan TE, et al. The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science. 2019;363:880–84.
Liu CC, Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9:106–18.
Hyman BT, Holtzman DM. Apolipoprotein E levels and Alzheimer risk. Ann Neurol. 2015;77:204–5.
Huynh TV, Davis AA, Ulrich JD, Holtzman DM. Apolipoprotein E and Alzheimer's disease: the influence of apolipoprotein E on amyloid-beta and other amyloidogenic proteins. J Lipid Res. 2017;58:824–36.
Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature. 2017;549:523–27.
Koo KYG, Schweizer TA, Fischer CE, Munoz DG. Abnormal sleep behaviours across the spectrum of Alzheimer's disease severity: influence of APOE genotypes and lewy bodies. Curr Alzheimer Res. 2019;16:243–50.
Hurtado-Alvarado G, Pavon L, Castillo-Garcia SA, Hernandez ME, Dominguez-Salazar E, Velazquez-Moctezuma J, et al. Sleep loss as a factor to induce cellular and molecular inflammatory variations. Clin Dev Immunol. 2013;2013:801341.
Bellesi M, de Vivo L, Chini M, Gilli F, Tononi G, Cirelli C. Sleep loss promotes astrocytic phagocytosis and microglial activation in mouse cerebral cortex. J Neurosci. 2017;37:5263–73.
Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer's disease. Lancet Neurol. 2015;14:388–405.
Petit D, Gagnon JF, Fantini ML, Ferini-Strambi L, Montplaisir J. Sleep and quantitative EEG in neurodegenerative disorders. J Psychosom Res. 2004;56:487–96.
Cox R, Hofman WF, Talamini LM. Involvement of spindles in memory consolidation is slow wave sleep-specific. Learn Mem. 2012;19:264–7.
Halasz P. K-complex, a reactive EEG graphoelement of NREM sleep: an old chap in a new garment. Sleep Med Rev. 2005;9:391–412.
McCarley RW. Neurobiology of REM and NREM sleep. Sleep Med. 2007;8:302–30.
Vazquez J, Baghdoyan HA. Basal forebrain acetylcholine release during REM sleep is significantly greater than during waking. Am J Physiol Regul Integr Comp Physiol. 2001;280:R598–601.
Vitiello MV. Recent advances in understanding sleep and sleep disturbances in older adults: growing older does not mean sleeping poorly. Curr Dir Psychol Sci. 2009;18:316–20.
Saper CB, Fuller PM, Pedersen NP, Lu J, Scammell TE. Sleep state switching. Neuron. 2010;68:1023–42.
Dement WC, Miles LE, Carskadon MA. "White paper" on sleep and aging. J Am Geriatr Soc. 1982;30:25–50.
Scullin MK, Bliwise DL. Sleep, cognition, and normal aging: integrating a half century of multidisciplinary research. Perspect Psychol Sci. 2015;10:97–137.
Wauquier A. Aging and changes in phasic events during sleep. Physiol Behav. 1993;54:803–6.
Landolt HP, Dijk DJ, Achermann P, Borbely AA. Effect of age on the sleep EEG: slow-wave activity and spindle frequency activity in young and middle-aged men. Brain Res. 1996;738:205–12.
Redline S, Kirchner HL, Quan SF, Gottlieb DJ, Kapur V, Newman A. The effects of age, sex, ethnicity, and sleep-disordered breathing on sleep architecture. Arch Intern Med. 2004;164:406–18.
Mander BA, Rao V, Lu B, Saletin JM, Lindquist JR, Ancoli-Israel S, et al. Prefrontal atrophy, disrupted NREM slow waves and impaired hippocampal-dependent memory in aging. Nat Neurosci. 2013;16:357–64.
Musiek ES, Xiong DD, Holtzman DM. Sleep, circadian rhythms, and the pathogenesis of Alzheimer disease. Exp Mol Med. 2015;47:e148.
Lim MM, Gerstner JR, Holtzman DM. The sleep-wake cycle and Alzheimer's disease: what do we know? Neurodegener Dis Manag. 2014;4:351–62.
Lyketsos CG, Lopez O, Jones B, Fitzpatrick AL, Breitner J, DeKosky S. Prevalence of neuropsychiatric symptoms in dementia and mild cognitive impairment: results from the cardiovascular health study. JAMA. 2002;288:1475–83.
Muangpaisan W, Intalapaporn S, Assantachai P. Neuropsychiatric symptoms in the community-based patients with mild cognitive impairment and the influence of demographic factors. Int J Geriatr Psychiatry. 2008;23:699–703.
Vitiello MV, Prinz PN, Williams DE, Frommlet MS, Ries RK. Sleep disturbances in patients with mild-stage Alzheimer's disease. J Gerontol. 1990;45:M131–8.
Roth T, Ancoli-Israel S. Daytime consequences and correlates of insomnia in the United States: results of the 1991 National Sleep Foundation Survey. II. Sleep. 1999;22:S354–8.
Schutte-Rodin S, Broch L, Buysse D, Dorsey C, Sateia M. Clinical guideline for the evaluation and management of chronic insomnia in adults. J Clin Sleep Med. 2008;4:487–504.
Bliwise DL, Tinklenberg J, Yesavage JA, Davies H, Pursley AM, Petta DE, et al. REM latency in Alzheimer's disease. Biol Psychiatry. 1989;25:320–8.
