
Abstract
Alcohol use disorder is a significant global burden. Stress has been identified as an etiological factor in the initiation and continuation of ethanol consumption. Understanding adaptations within stress circuitry is an important step toward novel treatment strategies. The effects of protracted abstinence following long-term ethanol self-administration on the central nucleus of the amygdala (CeA) and the hypothalamic paraventricular nucleus (PVN) were evaluated in male rhesus monkeys. Using whole-cell patch-clamp electrophysiology, inhibitory GABAergic transmission in the CeA and excitatory glutamatergic transmission in the PVN were measured. CeA neurons from abstinent drinkers displayed an elevated baseline spontaneous inhibitory postsynaptic current (sIPSC) frequency compared with controls, indicating increased presynaptic GABA release. Application of acute ethanol significantly increased the frequency of sIPSCs in controls, but not in abstinent drinkers, suggesting a tolerance to ethanol-enhanced GABA release in abstinent rhesus monkeys with a history of chronic ethanol self-administration and repeated abstinence. In the PVN, the frequency of spontaneous excitatory postsynaptic currents (sEPSC) was elevated in abstinent drinkers compared with controls, indicating increased presynaptic glutamate release. Notably, acute ethanol decreased presynaptic glutamate release onto parvocellular PVN neurons in both controls and abstinent drinkers, suggesting a lack of tolerance to acute ethanol among PVN neurons. These results are the first to demonstrate distinct synaptic adaptations and ethanol sensitivity in both the extrahypothalamic and hypothalamic stress circuits in abstinent rhesus males. Importantly, our findings describe adaptations in stress circuitry present in the brain at a state during abstinence, just prior to relapse to ethanol drinking.
Similar content being viewed by others


INTRODUCTION
There is strong evidence that the mammalian response to stress is an orchestration of endocrine, neural, and behavioral processes that, in the face of chronic ethanol, can become maladaptive and propagate further escalations of ethanol intake [1, 2]. This relationship between stress and ethanol is bidirectional. Long-term ethanol consumption results in dysregulation of stress systems and contributes to risk for relapse to alcohol drinking [3,4,5,6].
Stress circuitry can be divided into extrahypothalamic and hypothalamic pathways. A fundamental component of the extrahypothalamic stress circuitry is the amygdala. The major output of the amygdala is the central nucleus (CeA), which receives innervation from other amygdala nuclei, prefrontal cortex, hypothalamus, and other regions [7, 8] and is composed mostly of γ-aminobutyric acid (GABA) projection neurons and interneurons [9, 10]. Both acute and chronic ethanol increase GABAergic signaling in the CeA of rodents, in part by increasing presynaptic GABA release [11,12,13]. The GABAergic system within the CeA plays an important role in anxiety-like behaviors and the increased ethanol intake that is observed following withdrawal in ethanol-dependent animals [14,15,16].
The hypothalamic stress circuitry is comprised of the hypothalamic paraventricular nucleus (PVN), the anterior pituitary, and the adrenal cortex, collectively known as the HPA axis. Information converges onto neuroendocrine parvocellular neurons in the PVN from across the brain in a hierarchical manner, with many stress-sensitive regions such as the hippocampus, bed nucleus of the stria terminalis (BNST), and amygdala being relayed to the PVN via other hypothalamic nuclei [17]. In response to stress, parvocellular PVN neurons initiate a cascade of circulating stress hormones, including cortisol, the primary glucocorticoid in primates. The PVN has a role in HPA axis activation following acute ethanol [18], and repeated exposure results in tolerance to the HPA axis activating the effects of ethanol [19]. Importantly, the extrahypothalamic and hypothalamic stress circuitry are functionally interconnected. Rodent-based studies suggest that the CeA can influence parvocellular PVN neurons via direct, albeit limited, GABAergic projections which inhibit PVN activity and indirectly through disinhibition of the bed nucleus of the stria terminalis (BNST) and peri-PVN regions [20, 21]. Although GABAergic connections between the PVN and CeA have been emphasized in previous studies of stress in rodents, we have shown a unique relationship between presynaptic glutamate, but not GABA, within parvocellular PVN neurons and ethanol self-administration in female rhesus macaques with a history of ethanol self-administration [22]. Within the PVN, the majority of synapses onto parvocellular neurons are GABAergic and glutamatergic [23, 24], both having regulatory roles in the release of CRF and activation of the HPA axis.
Repeated forced abstinence enhances alcohol drinking in mice, and relapse following abstinence is a diagnostic criterion in alcohol use disorder [25,26,27]. The monkeys here underwent three forced abstinence periods and demonstrated elevated ethanol self-administration when ethanol was reintroduced following the first and second abstinence phases [28]. The macaque model of ethanol self-administration provides an important translational link between rodent and human studies. Notably, the macaque more closely resembles ethanol consumption [29,30,31], ethanol metabolism, endocrine physiology [see Ref. 5], and brain structure [32] of humans than rodents. These are critical advantages for understanding AUD, which involves volitional, self-selection of daily ethanol intakes, and the involvement of central and peripheral systems. Using the monkey model, we have reported altered synaptic signaling in macaques during chronic ethanol drinking in multiple brain regions including area [32] of the prefrontal cortex, striatum, PVN, nucleus accumbens, and the BNST [22, 33,34,35,36,37], as well as in the striatum and inferior olive during forced abstinence [33, 38, 39]. Stress circuitry adaptations to the effects of extended ethanol consumption and repeated forced abstinence are unexplored in the monkey model. Given the importance of stress in initiating and maintaining ethanol-drinking behavior, and literature demonstrating the sensitivity to acute and chronic ethanol within stress circuitry, understanding the adaptations within these regions will be critical in developing treatment and intervention strategies. The purpose of this study was to evaluate synaptic transmission and the effects of acute ethanol in the CeA and PVN of abstinent male rhesus macaques following a long history of chronic ethanol self-administration and repeated forced abstinence. Specifically, we focused on CeA inhibitory GABAergic transmission and PVN excitatory glutamatergic transmission, as these neurotransmitter systems are known to be particularly sensitive to the effects of ethanol [11,12,13,14,15,16, 22]. These results are the first to demonstrate that distinct neuronal adaptations and differences in ethanol sensitivity emerge in both the extrahypothalamic and hypothalamic stress regions that likely act in conjunction to contribute to the propensity to relapse to heavy drinking following abstinence.
MATERIALS AND METHODS
Animal information
Adult male rhesus macaques (Macaca mulatta) were housed indoors as previously reported [28]. Monkeys were assigned as control (n = 4) or ethanol drinker (n = 8), matched by weight. Monkeys were 4.0–5.5 years old at the onset of the protocol and 4.7–6.2 years old at the time of the first ethanol intake. Detailed drinking characteristics and behavioral signs of dependence on this cohort of monkeys have been recently published [28]. Please note that Allen et al. [28] use the last three digits of the IDs presented in this paper, but these are the same animals. All procedures were conducted in accordance with the Guide for the Care and Use of Laboratory Animals and the NIH guidelines for the care and use of laboratory animal resources and approved by the Oregon National Primate Research Center Institutional Animal Care and Use Committee. Further details on the voluntary ethanol consumption of this cohort (Rhesus 10) and additional data and publications can be found through the Monkey Alcohol and Tissue Research Resource (MATRR.com). The IDs in this paper correspond to the monkey IDs on MATRR.
