
Abstract
Fungi are important yet understudied contributors to the microbial communities of the gastrointestinal tract. Starting at birth, the intestinal mycobiome undergoes a period of dynamic maturation under the influence of microbial, host, and extrinsic influences, with profound functional implications for immune development in early life, and regulation of immune homeostasis throughout life. Candida albicans serves as a model organism for understanding the cross-talk between fungal colonization dynamics and immunity, and exemplifies unique mechanisms of fungal-immune interactions, including fungal dimorphism, though our understanding of other intestinal fungi is growing. Given the prominent role of the gut mycobiome in promoting immune homeostasis, emerging evidence points to fungal dysbiosis as an influential contributor to immune dysregulation in a variety of inflammatory and infectious diseases. Here we review current knowledge on the factors that govern host-fungi interactions in the intestinal tract and immunological outcomes in both mucosal and systemic compartments.
Similar content being viewed by others


Introduction
The microbiome is a multi-kingdom community of microorganisms that colonizes all environmentally exposed epithelial and mucosal surfaces of the body, with its greatest biomass and diversity within the gastrointestinal (GI) tract.1,2 The fungal microbiome, also known as the mycobiome, is a fundamental component of this community and its influence on host biology.3 The intestinal mycobiome begins at birth and undergoes dynamic maturation alongside the bacterial and viral communities of the gut.4,5,6,7 Recent advances in sequencing technology and curated taxonomic databases have uncovered the richness and diversity of fungal colonizers, yet the majority of research on fungal-immune interactions to date have focused on a select number of organisms. For example, Candida albicans is the most extensively studied fungal species, from which we have gained a depth of knowledge of the mechanisms of fungal colonization dynamics and fungal-immune interactions in health and disease.5,8 As well, research has begun to shed light on the role of other members of the mycobiome, their cumulative impact on microbiome ecology, and their roles in shaping the immune system of the gut and extraintestinal compartments. With increasing sophistication of multi-omics analysis in human cohorts and gnotobiotic animal modeling, our understanding of the causal mechanisms of fungal dysbiosis in immune, inflammatory, and infectious diseases is expanding.
Herein, we review the impact of intestinal fungal colonization on mucosal immunity in the gut, as well as its far-reaching impact on immune development and function in extraintestinal sites. Beginning with the establishment and development of the fungal microbiome in early life, we review the mechanisms governing the dynamic relationship between gut fungi, bacterial species, and their role in shaping the developing immune system. Using C. albicans as a model organism, we explore the unique impact of dimorphic growth on colonization and persistence within the GI tract, and the implication of yeast versus hyphal growth morphologies on immune homeostasis during eubiosis (the microbiome state associated with host health), and disease pathogenesis during dysbiosis (an altered microbiome state that contributes to host disease). We describe the mechanisms by which fungal dysbiosis contributes to chronic inflammatory disease, with a focus on recent evidence of a key role for fungal dysbiosis in the pathogenesis of inflammatory bowel disease (IBD), as well as chronic mucosal inflammation in extraintestinal organs like the lungs (e.g., asthma). Finally, we review evidence implicating gut fungi as regulators of systemic anti-pathogen immunity, including the emerging mechanism of trained immunity against a variety of infections.
Fungal colonization dynamics in the gastrointestinal tract
Early life establishment of the mycobiome
Amongst the microorganisms that colonize the GI tract, fungi are much less abundant than bacteria, with recent estimates suggesting that fungi make up 1–3% of the total microbes in human feces.9 Despite their modest input to the biomass of the gut microbiota, fungal colonizers make important contributions to the development and regulation of local and systemic immune responses in the host early in life.10 Similar to other body sites, the gut mycobiome starts developing from birth, as fungi have been detected in the gut of neonates as early as one day after delivery.11 Analysis of the fungal ecology of paired samples between mothers and neonates supports vertical transmission as an ecological mechanism for the establishment of the neonatal mycobiome.4 Using DNA fingerprinting to track C. albicans transfer from mother to child at birth, Bliss et al. detected vertical transmission in 65% of infants within the first week of life.12 Furthermore, distinct clusters of mycobiome composition have been identified by fungal 18S rRNA sequencing between infants born via Cesarean-section or vaginal delivery.11 Collectively, these studies implicate vertical transmission as an important determinant of pioneer fungal colonization in the gut. An additional maternally derived source of fungal exposure in early life is breast milk. Analysis of the fungal constituents in breast milk revealed the presence of Candida, Alternaria, Rhodotorula, Malassezia, Davidiella, Sistotrema, and Penicillium, all of which are known to colonize the intestinal tract.13,14,15 Furthermore, culture-based analysis of breast milk microbes confirmed the presence of viable yeast cells, with a high concordance between fungal isolates from breast milk samples and infant feces.16 Thus, there is supportive evidence for maternal sources as a primary route of acquisition of pioneer fungal microbes.