Bonanni E, Maestri M, Tognoni G, Fabbrini M, Nucciarone B, Manca ML, et al. Daytime sleepiness in mild and moderate Alzheimer's disease and its relationship with cognitive impairment. J Sleep Res. 2005;14:311–7.
Anderson KN, Bradley AJ. Sleep disturbance in mental health problems and neurodegenerative disease. Nat Sci Sleep. 2013;5:61–75.
Tractenberg RE, Singer CM, Kaye JA. Symptoms of sleep disturbance in persons with Alzheimer's disease and normal elderly. J Sleep Res. 2005;14:177–85.
Spira AP, Gamaldo AA, An Y, Wu MN, Simonsick EM, Bilgel M, et al. Self-reported sleep and beta-amyloid deposition in community-dwelling older adults. JAMA Neurol. 2013;70:1537–43.
Hahn EA, Wang HX, Andel R, Fratiglioni L. A change in sleep pattern may predict Alzheimer disease. Am J Geriatr Psychiatry. 2014;22:1262–71.
Lim AS, Kowgier M, Yu L, Buchman AS, Bennett DA. Sleep fragmentation and the risk of incident Alzheimer's disease and cognitive decline in older persons. Sleep. 2013;36:1027–32.
Lucey BP, McCullough A, Landsness EC, Toedebusch CD, McLeland JS, Zaza AM, et al. Reduced non-rapid eye movement sleep is associated with tau pathology in early Alzheimer's disease. Sci Transl Med. 2019;11:eaau6550.
Holth J, Patel T, Holtzman DM. Sleep in Alzheimer's disease—beyond amyloid. Neurobiol Sleep Circadian Rhythms. 2017;2:4–14.
Iraizoz I, Guijarro JL, Gonzalo LM, de Lacalle S. Neuropathological changes in the nucleus basalis correlate with clinical measures of dementia. Acta Neuropathol. 1999;98:186–96.
Brunnstrom H, Friberg N, Lindberg E, Englund E. Differential degeneration of the locus coeruleus in dementia subtypes. Clin Neuropathol. 2011;30:104–10.
Wilson RS, Nag S, Boyle PA, Hizel LP, Yu L, Buchman AS, et al. Neural reserve, neuronal density in the locus ceruleus, and cognitive decline. Neurology. 2013;80:1202–8.
Sanchez-Espinosa MP, Atienza M, Cantero JL. Sleep deficits in mild cognitive impairment are related to increased levels of plasma amyloid-beta and cortical thinning. Neuroimage. 2014;98:395–404.
Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, Delon MR. Alzheimer's disease and senile dementia: loss of neurons in the basal forebrain. Science. 1982;215:1237–9.
Baghdoyan HA, Rodrigo-Angulo ML, McCarley RW, Hobson JA. Site-specific enhancement and suppression of desynchronized sleep signs following cholinergic stimulation of three brainstem regions. Brain Res. 1984;306:39–52.
Holth JK, Mahan TE, Robinson GO, Rocha A, Holtzman DM. Altered sleep and EEG power in the P301S tau transgenic mouse model. Ann Clin Transl Neurol. 2017;4:180–90.
Brayet P, Petit D, Frauscher B, Gagnon JF, Gosselin N, Gagnon K, et al. Quantitative EEG of rapid-eye-movement sleep: a marker of amnestic mild cognitive impairment. Clin EEG Neurosci. 2016;47:134–41.
Rodrigues Brazete J, Gagnon JF, Postuma RB, Bertrand JA, Petit D, Montplaisir J. Electroencephalogram slowing predicts neurodegeneration in rapid eye movement sleep behavior disorder. Neurobiol Aging. 2016;37:74–81.
Galbiati A, Carli G, Hensley M, Ferini-Strambi L. REM sleep behavior disorder and Alzheimer's disease: definitely no relationship? J Alzheimers Dis. 2018;63:1–11.
Peever J, Luppi PH, Montplaisir J. Breakdown in REM sleep circuitry underlies REM sleep behavior disorder. Trends Neurosci. 2014;37:279–88.
Gagnon JF, Vendette M, Postuma RB, Desjardins C, Massicotte-Marquez J, Panisset M, et al. Mild cognitive impairment in rapid eye movement sleep behavior disorder and Parkinson's disease. Ann Neurol. 2009;66:39–47.
Liguori C. Orexin and Alzheimer's disease. Curr Top Behav Neurosci. 2017;33:305–22.
Chang HC, Guarente L. SIRT1 mediates central circadian control in the SCN by a mechanism that decays with aging. Cell. 2013;153:1448–60.
Witting W, Kwa IH, Eikelenboom P, Mirmiran M, Swaab DF. Alterations in the circadian rest-activity rhythm in aging and Alzheimer's disease. Biol Psychiatry. 1990;27:563–72.
Prinz PN, Peskind ER, Vitaliano PP, Raskind MA, Eisdorfer C, Zemcuznikov N, et al. Changes in the sleep and waking EEGs of nondemented and demented elderly subjects. J Am Geriatr Soc. 1982;30:86–93.
Coogan AN, Schutova B, Husung S, Furczyk K, Baune BT, Kropp P, et al. The circadian system in Alzheimer's disease: disturbances, mechanisms, and opportunities. Biol Psychiatry. 2013;74:333–9.
Hu K, Van Someren EJ, Shea SA, Scheer FA. Reduction of scale invariance of activity fluctuations with aging and Alzheimer's disease: involvement of the circadian pacemaker. Proc Natl Acad Sci USA. 2009;106:2490–4.