Self-administration and abstinence
Drinking panels replaced one wall of each cage and dispensed food and fluids, as previously described [28, 40]. Schedule-induced polydipsia was used to induce ethanol self-administration in daily 16-h sessions followed by daily 22-h open-access self-administration, as previously described [28, 40]. Because repeated abstinence is a hallmark of alcohol use disorder (AUD) [41], we used an extended self-administration protocol with repeated forced abstinent phases and resumed ethanol self-administration phases. Specifically, after 425 consecutive daily sessions (~14 months) of open access, ethanol was replaced with water during three forced abstinence phases, each lasting for 28–44 days. This time span was chosen to match human abstinence-based treatment protocols. Ethanol reintroduction following the first and second abstinence (Fig. 1a) reliably led to relapse drinking in all monkeys. Three months of relapse drinking following forced abstinence was chosen based on our previous analysis [29]. This time frame is sufficient to observe a separation of an initial elevation in ethanol consumption during the first month, from the sustained consumption seen only in the heavy and very-heavy drinking monkeys throughout the 3-month post-abstinence period. Necropsy occurred at the end of the third abstinence phase (35–44 days after the last ethanol session) to capture the state of the brain just prior to relapse.

a Experimental timeline. Daily 22-h sessions with water and ethanol concurrently available began following induction. Following 425 days of open-access self-administration, animals experienced the first of three abstinence phases, each lasting for 28–44 days. Ethanol was available during two post-abstinence (post-abs) periods lasting for 84 and 104 days, respectively. Necropsy was performed after 35–44 days of abstinence. The effect of abstinence was evaluated by comparing intake in 7-day bins before (B1, B2, and B3) and after abstinence (Δ1 and Δ2), indicated by blue and yellow. b Schematic and photograph of a 4-mm coronal block containing the central nucleus of the amygdala (CeA) and hypothalamic paraventricular nucleus (PVN). Dashed lines indicate the dissections of the CeA and PVN used for electrophysiology. Other prominent regions and landmarks include the caudate (Cd), putamen (Pu), anterior commissure (ac), and internal capsule (ic). c Representative current clamp traces showing a characteristic electrophysiological response to a depolarizing current step. Magnocellular neurons in the PVN are distinguished by a delay to action potential onset following the hyperpolarizing current step, indicated by the arrow (left), which is absent in parvocellular PVN neurons (right). d Average daily intake (104 sessions) and e blood ethanol concentration (21 samples/animal) during post-abstinence 2. f Summary of average daily intake during each 7-day bin pre- and post-abstinence periods and g intake normalized as percent of pre-abstinence intake. VHD: very-heavy drinkers: daily ethanol intake average ≥ 3 g/kg, with ≥ 10% of drinking days exceeding 4 g/kg; HD: heavy drinkers: 3 g/kg for ≥ 20% of open-access days; LD: light drinker: do not meet the criteria for any of the previous categories. *p<0.05, **p<0.001
Tissue preparation and electrophysiological recordings
Necropsy and tissue collection procedures have been previously reported [33, 38]. Coronal sections (250 µm) of CeA and PVN were simultaneously sectioned using a VTI200S vibratome (Leica, Buffalo Grove, IL) from the same coronal brain block (Fig. 1b), as previously described [33, 38]. Slices were transferred to recording chambers on upright microscopes (SliceScope, Scientifica, Clarksburg, NJ). The tissue was continuously perfused with aCSF maintained at 28–32oC (Automatic Temperature Controller, Warner Instruments, Hamden, CT). A 40x water immersion objective and IR-DIC optics were used to identify neurons and guide the placement of the recording pipette. The intracellular solution for CeA recordings was composed of (in mM) KCl, 145; EGTA, 5; MgCl2, 5; HEPES, 10; Na-ATP, 2; Na-GTP, 0.2. The intracellular solution for PVN recordings was composed of (in mM) K-gluconate, 126; KCl, 4; HEPES, 10; Mg-ATP, 4; Na-GTP, 0.3; phosphocreatine, 10; and 1 mg/kg Dextran Cascade Blue (Molecular Probes, Grand Island, NY). Recording pipettes were pulled from borosilicate glass capillaries with a resistance of 3–5 MΩ. Recordings were made using a MultiClamp 700B amplifier (Molecular Devices, Foster City, CA). Voltage-clamp recordings (Vhold = −55–60 mV) were performed in a gap-free acquisition mode with a sampling rate per signal of 10 kHz (1.14 MB/min). Series resistance was < 20 MΩ and was continuously monitored with a hyperpolarizing 10-mV pulse. Recordings of spontaneous inhibitory postsynaptic currents (sIPSCs) were performed in the presence of the glutamate receptor blockers 6,7-dinitroquinoxaline-2,3-dione (DNQX, 20 µM) and DL-2-amino-5-phosphonovalerate (AP-5, 50 µM) and the GABAB receptor antagonist CGP55845A (1 µM). Recordings of spontaneous excitatory postsynaptic currents (sEPSCs) were performed in the presence of picrotoxin (100 µM). Drugs were dissolved in aCSF and applied by bath perfusion. Current-clamp recordings (Vm = -55 mV for PVN and −70 mV for CeA) of action potential firing were performed in sweeps using current steps beginning near −100 pA and progressively depolarizing with each step. Following a hyperpolarizing current step, putative magnocellular neurons (herein referred to as magnocellular) display a transient outward rectification, resulting in a delay to action potential onset, and no low-threshold spikes (Fig. 1c, left). Alternatively, putative parvocellular neurons (herein referred to as parvocellular) generate non-bursting low-threshold spikes and do not display a transient outward rectification current (Fig. 1c, right) [42,43,44,45]. Cells were excluded from analysis if no action potentials were generated in response to the current-clamp protocol or if the series resistance increased to over 20 MΩ during the course of the experiment. In a subset of neurons, acute ethanol (20 mM in PVN and 44 mM in CeA) was bath-applied. Recordings were performed in a random alternating fashion depending on day and time of preparation. Control animals were randomly alternated with drinkers to minimize confounds of time of sample preparation or recording. CeA and PVN sections were prepared in parallel, and recordings were performed over the same period of time. Recordings were performed over a period of 20–60min, in some cases including a 10–20-min wash-in of acute ethanol. In the CeA, a total of 15 cells were recorded from 12 slices taken from four individual animals classified as controls; a total of 23 cells were recorded from 20 slices taken from eight individual animals classified as abstinent drinkers. In the PVN, a total of 14 cells from 13 slices were taken from four individual animals classified as controls; a total of 37 cells from 31 slices were taken from eight individual animals classified as abstinent drinkers.