Beyond maternally derived factors, the early-life mycobiome is influenced by a variety of host-intrinsic (e.g., genetics, sex, and age) and extrinsic (e.g., drugs, food, and hygiene) factors during infancy, which have been the topic of recent comprehensive reviews.17,18 These factors help shape a community that shifts during the first year of human life. Wampach and colleagues performed 18S amplicon sequencing of the mycobiome in 15 healthy term Luxembourgish children in the first year of life and observed considerable fungal diversity, with 45 fungal operational taxonomic units (OTUs) detected, and high representation of Saccharomyces cerevisiae reported in 9 sequenced samples from 1-day-old babies.11 Larger studies of the fecal mycobiome have reported a reduction in alpha-diversity and shifts in major taxa throughout infant development.19,20 A longitudinal study of 308 US infants found that mycobiome composition, surveyed through ITS2 sequencing, significantly changed over time, with shifts in fungal beta-diversity (community composition) linked to participant age, and a reduction in fungal alpha-diversity (community richness and evenness) with increasing infant age.20 The fecal mycobiome shifted from being dominated by Malassezia and Saccharomyces in neonates (~1 month old) to Saccharomyces and Candida in infants (11 months old).20 The latter approximates the mycobiome composition in healthy adults,5,21 suggesting a more mature fungal community composition after 1 year of age.
One common feature in mycobiome studies is a very high degree of interindividual variability, with only a few core genera consistently represented in most individuals including Candida, Saccharomyces, and Malassezia.4,5,11 Beyond these abundant and consistent taxa, a high degree of variability is seen in the representation of other taxa including yeasts (e.g., Rhodotorula, Pichia, and Trichosporon), filamentous fungi (e.g., Rhizopus, Phoma, Penicillium, and Phanerochaete), and clinically relevant opportunistic fungi (e.g., Aspergillus, Cryptococcus, and Cladosporium), among many other genera.4,11,20,22,23 This suggests that the recently reported strong immune modulation induced by the mycobiome,10,24,25 may be driven by the most abundant or commonly detected taxa (e.g., Candida and Saccharomyces), with infrequently detected or less abundant taxa potentially being less influential in immune development. However, extrinsic factors leading to microbiome perturbations, such as antibiotic or antifungal treatment, provide an opportunity for rare fungal species to emerge as immunologically dominant taxa, potentially contributing to the development of immune-mediated disorders, including allergic airway diseases.26,27,28
Intrinsic and extrinsic regulators of the gut mycobiome
Factors that influence bacterial microbiome development have begun to be evaluated in terms of their influence on the gut mycobiome. These can be categorized into intrinsic factors, such as transkingdom ecological interactions, and extrinsic factors, such as antimicrobials and diet.
Ecological interactions
Recent investigations into the ecological mechanisms regulating transkingdom interactions have revealed key roles for bacterial products as determinants of fungal growth, colonization, and virulence in the gut. For example, short-chain fatty acids (SCFA) produced by commensal bacteria can act on neighboring fungi to influence their replication and morphogenesis. The SCFA butyrate inhibits C. albicans germ tube formation,29 which is required for epithelial adhesion and invasion.30 Acetate, butyrate, and propionate have all been shown to inhibit growth kinetics and hyphae formation of C. albicans and Pichia kudriavzevii in vitro, which can impair stable colonization of the gut.31,32 Other bacterial metabolites such as lactic acid produced by Lactobacillus spp. suppress C. albicans colonization of the gut in vivo,33 while other Lactobacillus derived compounds have roles in reducing viability and filamentation of C. albicans.34,35
Additional transkingdom modulation of fungi in the gut is mediated by bacterial cell wall components. Liberation of peptidoglycan from bacteria in response to antibiotics potentiated hyphae formation, mucosal invasion, and extraintestinal dissemination by C. albicans.36,37 Furthermore, certain strains of Escherichia coli can release soluble factors that directly kill C. albicans,38 adding to the well-known role of this bacterium in inhibiting C. albicans replication, biofilm formation, and intestinal colonization.39,40,41 While several microbial mechanisms that influence mycobiome composition have been described, the metabolic complexity within the gut microbiome likely harnesses additional unknown mechanisms that help establish and maintain this microbial ecosystem during eubiosis.