Harper DG, Stopa EG, Kuo-Leblanc V, McKee AC, Asayama K, Volicer L, et al. Dorsomedial SCN neuronal subpopulations subserve different functions in human dementia. Brain. 2008;131:1609–17.
Swaab DF, Fliers E, Partiman TS. The suprachiasmatic nucleus of the human brain in relation to sex, age and senile dementia. Brain Res. 1985;342:37–44.
Zhou JN, Hofman MA, Swaab DF. VIP neurons in the human SCN in relation to sex, age, and Alzheimer's disease. Neurobiol Aging. 1995;16:571–6.
Huynh TV, Holtzman DM. In search of an identity for amyloid plaques. Trends Neurosci. 2018;41:483–6.
Glenner GG, Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–90.
Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA. 1985;82:4245–9.
Tanzi RE, Gusella JF, Watkins PC, Bruns GA, St George-Hyslop P, Van Keuren ML, et al. Amyloid beta protein gene: cDNA, mRNA distribution, and genetic linkage near the Alzheimer locus. Science. 1987;235:880–4.
Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, et al. The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–6.
Head E, Lott IT, Wilcock DM, Lemere CA. Aging in Down syndrome and the development of Alzheimer's disease neuropathology. Curr Alzheimer Res. 2016;13:18–29.
Golde TE, Eckman CB, Younkin SG. Biochemical detection of Abeta isoforms: implications for pathogenesis, diagnosis, and treatment of Alzheimer's disease. Biochim Biophys Acta. 2000;1502:172–87.
Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–66.
Bekris LM, Yu CE, Bird TD, Tsuang DW. Genetics of Alzheimer disease. J Geriatr Psychiatry Neurol. 2010;23:213–27.
Theuns J, Del-Favero J, Dermaut B, van Duijn CM, Backhovens H, Van den Broeck MV, et al. Genetic variability in the regulatory region of presenilin 1 associated with risk for Alzheimer's disease and variable expression. Hum Mol Genet. 2000;9:325–31.
Citron M, Westaway D, Xia W, Carlson G, Diehl T, Levesque G, et al. Mutant presenilins of Alzheimer's disease increase production of 42-residue amyloid beta-protein in both transfected cells and transgenic mice. Nat Med. 1997;3:67–72.
Kumar-Singh S, Theuns J, Van Broeck B, Pirici D, Vennekens K, Corsmit E, et al. Mean age-of-onset of familial alzheimer disease caused by presenilin mutations correlates with both increased Abeta42 and decreased Abeta40. Hum Mutat. 2006;27:686–95.
Wolfe MS. Unlocking truths of gamma-secretase in Alzheimer's disease: what is the translational potential? Future Neurol. 2014;9:419–29.
Szaruga M, Veugelen S, Benurwar M, Lismont S, Sepulveda-Falla D, Lleo A, et al. Qualitative changes in human gamma-secretase underlie familial Alzheimer's disease. J Exp Med. 2015;212:2003–13.
Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med. 1996;2:864–70.
Hecimovic S, Wang J, Dolios G, Martinez M, Wang R, Goate AM. Mutations in APP have independent effects on Abeta and CTFgamma generation. Neurobiol Dis. 2004;17:205–18.
Gu L, Guo Z. Alzheimer's Abeta42 and Abeta40 peptides form interlaced amyloid fibrils. J Neurochem. 2013;126:305–11.
Oh ES, Savonenko AV, King JF, Fangmark Tucker SM, Rudow GL, Xu G, et al. Amyloid precursor protein increases cortical neuron size in transgenic mice. Neurobiol Aging. 2009;30:1238–44.
Rice HC, de Malmazet D, Schreurs A, Frere S, Van Molle I, Volkov AN, et al. Secreted amyloid-beta precursor protein functions as a GABABR1a ligand to modulate synaptic transmission. Science. 2019;363:pii: eaao4827.
Holtzman DM, Bales KR, Paul SM, DeMattos RB. Abeta immunization and anti-Abeta antibodies: potential therapies for the prevention and treatment of Alzheimer's disease. Adv Drug Deliv Rev. 2002;54:1603–13.
Bharadwaj PR, Dubey AK, Masters CL, Martins RN, Macreadie IG. Abeta aggregation and possible implications in Alzheimer's disease pathogenesis. J Cell Mol Med. 2009;13:412–21.
Buckner RL, Snyder AZ, Shannon BJ, LaRossa G, Sachs R, Fotenos AF, et al. Molecular, structural, and functional characterization of Alzheimer's disease: evidence for a relationship between default activity, amyloid, and memory. J Neurosci. 2005;25:7709–17.
Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, et al. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med. 2012;367:795–804.
Ryman DC, Acosta-Baena N, Aisen PS, Bird T, Danek A, Fox NC, et al. Symptom onset in autosomal dominant Alzheimer disease: a systematic review and meta-analysis. Neurology. 2014;83:253–60.
Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, et al. Decreased clearance of CNS beta-amyloid in Alzheimer's disease. Science. 2010;330:1774.
Simic G, Babic Leko M, Wray S, Harrington C, Delalle I, Jovanov-Milosevic N, et al. Tau protein hyperphosphorylation and aggregation in Alzheimer's disease and other tauopathies, and possible neuroprotective strategies. Biomolecules. 2016;6:6.