Drugs and chemicals
DNQX, AP-5, and CGP55845A were purchased from Tocris Bioscience. Picrotoxin was purchased from Sigma-Aldrich. Ethanol (95%) was purchased from Fisher Scientific.
Statistical analysis
The frequency, amplitude, rise time, and decay time of spontaneous events were analyzed and visually confirmed using a semi-automated threshold-based mini detection software (Mini Analysis, Synaptosoft Inc.). All detected events were used for frequency analysis, but superimposed events were eliminated for amplitude and decay kinetic analysis. Data were averaged (n = 2–8 cells/animal). All electrophysiological data were collected and analyzed prior to knowledge of group assignment. In the CeA, sIPSC event characteristics were averaged from a minimum of 60 events before and after bath application of ethanol (the time period of analysis varied as a product of individual event frequency). Due to their principal role in activation of the HPA axis and the observed changes in presynaptic glutamate specifically onto parvocellular neurons in chronically drinking macaques [22], sEPSCs were recorded only from parvocellular PVN neurons. sEPSCs were analyzed over 3 min and excluded from analysis if fewer than 100 events occurred. Two-tailed independent t tests were used for group comparisons, paired t tests were used to evaluate within-subject effects, and a one-way ANOVA with Bonferroni post hoc analysis was used for comparisons between three or more groups. The effect of acute ethanol was calculated as (acute ethanol/baseline) × 100. The relationship between event frequency and cortisol was evaluated using Pearson’s correlation. Analyses were performed using Prism (GraphPad Software, La Jolla, CA). Alpha ≤ 0.05 was considered significant. Data are presented as mean ± SEM.
Additional details on control subjects and blood collection methods can be found in the supplementary materials.
RESULTS
Post-abstinence escalation of ethanol intake
Abstinence resulted in a significant but transient increase in ethanol, but not water, self-administration in these subjects [28]. Based upon previously established categorical drinking criteria [29], during the final 3 months of open access, the drinkers self-selected into (n = 3) very-heavy drinkers (VHD: daily ethanol intake average ≥ 3 g/kg, with ≥ 10% of drinking days exceeding 4 g/kg), (n = 4) heavy drinkers (HD: 3 g/kg for ≥ 20% of open-access days), (n = 0) binge drinkers (BD: intakes of ≥ 2 g/kg for ≥ 55% of drinking days and have a BEC ≥ 80 mg/dl at least once per year), and a (n = 1) light drinker (LD: do not meet the criteria for any of the previous categories) (Fig. 1). Using 17 g of ethanol as a human drink equivalent, this translates to 0.25 g/kg in an average 70-kg human. Thus, 3.0 g/kg/day is a 12 drink/day equivalent. During the final open-access phase, a maximum average daily intake of 5.4 g/kg/day (or an almost 22-drink equivalent) was reached (Fig. 1d), with average BECs acquired at 7 h into the daily session (Fig. 1e). Because of the variation throughout open access following abstinence, we evaluated ethanol intake following abstinence as the average daily intake 7 days before and after each abstinence period (Fig. 1a). Average daily intake during 7-day bins is illustrated in Fig. 1f. A one-way ANOVA with repeated measures revealed a significant effect of the experimental phase (F(4,53) = 15.71, p < 0.0001). Tukey’s post hoc analysis revealed significant differences in average daily intake before and after the second abstinence (B2 to Δ2; p = 0.0008) and prior to the second and third (B2 to B3; p = 0.013) abstinence periods. Ethanol intake did not differ before and after the first abstinence (B1 to Δ1; p = 0.15).
These data are summarized as a percent change from baseline (Fig. 1g). Following the first abstinence, 7 of 8 animals increased their ethanol intake (Δ1: 31.41 ± 32.52%), and following the second abstinence, this increased to 8 of 8 (Δ2: 71.11 ± 39.16%). A paired Student’s t test confirmed that the magnitude of increase was larger following the second abstinence (t(7) = 2.42, p = 0.046). These data illustrate the post-abstinence spike in ethanol consumption.
Membrane properties
The CeA and PVN were identified according to established anatomy (Fig. 1b) [46]. CeA recordings were performed in what was best approximated as the medial subdivision of the CeA; however, as there are no distinct neuroanatomical boundaries between the medial and lateral CeA, it is not possible to definitively say that our recordings were restricted solely to the medial subdivision. It was also difficult to distinguish neuronal cell types within the heterogeneous neuronal population of the CeA, and different cell types by subdivision may contribute to variability in responses. Within the PVN, however, parvocellular and magnocellular neurons were identified due to the absence or presence of a delay to action potential onset following a hyperpolarizing current step, respectively (Fig. 1c) [42, 43]. Within the CeA, resting membrane potential was not significantly different in neurons from controls (−59.7 ± 2.1 mV) or abstinent drinkers (−55.7 ± 2.1 mV; t(10) = 1.19, p = 0.26). The threshold to fire did not differ between controls (−55.0 ± 2.0 mV) and abstinent drinkers (−51.8 ± 1.1 mV; t(10) = 1.54, p = 0.15). Among parvocellular neurons, no effect of group was found in membrane resistance (controls: 1.48 ± 0.22 GΩ abstinent drinkers: 1.36 ± 0.16 GΩ; t(10) = 0.42, p = 0.68), capacitance (controls: 16.5 ± 1.7 pF; abstinent drinkers: 18.5 ± 1.4 pF; t(10) = 0.92, p = 0.34), or resting membrane potential (controls: −39 ± 3 mV; abstinent drinkers: −41 ± 1 mV; t(10) = 0.85, p = 0.44). Due to their principal role in regulating the HPA axis, the parvocellular neurons were the focus of the studies presented here. Membrane properties for magnocellular neurons are reported in the Supplementary Materials. Membrane resistance and membrane capacitance were not recorded for the CeA. These data indicate that a history of chronic ethanol drinking with repeated periods of forced abstinence does not alter baseline excitability of CeA neurons nor membrane properties of parvocellular PVN neurons.
Spontaneous synaptic transmission
Enhancement in GABAergic transmission has been reported in CeA of rodents after chronic ethanol exposure [13, 16], thus sIPSCs were recorded from CeA neurons (Fig. 2a). CeA neurons from abstinent drinkers had higher sIPSC frequency (4.3 ± 0.6 Hz) compared with controls (2.2 ± 0.2 Hz; t(10) = 2.46, p = 0.034; Fig. 2b). No significant differences were found in the amplitude of sIPSCs between controls (104.1 ± 19.0 pA) and abstinent drinkers (91.8 ± 8.4 pA; t(10) = 0.70, p = 0.50; Fig. 2c). There were also no differences in sIPSC rise (controls: 2.2 ± 0.2 ms, abstinent drinkers: 2.8 ± 0.2 ms; t(10) = 1.39, p = 0.19) or decay time (controls: 6.7 ± 0.7 ms, abstinent drinkers: 7.8 ± 0.8 ms; t(10) = 0.91, p = 0.39).