Antimicrobials
Although aimed to treat bacterial infections, antibiotic administration also has the collateral effect of disrupting the microbiota, promoting major compositional, functional, and symbiotic changes to both bacterial and fungal communities.42,43,44 For example, depletion of SCFA-producing bacteria with antibiotics results in increased C. albicans colonization.31,45 Similar to antibiotic treatment, antifungals can also induce dysbiosis in the mycobiome. Prolonged fluconazole administration to mice induces the expansion of filamentous fungi Aspergillus, Wallemia, and Epicoccum spp. and a decrease in Candida sp..46 Also, a study of allogenic hematopoietic cell transplant patients receiving prophylactic micafungin reported intestinal fungal expansion and increased blood stream infection by species of the C. parapsilosis complex.47 These effects likely underscore intra- and inter-kingdom interactions in regulating mycobiome composition, with antimicrobial disruption directly impacting ecological interactions maintained during eubiosis.
Diet
Dietary factors have started to be evaluated in mice and humans. A high-fat diet resulted in a higher abundance of C. albicans in mice, in contrast to a standard diet, which favored the growth of S. cerevisiae.48 There is inter-experimental variability in the findings of human studies which have looked at the effect of western diets, animal-based diets, or vegetarian and plant-based diets.18 Nonetheless, these studies provide strong evidence that diet influences the community structure and composition of the mycobiome. On that matter, obesity is also associated with a shift in mycobiome composition. Obese individuals have a consistent decrease in fungal diversity compared to healthy controls, with Mucor spp. being negatively associated with obesity in adults, and S. cerevisiae and C. albicans being reduced in the gut mycobiome of obese children.49,50 However, it is currently unknown if mycobiome shifts are a result of an obesogenic diet, or contribute to the disease process itself.
Fungal morphogenesis and microbe-host interactions
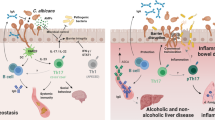
A unique characteristic of the mycobiome that distinguishes it from the bacterial microbiome is the ability of some fungi, such as Candida spp., to exist in several morphological growth forms. This adds a fascinating layer of complexity to host-mycobiome dynamics, in which fungi respond to the milieu of environmental signals in the gastrointestinal environment through metabolic shifts, similar to bacteria,51 but also through the evolutionary selection of morphological adaptations which ultimately support the establishment and persistence of these fungi within their niche.52,53 Several host-associated fungi impressively overhaul their transcriptional and phenotypic programs to achieve competitive fitness in the gut microenvironment. This is best exemplified by the dimorphic growth characteristic of C. albicans (Fig. 1a), which can grow as a unicellular yeast or as filamentous hyphae in response to a variety of gut stimuli (e.g., pH, O2, CO2 and nutrients).54,55 It is now well established that the morphological growth program of C. albicans engages in profound bi-directional interactions with the gut microenvironment of the host, with critical implications for commensalism and pathogenic potential52,55,56 (Fig. 1b, c).