Brion JP, Hanger DP, Bruce MT, Couck AM, Flament-Durand J, Anderton BH. Tau in Alzheimer neurofibrillary tangles. N- and C-terminal regions are differentially associated with paired helical filaments and the location of a putative abnormal phosphorylation site. Biochem J. 1991;273:127–33.
Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc Natl Acad Sci USA. 1975;72:1858–62.
Kadavath H, Hofele RV, Biernat J, Kumar S, Tepper K, Urlaub H, et al. Tau stabilizes microtubules by binding at the interface between tubulin heterodimers. Proc Natl Acad Sci USA. 2015;112:7501–6.
Prezel E, Elie A, Delaroche J, Stoppin-Mellet V, Bosc C, Serre L, et al. Tau can switch microtubule network organizations: from random networks to dynamic and stable bundles. Mol Biol Cell. 2018;29:154–65.
Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, et al. Association of missense and 5'-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–5.
Spillantini MG, Bird TD, Ghetti B. Frontotemporal dementia and Parkinsonism linked to chromosome 17: a new group of tauopathies. Brain Pathol. 1998;8:387–402.
Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, et al. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43:815–25.
Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–59.
Sergeant N, Delacourte A, Buee L. Tau protein as a differential biomarker of tauopathies. Biochim Biophys Acta. 2005;1739:179–97.
Lebouvier T, Pasquier F, Buee L. Update on tauopathies. Curr Opin Neurol. 2017;30:589–98.
Hernandez F, Avila J. Tauopathies. Cell Mol Life Sci. 2007;64:2219–33.
Spillantini MG, Goedert M. Tau pathology and neurodegeneration. Lancet Neurol. 2013;12:609–22.
Rudrabhatla P, Jaffe H, Pant HC. Direct evidence of phosphorylated neuronal intermediate filament proteins in neurofibrillary tangles (NFTs): phosphoproteomics of Alzheimer’s NFTs. FASEB J. 2011;25:3896–905.
Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology. 1992;42:631–9.
Bierer LM, Hof PR, Purohit DP, Carlin L, Schmeidler J, Davis KL, et al. Neocortical neurofibrillary tangles correlate with dementia severity in Alzheimer’s disease. Arch Neurol. 1995;52:81–8.
Guillozet AL, Weintraub S, Mash DC, Mesulam MM. Neurofibrillary tangles, amyloid, and memory in aging and mild cognitive impairment. Arch Neurol. 2003;60:729–36.
Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011;70:960–9.
Braak H, Del Tredici K. The pathological process underlying Alzheimer's disease in individuals under thirty. Acta Neuropathol. 2011;121:171–81.
Saper CB, Scammell TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature. 2005;437:1257–63.
Gotz J, Gladbach A, Pennanen L, van Eersel J, Schild A, David D, et al. Animal models reveal role for tau phosphorylation in human disease. Biochim Biophys Acta. 2010;1802:860–71.
Tacik P, Sanchez-Contreras M, Rademakers R, Dickson DW, Wszolek ZK. Genetic disorders with tau pathology: a review of the literature and report of two patients with tauopathy and positive family histories. Neurodegener Dis. 2016;16:12–21.
Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron. 1989;3:519–26.
Panda D, Samuel JC, Massie M, Feinstein SC, Wilson L. Differential regulation of microtubule dynamics by three- and four-repeat tau: implications for the onset of neurodegenerative disease. Proc Natl Acad Sci USA. 2003;100:9548–53.
Liu C, Gotz J. Profiling murine tau with 0N, 1N and 2N isoform-specific antibodies in brain and peripheral organs reveals distinct subcellular localization, with the 1N isoform being enriched in the nucleus. PLoS ONE. 2013;8:e84849.
Goedert M, Hasegawa M. The tauopathies: toward an experimental animal model. Am J Pathol. 1999;154:1–6.
Musiek ES, Holtzman DM. Three dimensions of the amyloid hypothesis: time, space and ‘wingmen’. Nat Neurosci. 2015;18:800–6.
Wolfe MS. The role of tau in neurodegenerative diseases and its potential as a therapeutic target. Science. 2012;2012:796024.
Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256:184–5.
Puzzo D, Lee L, Palmeri A, Calabrese G, Arancio O. Behavioral assays with mouse models of Alzheimer's disease: practical considerations and guidelines. Biochem Pharm. 2014;88:450–67.
Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, et al. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–4.
He Z, Guo JL, McBride JD, Narasimhan S, Kim H, Changolkar L, et al. Amyloid-beta plaques enhance Alzheimer's brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation. Nat Med. 2018;24:29–38.
Ricciarelli R, Fedele E. The amyloid cascade hypothesis in Alzheimer's disease: it's time to change our Mind. Curr Neuropharmacol. 2017;15:926–35.
Kametani F, Hasegawa M. Reconsideration of amyloid hypothesis and tau hypothesis in Alzheimer's disease. Front Neurosci. 2018;12:25.
Bryan KJ, Lee H, Perry G, Smith MA, Casadesus G. Transgenic mouse models of Alzheimer's disease: behavioral testing and considerations. In: Buccafusco JJ, (Ed.). Frontiers in Neuroscience, Methods of behavior analysis in neuroscience. Boca Raton, FL: CRC Press; 2009.