a Representative voltage-clamp traces from the CeA of an ethanol-naive control (left) and abstinent ethanol drinker (right). Summary of frequency b and amplitude c of sIPSCs recorded from the CeA. d Representative voltage-clamp traces from the PVN of an ethanol-naive control (left) and abstinent ethanol drinker (right). Summary of frequency e and amplitude f of sEPSCs recorded from the PVN. *p < 0.05
sEPSCs were recorded from parvocellular PVN neurons (Fig. 2d). The frequency of sEPSCs was higher in abstinent drinkers (4.79 ± 0.97 Hz) compared with controls (2.05 ± 0.54 Hz; t(10) = 2.46, p = 0.03, Fig. 2e). No differences were found in event characteristics (amplitude: controls: 30.25 ± 3.51 pA, abstinent drinkers: 30.31 ± 1.44 pA; t(10) = 0.04, p = 0.97, Fig. 2f; rise: controls 1.66 ± 0.04 ms, abstinent drinkers: 1.67 ± 0.04 ms; t(10) = 0.21, p = 0.84; decay: controls 1.86 ± 0.08 ms, abstinent drinkers: 1.84 ± 0.15 ms; t(10) = 0.09, p = 0.93). Collectively, these data show an increase in presynaptic GABA and glutamate release in the CeA and PVN, respectively, during protracted abstinence with no evidence for altered postsynaptic receptor availability or composition.
Relationship between presynaptic transmission, cortisol, and ethanol intake
In this model, forced abstinence results in a robust increase in circulating cortisol, as illustrated in detail in previous publications on both these same animals [28] and on a previous abstinent cohort [38]. This indicates that the elevated cortisol levels are reflective of the abstinent state rather than a reflection of individual variation. We sought to determine if there was a relationship between presynaptic neurotransmitter release and circulating stress hormones. No relationship was found between sIPSC frequency in the CeA and circulating cortisol (R2 = 0.22, p = 0.12; Fig. 3a) or ACTH (R2 = 0.11, p = 0.28) during abstinence. In contrast, and in agreement with glutamate’s role in activating the HPA axis [47], the sEPSC frequency positively correlated with circulating cortisol (R2 = 0.49, p = 0.012, Fig. 3b), but not with ACTH (R2 = 0.22, p = 0.13) during the final abstinence.

Correlation between cortisol during the final abstinence and CeA sIPSC frequency a or PVN sEPSC frequency b. Summary of baseline frequency by subject and by individual cell analysis across post-abstinence drinking category for the CeA (c, d) and PVN (e, f); (n = 4 ethanol-naive controls; LD = light drinker, n = 1; HD = heavy drinker, n = 4; VHD = very-heavy drinker, n = 3). *p < 0.05
A one-way ANOVA revealed a trend (p = 0.08) between sIPSC frequency in the CeA and post-abstinence drinking category (Fig. 3c). Due to the cellular heterogeneity of the CeA, averaging by subject may mask relevant correlations. Thus, data were also examined by individual cells recorded from subjects in each drinking category (i.e., not averaged by animal), and a significant interaction was observed (F(3,34) = 3.33, p = 0.03; Fig. 3d). These findings prompted questions of synaptic adaptations to ethanol that were long-lasting and likely to influence the probability and extent of relapse drinking. In contrast, there was no significant relationship between drinking category and sEPSC frequency in the PVN when the data were averaged by animal (p = 0.18; Fig. 3e). However, similar to the CeA, the parvocellular PVN is a heterogeneous population, and when the data were analyzed by individual cells, a significant interaction was observed (F(3,47) = 2.95, p = 0.042; Fig. 3f).
Acute ethanol in the CeA
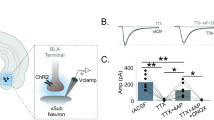
The CeA is mainly a GABAergic nucleus [7,8,9,10, 12, 13]. Our published rodent studies have shown that acute ethanol (44 mM, a maximal and reversable dose, equivalent to 202 mg/dl) significantly increases GABAergic transmission [12]. While glutamatergic activity in the CeA is also sensitive to acute and chronic ethanol [13, 48], in the present study, we chose to investigate GABAergic transmission as the effects of ethanol on GABAergic transmission are more robust, and play a major role as a regulator of the overall CeA activity in the context of chronic ethanol exposure, alcohol dependence, and abstinence [12, 13, 16, 49]. Therefore, we bath-applied 44 mM ethanol to CeA slices collected from control and abstinent drinkers (Figs. 4a, d, respectively). Acute ethanol significantly increased sIPSC frequency in controls (2.1 ± 0.2 Hz to 3.4 ± 0.6 Hz; t(3) = 3.34, p = 0.044, Fig. 4b), but not abstinent drinkers (4.6 ± 0.7 Hz to 4.6 ± 0.6 Hz; t(7) = 0.09, p = 0.93; Fig. 4e). Acute ethanol did not significantly alter sIPSC amplitude in controls (t(3) = 1.85, p = 0.16; Fig. 4c) or abstinent drinkers (t(7) = 1.84, p = 0.11; Fig. 4f). Similarly, acute ethanol did not alter sIPSC rise (controls: 2.2 ± 0.1 ms to 2.7 ± 0.2 ms; t(3) = 2.9, p = 0.063; abstinent drinkers: 2.7 ± 0.3 ms to 2.7 ± 0.2 ms; t(7) = 0.16, p = 0.89) or decay time (controls: 6.4 ± 0.4 ms to 7.0 ± 0.6 ms; p = 0.23; t(3) = 1.51; abstinent drinkers: 6.9 ± 0.8 ms to 7.3 ± 0.8 ms; t(7) = 1.2, p = 0.27).

a Representative voltage-clamp recordings of sIPSCs during pre-ethanol baseline (left) and acute ethanol (right; EtOH, 44 mM) in a CeA neuron from an ethanol-naive control. Summary of the average pre-ethanol application baseline and acute ethanol effects on sIPSC frequency b and amplitude c in CeA neurons from ethanol-naive controls. d Representative voltage-clamp recording of sIPSCs during pre-ethanol application baseline (left) and acute ethanol (right; EtOH 44 mM) in a CeA neuron from an abstinent ethanol drinker. Summary of the average baseline sIPSC frequency e and sIPSC amplitude f in CeA neurons from abstinent ethanol drinkers. Summary of normalized sIPSC frequency g and amplitude h with acute ethanol (EtOH, 44 mM) in the CeA of ethanol-naive controls and abstinent ethanol drinkers. *p < 0.05, #p < 0.01
When sIPSC frequency in CeA neurons was normalized to pre-ethanol application, acute ethanol increased sIPSC frequency to 158.8 ± 16.14% (t(3) = 3.67, p = 0.03) and 103.8 ± 6.1% (t(7) = 0.62, p = 0.55) in controls and abstinent ethanol drinkers, respectively. Controls exhibited a significant increase in sIPSC frequency compared with abstinent drinkers (t(10) = 3.96, p = 0.003; Fig. 4g). No change in sIPSC amplitude was observed following normalization (81.2 ± 11.4% and 91.5 ± 6.6%, in controls and abstinent drinkers, respectively), and no group difference was found in sIPSC amplitude (t(10) = 0.84, p = 0.42; Fig. 4h). These findings suggest that the sensitivity of GABAergic synapses to acute ethanol was diminished in long-term ethanol abstinence, potentially due to long-term elevations in GABA release.