a Antibiotic-treatment and microbiome perturbations lead to fungal overgrowth and morphological changes in fungal colonizers. C. albicans morphological switch is mediated by master transcription factors (e.g., Efg1 and Wor1) that orchestrate gene expression and the transition from commensal yeast form to pathological hyphal growth.52,53 Commensal bacteria and secreted metabolic products (e.g., SCFA) suppress hyphae formation and can induce commensalism.29,31,32,35 Community disruption and release of bacterial cell wall components (e.g., peptidoglycans) allows for hyphal growth, associated with intestinal inflammation and extraintestinal dissemination.36,37 b During eubiosis, fungal interactions with the mucosal immune system induce tolerogenic and homeostatic immune responses. Fungal sensing by tolerogenic DCs induce IL-10 secretion and Tregs in intestinal lamina propria.82,83 Intestinal pathobionts also induce class switching and systemic release of antifungal IgG.24 Similarly, induced Th17 responses, and secreted cytokines IL-17 and IL-22 further contribute to barrier integrity by supporting mucus production, antimicrobial peptides (e.g., β-defensins), and immunoregulatory cytokines (e.g., TSLP and IL-6) released by epithelial cells.104,108,142,143,144 c Following microbiome disruption, hyphae formation allows for epithelial adhesion and cell damage, eliciting mucosal inflammation. Hyphae detection by inflammatory DCs induces Th2 responses and eosinophilia.82 Intestinal fungi uptake by cDC2 and CX3CR1+ MNP induces antibody class switch and secretory IgA towards the mycobiome.60 Hyphal cell recognition by NLRP3 inflammasome releases pro-inflammatory cytokines (e.g., IL-1β and IL-18) that support Th1 and Th17 differentiation.112,114,115 Additionally, candidalysin, the cytotoxic peptide secreted by C. albicans hyphal cells, induces epithelial damage and release of alarmins, further supporting antifungal inflammatory responses and neutrophilia in the intestinal lamina propria.103,104,108,112 SCFA short-chain fatty acids; PPG peptidoglycans; AMP antimicrobial peptides; DC dendritic cells; MNP mononuclear phagocytes; Treg regulatory T cells; Th T helper cell responses (type1, type 2, and type 17); TSLP thymic stromal lymphopoietin; Eos, eosinophils; Neut neutrophils; NLRP3 NOD-and pyrin domain-containing protein 3.
The yeast form of C. albicans is generally associated with enhanced fitness for colonization and commensalism, whereas the hyphal form is associated with virulence and invasiveness. Mutated strains of C. albicans that are genetically locked in a hyphal morphotype display reduced fitness to colonize the gut.57 Furthermore, passaging of C. albicans through the gut of mice favors the development of a yeast-locked phenotype with enhanced fitness for gut colonization that is mediated by the accumulation of mutations in genes that regulate the hyphal morphogenesis program such as Efg1 and Flo8.53 Alternatively, passage through the mouse gut induces a unique host-adapted morphogenesis program termed GUT (gastrointestinal induced transition), through upregulation of Wor1 expression, that is physiologically and metabolically adapted for intestinal colonization and growth.52
The enhanced fitness of yeast cells is also facilitated by selective pressures from the gut microenvironment. For example, the complexity of the bacterial community can influence C. albicans growth morphology, with yeast forms predominating in gnotobiotic mice monocolonized with C. albicans, whereas colonization of conventional mice results in a mixture of yeast and hyphal forms.31,58 Mucosal immune responses can also provide selective pressure that favors colonization by yeast morphotypes. Recent work showed that mucosal IgA specific to hyphal-associated epitopes neutralized hyphal cells and favored the yeast morphology.59,60 Collectively, these findings exemplify the multi-directional cross-talk between fungi, bacteria, and host immunity in the gut microenvironment to regulate fungal colonization dynamics.
In addition to C. albicans, other fungal colonizers exhibit complex morphogenetic programs, but the implications on gut colonization and symbiotic relationships with the host have not been explored. S. cerevisiae is a common member of the gut mycobiome unable to form true hyphae and is often considered a benign microbe, but has been shown to form pseudohyphae, an intermediate growth form between the yeast and hyphal morphologies associated with invasive infections.61,62,63 Malassezia spp. are common members of the skin mycobiome and are predominantly found as yeast cells.64 Notably, some Malassezia species (M. furfur, M. globosa, M. restricta, and M. sympodialis) can form hyphae in association with a shift to pathogenesis.65,66 Cryptococcus neoformans, a common pathogenic fungus not typically found as a commensal,67 can proliferate during dysbiosis associated with Crohn’s disease (CD), leading to adverse disease outcomes.68 C. neoformans can also undergo a yeast to hyphal switch during mating, yet the yeast form is most injurious to the host.69,70 Altogether, the morphological growth and secreted factors of host-associated fungi is an integral factor to consider when studying fungal alterations and dysbiosis. Continued research in this field is necessary to further characterize the regulation of polymorphism and the eubiotic morphogenic status of fungi in the microbiome.