Ostrowitzki S, Deptula D, Thurfjell L, Barkhof F, Bohrmann B, Brooks DJ, et al. Mechanism of amyloid removal in patients with Alzheimer disease treated with gantenerumab. Arch Neurol. 2012;69:198–207.
Giacobini E, Gold G. Alzheimer disease therapy—moving from amyloid-beta to tau. Nat Rev Neurol. 2013;9:677–86.
Doody RS, Thomas RG, Farlow M, Iwatsubo T, Vellas B, Joffe S, et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer's disease. N Engl J Med. 2014;370:311–21.
Brown BM, Rainey-Smith SR, Villemagne VL, Weinborn M, Bucks RS, Sohrabi HR, et al. The relationship between sleep quality and brain amyloid burden. Sleep. 2016;39:1063–8.
Branger P, Arenaza-Urquijo EM, Tomadesso C, Mezenge F, Andre C, de Flores R, et al. Relationships between sleep quality and brain volume, metabolism, and amyloid deposition in late adulthood. Neurobiol Aging. 2016;41:107–14.
Sprecher KE, Bendlin BB, Racine AM, Okonkwo OC, Christian BT, Koscik RL, et al. Amyloid burden is associated with self-reported sleep in nondemented late middle-aged adults. Neurobiol Aging. 2015;36:2568–76.
Ooms S, Overeem S, Besse K, Rikkert MO, Verbeek M, Claassen JA. Effect of 1 night of total sleep deprivation on cerebrospinal fluid beta-amyloid 42 in healthy middle-aged men: a randomized clinical trial. JAMA Neurol. 2014;71:971–7.
Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, et al. Sleep drives metabolite clearance from the adult brain. Science. 2013;342:373–7.
Nedergaard M. Neuroscience. Garbage truck of the brain. Science. 2013;340:1529–30.
Schneider F, Baldauf K, Wetzel W, Reymann KG. Behavioral and EEG changes in male 5xFAD mice. Physiol Behav 2014;135:25–33.
Huitron-Resendiz S, Sanchez-Alavez M, Gallegos R, Berg G, Crawford E, Giacchino JL, et al. Age-independent and age-related deficits in visuospatial learning, sleep-wake states, thermoregulation and motor activity in PDAPP mice. Brain Res. 2002;928:126–37.
Colby-Milley J, Cavanagh C, Jego S, Breitner JC, Quirion R, Adamantidis A. Sleep-wake cycle dysfunction in the TgCRND8 mouse model of Alzheimer's disease: from early to advanced pathological stages. PLoS ONE. 2015;10:e0130177.
Zhang B, Veasey SC, Wood MA, Leng LZ, Kaminski C, Leight S, et al. Impaired rapid eye movement sleep in the Tg2576 APP murine model of Alzheimer's disease with injury to pedunculopontine cholinergic neurons. Am J Pathol. 2005;167:1361–9.
Sethi M, Joshi SS, Webb RL, Beckett TL, Donohue KD, Murphy MP, et al. Increased fragmentation of sleep-wake cycles in the 5XFAD mouse model of Alzheimer's disease. Neuroscience. 2015;290:80–9.
Standaert DG, Lee VM, Greenberg BD, Lowery DE, Trojanowski JQ. Molecular features of hypothalamic plaques in Alzheimer's disease. Am J Pathol. 1991;139:681–91.
Parvizi J, Van Hoesen GW, Damasio A. Selective pathological changes of the periaqueductal gray matter in Alzheimer's disease. Ann Neurol. 2000;48:344–53.
Busche MA, Kekus M, Adelsberger H, Noda T, Forstl H, Nelken I, et al. Rescue of long-range circuit dysfunction in Alzheimer's disease models. Nat Neurosci. 2015;18:1623–30.
Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, Holtzman DM. Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol. 2007;64:343–9.
Mattsson N, Zetterberg H, Hansson O, Andreasen N, Parnetti L, Jonsson M, et al. CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. JAMA. 2009;302:385–93.
Brier MR, Gordon B, Friedrichsen K, McCarthy J, Stern A, Christensen J, et al. Tau and Abeta imaging, CSF measures, and cognition in Alzheimer's disease. Sci Transl Med. 2016;8:338ra66.
Stratmann K, Heinsen H, Korf HW, Del Turco D, Ghebremedhin E, Seidel K, et al. Precortical phase of Alzheimer's dsease (AD)-related tau cytoskeletal pathology. Brain Pathol. 2016;26:371–86.
Lim AS, Ellison BA, Wang JL, Yu L, Schneider JA, Buchman AS, et al. Sleep is related to neuron numbers in the ventrolateral preoptic/intermediate nucleus in older adults with and without Alzheimer's disease. Brain. 2014;137:2847–61.
Parvizi J, Van Hoesen GW, Damasio A. The selective vulnerability of brainstem nuclei to Alzheimer's disease. Ann Neurol. 2001;49:53–66.
Platt B, Drever B, Koss D, Stoppelkamp S, Jyoti A, Plano A, et al. Abnormal cognition, sleep, EEG and brain metabolism in a novel knock-in Alzheimer mouse, PLB1. PLoS ONE. 2011;6:e27068.
Jyoti A, Plano A, Riedel G, Platt B. Progressive age-related changes in sleep and EEG profiles in the PLB1Triple mouse model of Alzheimer's disease. Neurobiol Aging. 2015;36:2768–84.
Yamada K, Holth JK, Liao F, Stewart FR, Mahan TE, Jiang H, et al. Neuronal activity regulates extracellular tau in vivo. J Exp Med. 2014;211:387–93.