Acute ethanol in the PVN
To the best of our knowledge, these are the first studies to investigate the electrophysiological effect of ethanol on parvocellular neurons of the PVN in any species. With no previous electrophysiological studies from which to select an appropriate dose of acute ethanol, we felt the best approach was to select a concentration near the average BEC measured during the final 3 months of open access (20 mM ethanol is equivalent to 92 mg/dl). Because this dose is physiologically relevant and was achieved by all animals over the course of the study, we believe that this provides a strong foundation for future studies. Glutamatergic sEPSCs were measured before and during bath application of 20 mM ethanol from slices collected from control and abstinent drinkers (Fig. 5a, d, respectively). Data from our laboratory on this same cohort of animals have shown that upon reintroduction of ethanol, cortisol quickly decreases, which we speculate may involve direct effects of an intoxicating dose of ethanol on the PVN [28]. In slices from control subjects, no differences were found in sEPSC frequency (3.17 ± 1.70 Hz to 1.94 ± 1.09 Hz; t(3) = 1.94, p = 0.19; Fig. 5b) or amplitude (33.49 ± 8.80 pA to 26.49 ± 7.17 pA; t(3) = 1.50, p = 0.27; Fig. 5c) with 20 mM acute ethanol. In abstinent drinkers, 20 mM ethanol to PVN neurons resulted in a significant decrease in sEPSC frequency (6.22 ± 2.19 Hz to 2.95 ± 1.08 Hz; t(7) = 2.49, p = 0.042; Fig. 5e) but not amplitude (32.66 ± 1.68 pA to 29.68 ± 2.76 pA; t(7) = 1.31, p = 0.23; Fig. 5f).

a Representative voltage-clamp recordings of sEPSC during pre-ethanol baseline (left) and acute ethanol (right; EtOH, 20 mM) in a PVN neuron from an ethanol-naive control. Summary of frequency b and amplitude c between pre-ethanol baseline and acute ethanol application in ethanol-naive controls. d Representative voltage-clamp recording of sEPSC from an abstinent drinker during pre-ethanol baseline (left) and following application of 20 mM ethanol (right; EtOH 20 mM). Summary of frequency e and amplitude f between pre-ethanol baseline and acute ethanol application in abstinent drinkers. Summary of normalized sEPSC frequency g and amplitude h with acute ethanol (EtOH, 20 mM) in the PVN of ethanol-naive controls and abstinent ethanol drinkers. *p < 0.05, #p < 0.01
When analyzed within-subject as the percent of pre-ethanol application (baseline), the frequency of sEPSCs from both controls and abstinent drinkers was significantly decreased following application of 20 mM ethanol (controls: 63 ± 7% of baseline; t(2) = 5.83, p = 0.028, abstinent drinkers: 47 ± 2% of baseline; t(7) = 3.84, p = 0.006; Fig. 5g) but no effect in the percent change in amplitude (controls: 79 ± 8% of baseline; t(2) = 1.50, p = 0.27, abstinent drinkers: 91 ± 2% of baseline; t(7) = 1.31, p = 0.24; Fig. 5h). Similarly, no differences were found in decay (controls: 1.56 ± 0.24 ms to 1.54 ± 0.32 ms; 99 ± 1% of baseline, t(2) = 0.12, p = 0.92; abstinent drinkers: 1.66 ± 0.20 ms to 1.70 ± 0.19 ms; 102 ± 1% of baseline; t(10) = 0.34, p = 0.72) with acute ethanol application. These data demonstrate that in both controls and abstinent drinkers, an intoxicating (i.e., >80 mg/dl) concentration of ethanol decreased presynaptic glutamate release onto parvocellular PVN neurons.
DISCUSSION
The ethanol self-administration protocol used here captures a hallmark of AUD, namely repeated cycles of heavy drinking, abstinence, and a return to heavy drinking [25]. This “relapse” drinking is a well-documented effect of abstinence across animal and human studies [1, 50, 51] and is a robust and lasting phenomenon under the abstinence protocol employed here [28, 38]. Aberrant stress regulation also associated with relapse to heavy drinking following a prolonged period of abstinence is a hallmark of AUD. In this non-human primate model, we have documented that baseline anxiety, baseline HPA response to the schedule-induced induction procedure, and total fluid intake does not predict future ethanol intakes [28, 29, 52,53,54]. Therefore, HPA axis dysfunction leading to relapse in this model appears to be the result of adaptations to chronic ethanol exposure and withdrawal rather than a response to stress at the initiation to daily drinking. Thus, these studies were designed to better understand the alterations in two key nodes, one extrahypothalamic and the other hypothalamic, involved in ethanol-induced dysregulation in the stress response. Further, we focused on measuring parallel (within animal) changes in synaptic adaptations in these two regions choosing glutamatergic transmission in the PVN and GABAergic transmission in the CeA based on previous work by us and others demonstrating the relevance of these specific neurotransmitters in the context of ethanol exposure [see refs.13, 22].
A key finding of these studies is that there is evidence for increased presynaptic neurotransmitter release within both the CeA and PVN. Specifically, GABA signaling in the CeA and glutamate signaling in the PVN were increased under basal conditions in abstinent drinkers. For GABA sIPSC frequency, the data agree with previous rodent studies showing increased inhibitory signaling in the CeA following chronic ethanol exposure at early (2–10 h) withdrawal [13] and late (5–7 days) withdrawal [49], and a recent study showing decreased GABA transporter (GAT-3) levels and impaired GABA clearance in the CeA of alcohol-preferring rodents and in humans [55]. Within the CeA, the apparent tolerance of presynaptic GABAergic transmission to ethanol observed in abstinent drinkers compared with controls further suggests involvement of inhibitory input to CeA neurons in the higher ethanol intakes during relapse drinking. Note that tolerance to ethanol in GABAergic transmission in CeA slices is in contrast to previous rat studies, where ethanol increases GABA signaling similarly as in CeA neurons of ethanol-naive and ethanol-dependent rodents [13, 16]. We speculate that the loss of an acute effect of ethanol in the CeA in abstinent monkeys may be due to the increased basal GABA release (Fig. 2b) and the extended (28-day) ethanol-abstinent protocol, which we have not tested in rodent models. This suggests that the increase in baseline GABA transmission is produced by long-lasting changes in CeA circuit activity and/or local synaptic adaptations.
For glutamatergic sEPSC, our previous ultrastructural data from parvocellular PVN neurons suggest that glutamate release onto neighboring parvocellular neurons is proportional to ethanol drinks/day after chronic daily drinking for 12 months [22]. The current data showing an increase in sEPSC frequency support this initial evidence of an increase in presynaptic glutamate release and demonstrate that heightened glutamatergic adaptations remain even during prolonged abstinence. Although the resting membrane potential of parvocellular neurons was quite depolarized compared with rodents [42, 43], the cells were spontaneously active and the integrity of the patch was continuously monitored, demonstrating that these neurons were healthy.