Mucosal mycobiome–immune interactions
As omnipresent members of the intestinal microbiome, fungi can elicit homeostatic and detrimental immune responses in the mammalian host24,71,72,73 (Fig. 1). The following sections describe the innate and adaptive mechanisms underlying the crosstalk between intestinal fungi and the gut mucosal immune system in both scenarios.
Intestinal fungi in mucosal immune homeostasis
Immune recognition of fungi in the intestine is initiated by the engagement of innate pattern recognition receptors (PRR), expressed by epithelial and myeloid cells, with their specific microbial associated molecular pattern (MAMP) ligands.74 Among PRR, C-type lectin receptors (CLR) play a primary role in antifungal immunity due to specificity for various carbohydrate-rich motifs present in fungal cell walls (reviewed in Refs. 3,74,75) Deficiency of the CLR dectin-1 (Clec7a) leads to recurrent mucosal infections and enhanced fungal dissemination by dampened immune responsiveness to pathobionts.76,77,78 Besides dectin-1, dectin-3 (Clec4d) and the soluble mannan-binding lectin (MBL) are also important CLR involved in fungal recognition and intestinal homeostasis.79,80 In addition, lamina propria C-X3-C motif chemokine receptor 1-positive (CX3CR1+) mononuclear phagocytes (MNP) initiate and drive immune responses towards fungal colonizers in murine models.24,81 Gut resident CX3CR1+ MNP upregulate expression of genes involved in fungal recognition, including CLR dectin-1, dectin-2 (Clec6a), and Mincle (Clec4e), and efficiently phagocytose C. albicans and other fungal species in vivo.24 Immune responses to fungi are also Toll-like receptor (TLR) dependent, demonstrated by TLR-induced tolerogenic responses in dendritic cells (DCs) and defective neutrophil adherence and killing of C. albicans in TLR1−/− and TLR2−/− mice.82,83,84 Deficiency in TLR2 further promotes increased colonization and extraintestinal dissemination of C. albicans in immunosuppressed mice.85,86 While other PRR (e.g., TLR4, NOD-like receptors) may influence antifungal responses, they may play a less influential role during homeostasis, and instead mediate inflammatory responses during dysbiosis.75,87,88,89,90,91,92
Intestinal fungi are also critical for the induction of innate and adaptive tolerogenic responses. Shortly after birth, fungal colonization supports immune ontogeny by inducing the recruitment of retinol dehydrogenase positive (RALDH+) DCs from the intestinal lamina propria to secondary lymphoid organs (SLO).25 Fungal colonization-induced RALDH+ DC trafficking supports gut-associated lymphoid tissue (GALT) development, inducing lymphocyte gut homing to Peyer’s Patches and mesenteric lymph nodes (mLN).25 Besides GALT formation, the known role of RALDH enzymes in the production of retinoic acid also supports tolerogenic responses by this DC population.93,94,95 Work by Bonifazi et al. also showed that Peyer’s Patch-isolated DCs induce tolerogenic responses to C. albicans by engagement of TLR4, inducing TIR-domain-containing adapter-inducing interferon-β (TRIF) and non-canonical nuclear factor-kappaB (NF-kB) pathway.82 Similarly, the yeast-derived fungal cell wall component zymosan induces tolerogenic DCs via engagement of TLR2.83 These tolerogenic DCs show higher secretion of IL-10 and reduced secretion of IL-6, IL-12(p40), IL-12(p70), and TNF-α pro-inflammatory cytokines in a mitogen-activated protein kinase (MAPK) signaling-dependent matter.83 Altogether, these studies support the role of pioneer fungal colonizers in inducing tolerogenic DCs and regulatory T effector (Teff) responses in the gut.