Pooler AM, Phillips EC, Lau DH, Noble W, Hanger DP. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 2013;14:389–94.
Ju YS, Ooms SJ, Sutphen C, Macauley SL, Zangrilli MA, Jerome G, et al. Slow wave sleep disruption increases cerebrospinal fluid amyloid-beta levels. Brain. 2017;140:2104–11.
Mander BA, Marks SM, Vogel JW, Rao V, Lu B, Saletin JM, et al. beta-amyloid disrupts human NREM slow waves and related hippocampus-dependent memory consolidation. Nat Neurosci. 2015;18:1051–7.
Winer JR, Mander BA, Helfrich RF, Maass A, Harrison TM, Baker SL, et al. Sleep as a potential biomarker of tau and beta-amyloid burden in the human brain. J Neurosci. 2019;39:6315–6324.
Kam K, Parekh A, Sharma RA, Andrade A, Lewin M, Castillo B, et al. Sleep oscillation-specific associations with Alzheimer's disease CSF biomarkers: novel roles for sleep spindles and tau. Mol Neurodegener. 2019;14:10.
Remmers JE, deGroot WJ, Sauerland EK, Anch AM. Pathogenesis of upper airway occlusion during sleep. J Appl Physiol Respir Environ Exerc Physiol. 1978;44:931–8.
Young T, Palta M, Dempsey J, Skatrud J, Weber S, Badr S. The occurrence of sleep-disordered breathing among middle-aged adults. N Engl J Med. 1993;328:1230–5.
Bixler EO, Vgontzas AN, Ten Have T, Tyson K, Kales A. Effects of age on sleep apnea in men: I. Prevalence and severity. Am J Respir Crit Care Med. 1998;157:144–8.
Nagayoshi M, Lutsey PL, Benkeser D, Wassel CL, Folsom AR, Shahar E, et al. Association of sleep apnea and sleep duration with peripheral artery disease: the Multi-Ethnic Study of Atherosclerosis (MESA). Atherosclerosis. 2016;251:467–75.
Phillips CL, Yee BJ, Marshall NS, Liu PY, Sullivan DR, Grunstein RR. Continuous positive airway pressure reduces postprandial lipidemia in obstructive sleep apnea: a randomized, placebo-controlled crossover trial. Am J Respir Crit Care Med. 2011;184:355–61.
Somers VK, White DP, Amin R, Abraham WT, Costa F, Culebras A, et al. Sleep apnea and cardiovascular disease: an American Heart Association/american College Of Cardiology Foundation Scientific Statement from the American Heart Association Council for High Blood Pressure Research Professional Education Committee, Council on Clinical Cardiology, Stroke Council, and Council On Cardiovascular Nursing. In collaboration with the National Heart, Lung, and Blood Institute National Center on Sleep Disorders Research (National Institutes of Health). Circulation. 2008;118:1080–111.
Pedrosa RP, Drager LF, Gonzaga CC, Sousa MG, de Paula LK, Amaro AC, et al. Obstructive sleep apnea: the most common secondary cause of hypertension associated with resistant hypertension. Hypertension. 2011;58:811–7.
Schwartz AR, Patil SP, Laffan AM, Polotsky V, Schneider H, Smith PL. Obesity and obstructive sleep apnea: pathogenic mechanisms and therapeutic approaches. Proc Am Thorac Soc. 2008;5:185–92.
Yaffe K, Laffan AM, Harrison SL, Redline S, Spira AP, Ensrud KE, et al. Sleep-disordered breathing, hypoxia, and risk of mild cognitive impairment and dementia in older women. JAMA 2011;306:613–9.
Chang WP, Liu ME, Chang WC, Yang AC, Ku YC, Pai JT, et al. Sleep apnea and the risk of dementia: a population-based 5-year follow-up study in Taiwan. PLoS ONE. 2013;8:e78655.
Osorio RS, Gumb T, Pirraglia E, Varga AW, Lu SE, Lim J, et al. Sleep-disordered breathing advances cognitive decline in the elderly. Neurology. 2015;84:1964–71.
Reitz C, Mayeux R. Alzheimer disease: epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem Pharm. 2014;88:640–51.
Ancoli-Israel S, Palmer BW, Cooke JR, Corey-Bloom J, Fiorentino L, Natarajan L, et al. Cognitive effects of treating obstructive sleep apnea in Alzheimer's disease: a randomized controlled study. J Am Geriatr Soc. 2008;56:2076–81.
Cooke JR, Ayalon L, Palmer BW, Loredo JS, Corey-Bloom J, Natarajan L, et al. Sustained use of CPAP slows deterioration of cognition, sleep, and mood in patients with Alzheimer's disease and obstructive sleep apnea: a preliminary study. J Clin Sleep Med. 2009;5:305–9.
Daulatzai MA. Death by a thousand cuts in Alzheimer's disease: hypoxia—the prodrome. Neurotox Res. 2013;24:216–43.
Wang Z, Zhang XJ, Li T, Li J, Tang Y, Le W. Valproic acid reduces neuritic plaque formation and improves learning deficits in APP(Swe) /PS1(A246E) transgenic mice via preventing the prenatal hypoxia-induced down-regulation of neprilysin. CNS Neurosci Ther. 2014;20:209–17.