Cortisol is elevated during periods of repeated abstinence and returns to pre-abstinence concentrations when ethanol is reintroduced [28, 38]. The present data show that glutamatergic activity onto parvocellular PVN neurons, the neurons responsible for activating the HPA axis, is positively correlated with cortisol during abstinence, thus providing a possible mechanism by which HPA axis activity is increased during abstinence. Furthermore, acute ethanol decreased glutamatergic activity in both abstinent drinkers and controls, suggesting that parvocellular PVN neurons were not tolerant to an intoxicating dose (> 80 mg/dl) of acute ethanol in prolonged abstinence. Rather, the ex vivo ethanol-induced decrease in glutamatergic activity in the PVN of abstinent drinkers brought the glutamatergic transmission into the range measured in control subjects. The elevation in cortisol and glutamatergic activity during abstinence, that is then restored by ethanol consumption or the application of an intoxicating dose of ethanol, respectively, suggests an allostatic shift in HPA function.
The contrasting findings of tolerance to acute ethanol in presynaptic GABA response to the CeA in the abstinent state and similar sensitivity to ethanol in presynaptic glutamatergic response in the PVN could be due to alternative adaptations specific to the brain region, the neurotransmitters measured, or the acute ethanol challenge dose. Because different concentrations of acute ethanol were used in CeA and PVN slices to address the response to an ethanol challenge, equating changes in sensitivity to ethanol in these two brain areas is not possible. However, both concentrations are within the range of average BEC measured during the last 3 months of open-access ethanol self-administration (Fig. 1e). In attempts to equate direct ethanol effects across these brain regions, it is important to acknowledge that these brain regions are not robustly connected. For example, the GABAergic projections from the CeA to the PVN are limited and involve intermediate regions, such as the BNST and/or peri-PVN. Thus, rather than a definitive coadaptation response, the results of the present study provide a critical foundation on which a deeper interconnected view of the hypothalamic and extrahypothalamic stress circuitries can be explored in future studies. For example, cortisol is the primary glucocorticoid and is increased by HPA axis activation. Cortisol acts via genomic and non-genomic mechanisms to influence both the CeA and PVN. However, the response to cortisol in these regions is generally opposite: reducing CRF mRNA in the PVN but increasing CRF mRNA in the CeA [56]. Future studies will be necessary to determine the influence of prolonged elevated cortisol during abstinence and the effects on both the CeA and PVN. Additionally, given that the CeA and PVN are both rich in CRF and CRF receptors, future studies are necessary to determine how the CRF system is engaged over the course of long-term ethanol self-administration and whether this influences signaling in these two key stress regions.
Overall, the present data add to a growing literature from the monkey model of ethanol self-administration that demonstrates disruption in circulating stress hormones, diurnal rhythms [5], and neurocircuitry across the brain including the orbitofrontal cortex [34], striatum [33, 38], and nucleus accumbens [37, 57]. As well, allostatic changes in other stress regions have been found in macaques. For example, the central and medial amygdala nuclei and the BNST are critical regions for integration of emotionally relevant, or psychogenic, stressors [20, 58] and AUD [8]. Importantly, the BNST serves as a relay between the amygdala and PVN [20, 21], and in this monkey model, there is also an increase in the frequency of sIPSCs in the BNST of rhesus males following ~12-months ethanol self-administration [35]. Inhibitory signaling to the BNST, in part, comes from the central and medial nuclei of the amygdala [21], key information in building an ethanol-stress circuitry that contributes to excessive, chronic alcohol consumption associated with AUD.
The foundation for the current studies rests firmly on both rodent and human literature. The human literature documenting disrupted HPA axis function following long-term alcohol consumption served as a guidance for exploring the peripheral and central mechanisms that control and mediate these responses [50, 59, 60]. Animal models, primarily rodents, have been instrumental in gaining an understanding of the mechanistic underpinnings of the interaction between stress and alcohol exposure [1, 61]. The monkey model takes advantage of the similarities between macaques and humans in the rates of absorption and metabolism of ethanol [40, 62], endocrine physiology [see Ref. 5], and brain structure [32] while controlling or measuring critical variables such as age at the first drink and daily intakes to bridge the gap between humans and animal models of this complex disorder. Within the scope of the current cohort, we present an unparalleled look at the state of two critical nuclei within stress circuitry prior to relapse to heavy drinking. It is important to note that although the monkey model closely captures many features of the complex human disorder, several limitations remain. One critical consideration is the balance between the strength of models that emphasize individual differences and the number of subjects needed for statistical power. In this regard, we have presented our electrophysiological data primarily as an average of cells per animal. This approach aims to reduce the variability of dependent measures within-subject while exploring differences between subjects.
In summary, these studies represent a unique effort to understand the consequences of long-term alcohol consumption across the brain. Further studies are required to identify the underlying mechanisms of ethanol allostasis and if these mechanisms can be targeted to provide a foundation for rational therapeutic development. As an initial step in that direction, the present data are the first to simultaneously investigate two nodes of stress circuitry from a primate model that captures the phenotypic cycles of sustained, very-heavy relapse drinking. The long-lasting adaptations in presynaptic GABA and glutamate release in the CeA and PVN, respectively, from the same animals, provide critical information on the state of the brain within and across individuals prior to relapse.
Funding and disclosure
This research was supported in part by grants from the National Institutes of Health: U01 AA013498, AA019431, AA013641, AA006420, AA015566,AA021491, AA 013510 and U01 AA013510. The authors have no disclosures. This is manuscript number 29722 from The Scripps Research Institute.
References
Becker HC. Effects of alcohol dependence and withdrawal on stress responsiveness and alcohol consumption. Alcohol Res. 2012;34:448–58.
Blaine SK, Seo D, Sinha R. Peripheral and prefrontal stress system markers and risk of relapse in alcoholism. Addict Biol. 2017;22:468–78.
Adinoff B, Krebaum SR, Chandler PA, Ye W, Brown MB, Williams MJ. Dissection of hypothalamic-pituitary-adrenal axis pathology in 1-month-abstinent alcohol-dependent men, part 2: response to ovine corticotropin-releasing factor and naloxone. Alcohol Clin Exp Res. 2005;29:528–37.
Breese GR, Sinha R, Heilig M. Chronic alcohol neuroadaptation and stress contribute to susceptibility for alcohol craving and relapse. Pharmacol Ther. 2011;129:149–71.
Jimenez VA, Grant KA. Studies using macaque monkeys to address excessive alcohol drinking and stress interactions. Neuropharmacology. 2017;122:127–35.
Tunstall BJ, Carmack SA, Koob GF, Vendruscolo LF. Dysregulation of brain stress systems mediates compulsive alcohol drinking. Curr Opin Behav Sci. 2017;13:85–90.