Beyond mucosal tolerogenic responses, fungal colonization influences the delicate network of the various Teff subsets in the intestinal mucosa, which are critical for intestinal homeostasis by preventing aberrant inflammation and maintaining barrier integrity.96,97 Eubiotic conditions favor constant luminal sampling that results in heterogeneous Teff responses to fungal colonizers24 (Fig. 1b). T helper 17 (Th17) cells are the most abundant effector response against C. albicans, which has been proposed to increase Th17 proportions in the colon through epithelial cell adherence.8,98 Although, C. albicans also induces lower proportions of Th1 and Th2.99 Additionally, C. albicans can induce differentiation of naive CD4+ T cells to a non-conventional Th1 subset, characterized by elevated production of IFN-γ and reduced IL-17A, IL-22, and IL-4.99 Mice colonized with a yeast-locked C. albicans strain (flo8 mutant) induced IL-10 secretion by DCs and macrophages, supporting survival and efflux of naive CD4+ T cells from the thymus in a dectin-2-dependent matter.100 This systemic priming with a flo8 mutant increased Th1-biased immune response in subsequent infection models with C. albicans and polymicrobial sepsis,100 supporting the role of yeast-induced immune priming in triggering Teff responses and preventing disseminated infections.
As with the bacterial microbiome, mycobiome–immune interactions in the gut are bidirectional (Fig. 1b). The mycobiome is shaped by selective pressure from mucosal immunoglobulins. As mentioned above, secretory IgA produced in response to intestinal colonization by C. albicans is preferentially directed against the hyphal morphotype, producing a negative selective pressure that favors colonization by the yeast form.59,60 Mucosal phagocytes also contribute to shaping the mycobiome through recognition of intestinal fungi and regulation of antifungal humoral responses. Interestingly, while systemic antifungal IgG responses seem to be dependent on CX3CR1+ MNP only, luminal IgA responses are significantly reduced in mice lacking both CX3CR1+ MNP and CD11c+CD11b+CD103+ conventional DC (cDC2), supporting the role of both cell populations in inducing IgA responses in GALT.9,24,60 It has been also shown that Cx3cr1−/− mice harbor dysbiotic mycobiome, with overrepresentation of the Ascomycota phylum, although no changes were observed in the absence of cDC2 cells,24 suggesting that CX3CR1+ MNP are critical to sustain fungal gut eubiosis. Further, antimicrobial peptides (AMP) released by intestinal epithelial cells contribute to colonization fitness as well as spatial distribution fungi in the gut lumen.101 These examples highlight the intricate communication between the immune system and the mycobiome, in which bidirectional interactions modulate homeostatic immune responses and intestinal eubiotic conditions.
Fungal dysbiosis and intestinal immunopathology
Mycobiome perturbations can constitute dysbiotic states resulting in pathological mucosal immune responses (Fig. 1c). Mechanistically, this is perhaps best exemplified by the consequences of antibiotic-induced dysbiosis for Candida-immune interactions. By disrupting homeostatic transkingdom interactions between commensal bacteria and Candida, antibiotic treatment enables hyphal morphogenesis. Hyphal Candida expresses adhesins and other virulence factors that induce epithelial damage, barrier disruption, immune activation, production of pro-inflammatory cytokines, and the recruitment of inflammatory cells.59,102,103,104 Fungal infection and epithelial cell activation/damage can release alarmins, which further activate antigen presenting cells and exacerbate innate and adaptive immune responses.105 Yeast cells differentially activate innate immune responses in epithelial cells, with hyphal recognition and increased fungal burden being required for second-phase MAPK signaling cascade and pro-inflammatory responses.104 Moyes et al. demonstrated that NF-κB activation in epithelial cells occurs independently of fungal morphology, yet the hyphal growth induces the secondary MAPK signaling pathway, leading to MKP1 and c-Fos activation, and stronger cytokine and chemokine production, including G-CSG, GM-CSF, IL-1, and IL-6.104,106 This enhanced pro-inflammatory response was only initiated past a certain hyphal burden, suggesting tightly regulated immune mechanisms able to differentially respond to colonizing versus pathogenic fungal forms. Additionally, intestinal epithelial cells exposed to hyphae result in increased secretion of IL-8, a pro-inflammatory cytokine with notorious chemoattractant property on neutrophils.107,108,109 Overall, differential signaling pathways mediate proinflammatory responses elicited by the hyphal morphology of C. albicans during conditions of community disruption.