Zhang X, Li L, Zhang X, Xie W, Li L, Yang D, et al. Prenatal hypoxia may aggravate the cognitive impairment and Alzheimer's disease neuropathology in APPSwe/PS1A246E transgenic mice. Neurobiol Aging. 2013;34:663–78.
Ju YE, Finn MB, Sutphen CL, Herries EM, Jerome GM, Ladenson JH, et al. Obstructive sleep apnea decreases central nervous system-derived proteins in the cerebrospinal fluid. Ann Neurol. 2016;80:154–9.
Liguori C, Mercuri NB, Izzi F, Romigi A, Cordella A, Sancesario G, et al. Obstructive sleep apnea is associated with early but possibly modifiable Alzheimer's disease biomarkers changes. Sleep. 2017;40:zsx011.
Ju YS, Zangrilli MA, Finn MB, Fagan AM, Holtzman DM. Obstructive sleep apnea treatment, slow wave activity, and amyloid-beta. Ann Neurol. 2019;85:291–95.
Bu G. Apolipoprotein E and its receptors in Alzheimer's disease: pathways, pathogenesis and therapy. Nat Rev Neurosci. 2009;10:333–44.
Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988;240:622–30.
Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261:921–3.
Bertram L, Tanzi RE. Thirty years of Alzheimer's disease genetics: the implications of systematic meta-analyses. Nat Rev Neurosci. 2008;9:768–78.
Kok E, Haikonen S, Luoto T, Huhtala H, Goebeler S, Haapasalo H, et al. Apolipoprotein E-dependent accumulation of Alzheimer disease-related lesions begins in middle age. Ann Neurol. 2009;65:650–7.
Morris JC, Roe CM, Xiong C, Fagan AM, Goate AM, Holtzman DM, et al. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol. 2010;67:122–31.
Verghese PB, Castellano JM, Holtzman DM. Apolipoprotein E in Alzheimer's disease and other neurological disorders. Lancet Neurol. 2011;10:241–52.
Kadotani H, Kadotani T, Young T, Peppard PE, Finn L, Colrain IM, et al. Association between apolipoprotein E epsilon4 and sleep-disordered breathing in adults. JAMA. 2001;285:2888–90.
Gottlieb DJ, DeStefano AL, Foley DJ, Mignot E, Redline S, Givelber RJ, et al. APOE epsilon4 is associated with obstructive sleep apnea/hypopnea: the Sleep Heart Health Study. Neurology. 2004;63:664–8.
Cosentino FI, Bosco P, Drago V, Prestianni G, Lanuzza B, Iero I, et al. The APOE epsilon4 allele increases the risk of impaired spatial working memory in obstructive sleep apnea. Sleep Med. 2008;9:831–9.
Lu Z, Wu X, Jin X, Peng F, Lin J. Apolipoprotein E varepsilon2/varepsilon3/varepsilon4 variant in association with obstructive sleep apnoea and lipid profile: A meta-analysis. J Int Med Res. 2016;44:3–14.
Thakre TP, Mamtani MR, Kulkarni H. Lack of association of the APOE epsilon 4 allele with the risk of obstructive sleep apnea: meta-analysis and meta-regression. Sleep. 2009;32:1507–11.
Drogos LL, Gill SJ, Tyndall AV, Raneri JK, Parboosingh JS, Naef A, et al. Evidence of association between sleep quality and APOE epsilon4 in healthy older adults: a pilot study. Neurology. 2016;87:1836–42.
Lim AS, Yu L, Kowgier M, Schneider JA, Buchman AS, Bennett DA. Modification of the relationship of the apolipoprotein E epsilon4 allele to the risk of Alzheimer disease and neurofibrillary tangle density by sleep. JAMA Neurol. 2013;70:1544–51.
Katsumoto A, Takeuchi H, Takahashi K, Tanaka F. Microglia in Alzheimer's disease: risk factors and inflammation. Front Neurol. 2018;9:978.
Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–8.
Parkhurst CN, Gan WB. Microglia dynamics and function in the CNS. Curr Opin Neurobiol. 2010;20:595–600.
Dickson DW, Farlo J, Davies P, Crystal H, Fuld P, Yen SH. Alzheimer's disease. A double-labeling immunohistochemical study of senile plaques. Am J Pathol. 1988;132:86–101.
Haga S, Akai K, Ishii T. Demonstration of microglial cells in and around senile (neuritic) plaques in the Alzheimer brain. An immunohistochemical study using a novel monoclonal antibody. Acta Neuropathol. 1989;77:569–75.
Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K, et al. Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol. 1998;152:307–17.
Stalder M, Phinney A, Probst A, Sommer B, Staufenbiel M, Jucker M. Association of microglia with amyloid plaques in brains of APP23 transgenic mice. Am J Pathol. 1999;154:1673–84.
Serrano-Pozo A, Muzikansky A, Gomez-Isla T, Growdon JH, Betensky RA, Frosch MP, et al. Differential relationships of reactive astrocytes and microglia to fibrillar amyloid deposits in Alzheimer disease. J Neuropathol Exp Neurol. 2013;72:462–71.
Gratuze M, Leyns CEG, Holtzman DM. New insights into the role of TREM2 in Alzheimer's disease. Mol Neurodegener. 2018;13:66.
Jay TR, Miller CM, Cheng PJ, Graham LC, Bemiller S, Broihier ML, et al. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer's disease mouse models. J Exp Med. 2015;212:287–95.