Duvarci S, Pare D. Amygdala microcircuits controlling learned fear. Neuron. 2014;82:966–80.
Gilpin NW, Herman MA, Roberto M. The central amygdala as an integrative hub for anxiety and alcohol use disorders. Biol Psychiatry. 2015;77:859–69.
Sun N, Cassell MD. Intrinsic GABAergic neurons in the rat central extended amygdala. J Comp Neurol. 1993;330:381–404.
Veinante P, Freund-Mercier MJ. Intrinsic and extrinsic connections of the rat central extended amygdala: an in vivo electrophysiological study of the central amygdaloid nucleus. Brain Res. 1998;794:188–98.
Herman MA, Contet C, Justice NJ, Vale W, Roberto M. Novel subunit-specific tonic GABA currents and differential effects of ethanol in the central amygdala of CRF receptor-1 reporter mice. J Neurosci. 2013;33:3284–98.
Roberto M, Madamba SG, Moore SD, Tallent MK, Siggins GR. Ethanol increases GABAergic transmission at both pre- and postsynaptic sites in rat central amygdala neurons. Proc Natl Acad Sci USA. 2003;100:2053–8.
Roberto M, Madamba SG, Stouffer DG, Parsons LH, Siggins GR. Increased GABA release in the central amygdala of ethanol-dependent rats. J Neurosci. 2004;24:10159–66.
Funk CK, Zorrilla EP, Lee MJ, Rice KC, Koob GF. Corticotropin-releasing factor 1 antagonists selectively reduce ethanol self-administration in ethanol-dependent rats. Biol Psychiatry. 2007;61:78–86.
Menzaghi F, Rassnick S, Heinrichs S, Baldwin H, Pich EM, Weiss F, et al. The role of corticotropin-releasing factor in the anxiogenic effects of ethanol withdrawal. Ann N Y Acad Sci. 1994;739:176–84.
Roberto M, Cruz MT, Gilpin NW, Sabino V, Schweitzer P, Bajo M, et al. Corticotropin releasing factor-induced amygdala gamma-aminobutyric acid release plays a key role in alcohol dependence. Biol Psychiatry. 2010;67:831–9.
Herman JP, Figueiredo H, Mueller NK, Ulrich-Lai Y, Ostrander MM, Choi DC, et al. Central mechanisms of stress integration: hierarchical circuitry controlling hypothalamo-pituitary-adrenocortical responsiveness. Front Neuroendocrinol. 2003;24:151–80.
Lee S, Selvage D, Hansen K, Rivier C. Site of action of acute alcohol administration in stimulating the rat hypothalamic-pituitary-adrenal axis: comparison between the effect of systemic and intracerebroventricular injection of this drug on pituitary and hypothalamic responses. Endocrinology. 2004;145:4470–9.
Richardson HN, Lee SY, O’Dell LE, Koob GF, Rivier CL. Alcohol self-administration acutely stimulates the hypothalamic-pituitary-adrenal axis, but alcohol dependence leads to a dampened neuroendocrine state. Eur J Neurosci. 2008;28:1641–53.
Crestani CC, Alves FH, Gomes FV, Resstel LB, Correa FM, Herman JP. Mechanisms in the bed nucleus of the stria terminalis involved in control of autonomic and neuroendocrine functions: a review. Curr Neuropharmacol. 2013;11:141–59.
Prewitt CM, Herman JP. Anatomical interactions between the central amygdaloid nucleus and the hypothalamic paraventricular nucleus of the rat: a dual tract-tracing analysis. J Chem Neuroanat. 1998;15:173–85.
Jimenez VA, Helms CM, Cornea A, Meshul CK, Grant KA. An ultrastructural analysis of the effects of ethanol self-administration on the hypothalamic paraventricular nucleus in rhesus macaques. Front Cell Neurosci. 2015;9:260.
Miklos IH, Kovacs KJ. GABAergic innervation of corticotropin-releasing hormone (CRH)-secreting parvocellular neurons and its plasticity as demonstrated by quantitative immunoelectron microscopy. Neuroscience. 2002;113:581–92.
van den Pol AN, Wuarin JP, Dudek FE. Glutamate, the dominant excitatory transmitter in neuroendocrine regulation. Science. 1990;250:1276–8.
American Psychiatric Association. Diagnostic and statistical manual of mental disorders: DSM-5: fifth edition. Arlington, VA: American Psychiatric Publishing; 2013.
Becker HC, Lopez MF. Increased ethanol drinking after repeated chronic ethanol exposure and withdrawal experience in C57BL/6 mice. Alcohol Clin Exp Res. 2004;28:1829–38.
Griffin WC 3rd, Lopez MF, Becker HC. Intensity and duration of chronic ethanol exposure is critical for subsequent escalation of voluntary ethanol drinking in mice. Alcohol Clin Exp Res. 2009;33:1893–900.
Allen DC, Gonzales SW, Grant KA. Effect of repeated abstinence on chronic ethanol self-administration in the rhesus monkey. Psychopharmacol (Berl). 2018;235:109–20.
Baker EJ, Farro J, Gonzales S, Helms C, Grant KA. Chronic alcohol self-administration in monkeys shows long-term quantity/frequency categorical stability. Alcohol Clin Exp Res. 2014;38:2835–43.
Baker EJ, Walter NA, Salo A, Rivas Perea P, Moore S, Gonzales S, et al. Identifying future drinkers: behavioral analysis of monkeys initiating drinking to intoxication is predictive of future drinking classification. Alcohol Clin Exp Res. 2017;41:626–36.
Mello NK, Mendelson JH. Drinking patterns during work-contingent and noncontingent alcohol acquisition. Psychosom Med. 1972;34:139–64.
Miranda-Dominguez O, Mills BD, Grayson D, Woodall A, Grant KA, Kroenke CD, et al. Bridging the gap between the human and macaque connectome: a quantitative comparison of global interspecies structure-function relationships and network topology. J Neurosci. 2014;34:5552–63.
Cuzon Carlson VC, Grant KA, Lovinger DM. Synaptic adaptations to chronic ethanol intake in male rhesus monkey dorsal striatum depend on age of drinking onset. Neuropharmacology. 2018;131:128–42.
Nimitvilai S, Uys JD, Woodward JJ, Randall PK, Ball LE, Williams RW, et al. Orbitofrontal neuroadaptations and cross-species synaptic biomarkers in heavy-drinking macaques. J Neurosci. 2017;37:3646–60.
Pleil KE, Helms CM, Sobus JR, Daunais JB, Grant KA, Kash TL. Effects of chronic alcohol consumption on neuronal function in the non-human primate BNST. Addict Biol. 2016;21:1151–67.
Siciliano CA, Calipari ES, Cuzon Carlson VC, Helms CM, Lovinger DM, Grant KA, et al. Voluntary ethanol intake predicts kappa-opioid receptor supersensitivity and regionally distinct dopaminergic adaptations in macaques. J Neurosci. 2015;35:5959–68.