Jay TR, Hirsch AM, Broihier ML, Miller CM, Neilson LE, Ransohoff RM, et al. Disease progression-dependent effects of TREM2 deficiency in a mouse model of Alzheimer's disease. J Neurosci. 2017;37:637–47.
Leyns CEG, Ulrich JD, Finn MB, Stewart FR, Koscal LJ, Remolina Serrano J, et al. TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc Natl Acad Sci USA. 2017;114:11524–29.
An H, Cho MH, Kim DH, Chung S, Yoon SY. Orexin impairs the phagocytosis and degradation of amyloid-beta fibrils by microglial cells. J Alzheimers Dis. 2017;58:253–61.
Sprecher KE, Koscik RL, Carlsson CM, Zetterberg H, Blennow K, Okonkwo OC, et al. Poor sleep is associated with CSF biomarkers of amyloid pathology in cognitively normal adults. Neurology. 2017;89:445–53.
Zissimopoulos J, Crimmins E, St Clair P. The value of delaying Alzheimer's disease onset. Forum Health Econ Policy. 2014;18:25–39.
Cummings J, Aisen PS, DuBois B, Frolich L, Jack CR Jr., Jones RW, et al. Drug development in Alzheimer's disease: the path to 2025. Alzheimers Res Ther. 2016;8:39.
Mangialasche F, Solomon A, Winblad B, Mecocci P, Kivipelto M. Alzheimer's disease: clinical trials and drug development. Lancet Neurol. 2010;9:702–16.
Montagne A, Zhao Z, Zlokovic BV. Alzheimer's disease: a matter of blood-brain barrier dysfunction? J Exp Med. 2017;214:3151–69.
Gomez-Gonzalez B, Hurtado-Alvarado G, Esqueda-Leon E, Santana-Miranda R, Rojas-Zamorano JA, Velazquez-Moctezuma J. REM sleep loss and recovery regulates blood-brain barrier function. Curr Neurovasc Res. 2013;10:197–207.
He J, Hsuchou H, He Y, Kastin AJ, Wang Y, Pan W. Sleep restriction impairs blood-brain barrier function. J Neurosci. 2014;34:14697–706.
Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat Rev Neurosci. 2011;12:723–38.
Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z, et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron. 2015;85:296–302.
Sekiguchi H, Iritani S, Fujita K. Bright light therapy for sleep disturbance in dementia is most effective for mild to moderate Alzheimer's type dementia: a case series. Psychogeriatrics. 2017;17:275–81.
Van Someren EJ, Swaab DF, Colenda CC, Cohen W, McCall WV, Rosenquist PB. Bright light therapy: improved sensitivity to its effects on rest-activity rhythms in Alzheimer patients by application of nonparametric methods. Chrono- Int. 1999;16:505–18.
Yang PY, Ho KH, Chen HC, Chien MY. Exercise training improves sleep quality in middle-aged and older adults with sleep problems: a systematic review. J Physiother. 2012;58:157–63.
Kandasamy G, Almaghaslah D, Sivanandy P, Arumugam S. Effectiveness of nasal continuous airway pressure therapy in patients with obstructive sleep apnea. Int J Health Plann Manag. 2019;34:e1200–e1207.
Van Egroo M, Narbutas J, Chylinski D, Villar Gonzalez P, Maquet P, Salmon E, et al. Sleep-wake regulation and the hallmarks of the pathogenesis of Alzheimer's disease. Sleep. 2019;42:zsz017.
Perrault AA, Khani A, Quairiaux C, Kompotis K, Franken P, Muhlethaler M, et al. Whole-night continuous rocking entrains spontaneous neural oscillations with benefits for sleep and memory. Curr Biol. 2019;29:402–11 e3.
Kompotis K, Hubbard J, Emmenegger Y, Perrault A, Muhlethaler M, Schwartz S, et al. Rocking promotes sleep in mice through rhythmic stimulation of the vestibular system. Curr Biol. 2019;29:392–401 e4.
Bayer L, Constantinescu I, Perrig S, Vienne J, Vidal PP, Muhlethaler M, et al. Rocking synchronizes brain waves during a short nap. Curr Biol. 2011;21:R461–2.
Acknowledgements
The authors would like to thank Jerrah Holth, Michael Strickland, Monica Xiong, Alec Moore, Geraldine Kress, and Jason Ulrich for assisting in editing and correcting the structure/grammar of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wang, C., Holtzman, D.M. Bidirectional relationship between sleep and Alzheimer’s disease: role of amyloid, tau, and other factors. Neuropsychopharmacol. 45, 104–120 (2020). https://doi.org/10.1038/s41386-019-0478-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-019-0478-5
This article is cited by
-
Sleep duration, sleep efficiency, and amyloid β among cognitively healthy later-life adults: a systematic review and meta-analysis
BMC Geriatrics (2024)
-
Neuronal and glial vulnerability of the suprachiasmatic nucleus in tauopathies: evidence from human studies and animal models
Molecular Neurodegeneration (2024)
-
Epilepsy and epileptiform activity in late-onset Alzheimer disease: clinical and pathophysiological advances, gaps and conundrums
Nature Reviews Neurology (2024)
-
Linking activity dyshomeostasis and sleep disturbances in Alzheimer disease
Nature Reviews Neuroscience (2024)
-
Role of sleep in neurodegeneration: the consensus report of the 5th Think Tank World Sleep Forum
Neurological Sciences (2024)