Siciliano CA, Calipari ES, Yorgason JT, Lovinger DM, Mateo Y, Jimenez VA, et al. Increased presynaptic regulation of dopamine neurotransmission in the nucleus accumbens core following chronic ethanol self-administration in female macaques. Psychopharmacol (Berl). 2016;233:1435–43.
Cuzon Carlson VC, Seabold GK, Helms CM, Garg N, Odagiri M, Rau AR, et al. Synaptic and morphological neuroadaptations in the putamen associated with long-term, relapsing alcohol drinking in primates. Neuropsychopharmacology. 2011;36:2513–28.
Welsh JP, Han VZ, Rossi DJ, Mohr C, Odagiri M, Daunais JB, et al. Bidirectional plasticity in the primate inferior olive induced by chronic ethanol intoxication and sustained abstinence. Proc Natl Acad Sci USA. 2011;108:10314–9.
Vivian JA, Green HL, Young JE, Majerksy LS, Thomas BW, Shively CA, et al. Induction and maintenance of ethanol self-administration in cynomolgus monkeys (Macaca fascicularis): long-term characterization of sex and individual differences. Alcohol Clin Exp Res. 2001;25:1087–97.
Schuckit MA, Tipp JE, Smith TL, Bucholz KK. Periods of abstinence following the onset of alcohol dependence in 1,853 men and women. J Stud Alcohol. 1997;58:581–9.
Hoffman NW, Tasker JG, Dudek FE. Immunohistochemical differentiation of electrophysiologically defined neuronal populations in the region of the rat hypothalamic paraventricular nucleus. J Comp Neurol. 1991;307:405–16.
Luther JA, Tasker JG. Voltage-gated currents distinguish parvocellular from magnocellular neurones in the rat hypothalamic paraventricular nucleus. J Physiol. 2000;523:193–209.
Stern JE. Electrophysiological and morphological properties of pre-autonomic neurones in the rat hypothalamic paraventricular nucleus. J Physiol. 2001;537(Pt 1):161–77.
Tasker JG, Dudek FE. Electrophysiological properties of neurones in the region of the paraventricular nucleus in slices of rat hypothalamus. J Physiol. 1991;434:271–93.
Paxinos G, Huang XF, Petrides M, Toga AW. The Rhesus Monkey Brain in stereotaxic coordinates. Second Edition. Elsevier, 2009. London, UK
Feldman S, Weidenfeld J. Hypothalamic mechanisms mediating glutamate effects on the hypothalamo-pituitary-adrenocortical axis. J Neural Transm (Vienna). 1997;104:633–42.
Roberto M, Bajo M, Crawford E, Madamba SG, Siggins GR. Chronic ethanol exposure and protracted abstinence alter NMDA receptors in central amygdala. Neuropsychopharmacology. 2006;31:988–96.
Herman MA, Contet C, Roberto M. A functional switch in tonic GABA currents alters the output of central amygdala corticotropin releasing factor receptor-1 neurons following chronic ethanol exposure. J Neurosci. 2016;36:10729–41.
Adinoff B, Iranmanesh A, Veldhuis J, Fisher L. Disturbances of the stress response: the role of the HPA axis during alcohol withdrawal and abstinence. Alcohol Health Res World. 1998;22:67–72.
Koros E, Piasecki J, Kostowski W, Bienkowski P. Development of alcohol deprivation effect in rats: lack of correlation with saccharin drinking and locomotor activity. Alcohol Alcohol. 1999;34:542–50.
Grant KA, Leng X, Green HL, Szeliga KT, Rogers LS, Gonzales SW. Drinking typography established by scheduled induction predicts chronic heavy drinking in a monkey model of ethanol self-administration. Alcohol Clin Exp Res. 2008;32:1824–38.
Helms CM, Gonzales SW, Green HL, Szeliga KT, Rogers LS, Grant KA. Diurnal pituitary-adrenal activity during schedule-induced polydipsia of water and ethanol in cynomolgus monkeys (Macaca fascicularis). Psychopharmacol (Berl). 2013;228:541–9.
McClintick MN, Grant KA. Aggressive temperament predicts ethanol self-administration in late adolescent male and female rhesus macaques. Psychopharmacol (Berl). 2016;233:3965–76.
Augier E, Barbier E, Dulman RS, Licheri V, Augier G, Domi E, et al. A molecular mechanism for choosing alcohol over an alternative reward. Science. 2018;360:1321–6.
Groeneweg FL, Karst H, de Kloet ER, Joels M. Rapid non-genomic effects of corticosteroids and their role in the central stress response. J Endocrinol. 2011;209:153–67.
Cervera-Juanes R, Wilhelm LJ, Park B, Grant KA, Ferguson B. Alcohol-dose-dependent DNA methylation and expression in the nucleus accumbens identifies coordinated regulation of synaptic genes. Transl Psychiatry. 2017;7:e994.
Jankord R, Herman JP. Limbic regulation of hypothalamo-pituitary-adrenocortical function during acute and chronic stress. Ann N Y Acad Sci. 2008;1148:64–73.
Blaine SK, Sinha R. Alcohol, stress, and glucocorticoids: From risk to dependence and relapse in alcohol use disorders. Neuropharmacology. 2017;122:136–47.
Mendelson JH, Stein S, McGuire MT. Comparative psychophysiological studies of alcoholic and nonalcoholic subjects undergoing experimentally induced ethanol intoxication. Psychosom Med. 1966;28:1–12.
Lovinger DM, Roberto M. Synaptic effects induced by alcohol. Curr Top Behav Neurosci. 2013;13:31–86.
Green KL, Szeliga KT, Bowen CA, Kautz MA, Azarov AV, Grant KA. Comparison of ethanol metabolism in male and female cynomolgus macaques (Macaca fascicularis). Alcohol Clin Exp Res. 1999;23:611–6.
Acknowledgements
We thank Tim Carlson and Samuel Shin for their assistance and hospitality, Steven Gonzales for exceptional technical support and data processing, and Dr. Jim Daunais for expert assistance in collecting the regions of interest.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Jimenez, V.A., Herman, M.A., Cuzon Carlson, V.C. et al. Synaptic adaptations in the central amygdala and hypothalamic paraventricular nucleus associated with protracted ethanol abstinence in male rhesus monkeys. Neuropsychopharmacol. 44, 982–993 (2019). https://doi.org/10.1038/s41386-018-0290-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-018-0290-7
This article is cited by
-
GDNF gene therapy for alcohol use disorder in male non-human primates
Nature Medicine (2023)
-
A paraventricular thalamus to insular cortex glutamatergic projection gates “emotional” stress-induced binge eating in females
Neuropsychopharmacology (2023)
-
Synaptic effects of IL-1β and CRF in the central amygdala after protracted alcohol abstinence in male rhesus macaques
Neuropsychopharmacology (2022)
-
Importance of sex and trauma context on circulating cytokines and amygdalar GABAergic signaling in a comorbid model of posttraumatic stress and alcohol use disorders
Molecular Psychiatry (2021)