While its precise role in GI colonization and extraintestinal dissemination remains to be explored in vivo, the hyphae-secreted peptide toxin candidalysin, induces cytotoxic damage in in vitro models of oral,103 vaginal,110 and intestinal epithelial cells,102 besides eliciting endothelial damage and supporting systemic fungal infection in mice.111 Candidalysin can also activate proinflammatory pathways in mucosal phagocytes such as NOD-and pyrin domain-containing protein 3 (NLRP3) inflammasome and caspase-1 activation.112 While the yeast form of C. albicans is unable to induce the inflammasome,113 hyphal sensing by the NLRP3 inflammasome induces the release of IL-18 and IL-1β by myeloid cells, and enhanced proinflammatory Teff responses (Th1 and Th17).112,114,115 In addition to promoting pathological inflammation, activation of the NLRP3 inflammasome by C. albicans can also promote immune evasion of this pathobiont by impairing phagocytosis and inducing cell death.112 Hyphal outgrowth of Candida during antibiotic-induced dysbiosis can also contribute to dysregulated intestinal inflammation through TLR pathways. Candida-sensing via TLR4 induces pro-inflammatory cytokine secretion in macrophages, including CXCL1, IL-1β, and TNF-ɑ.91 TLR4 is also important for controlling Candida dissemination and neutrophil recruitment to sites of infection.91,92 TLR2 engagement in inflammatory DCs activates the myeloid differentiation primary response 88 (MyD88) and canonical nuclear NF-κB signaling, also leading to Th2 (CD4+GATA3+) cell accumulation in mLN.82 The authors also reported a higher activation of reactive oxygen species in hyphae-pulsed DCs, potentially involved in the activation of the Jun N-terminal kinases 1 and 2 (JNK 1/2) signaling pathway and Th2 induction by these inflammatory DCs.82 Altogether, differential activation of DCs by yeast and hyphal forms of C. albicans can mediate distinct immune responses in intestinal tissue by inducing homeostatic and inflammatory DC functions, respectively.
Fungal dysbiosis also elicits regulatory mechanisms that aid in fungal immune control. During candidiasis, there is an expansion of Tregs in murine intestinal tissue and draining mLN, which dampens Th1/Th2 responses while boosting antifungal Th17 immunity.82,116,117,118,119 Interestingly, fungal interaction with indoleamine-2,3-deoxigenase (IDO)+ DCs induced Treg responses that controlled overt Th1/Th17 activity. However, yeast cells were not able to stimulate IDO expression, suggesting hyphal cells are uniquely capable of inducing Th17 effector responses via the expansion of Tregs.116 Similarly, differential interaction of yeast and hyphae cells with Peyer’s Patch DCs affect local antifungal responses. Yeast cell interaction with proinflammatory DCs induced Th2 differentiation, while hyphae promoted Th1/Treg differentiation to control fungal growth,82 further demonstrating the intricate role of fungal morphology on ensuing immune regulatory responses.
Emerging evidence implicates mycobiome perturbations as an important contributor to chronic intestinal inflammation. For example, antifungal-induced expansion of the fungi Aspergillus amstelodami, Epicoccum nigrum, and Wallemia sebi exacerbated experimental models of colitis.46 However, the collateral antimicrobial-induced shifts to the bacterial microbiome may confound these results.46 Mycobiome dysbiosis may also contribute to IBD pathogenesis in humans. Increased absolute abundance of C. albicans has been associated with IBD flares,120 and its relative abundance is also predictive of clinical remission after fecal microbiota transfer (FMT) treatment, which led to decreased Candida sp. abundance.121 This work revealed the role of mycobiome dysbiosis in chronic colitis development and resolution, likely through a combination of immune and ecological mechanisms.
Further evidence supporting the causal contribution of fungal immune recognition mechanisms in IBD pathogenesis stems from genome wide association studies (GWAS). These include the association between Clec7a haplotype rs2078178-rs16910631 and increased ulcerative colitis (UC) severity, and the CX3CR1 T280M polymorphism linked to impaired antifungal antibody responses in CD patients.24,122 Microbial-genotype association studies have further elucidated fungal recognition immune functions with specific fungal taxa that modulate disease severity. For instance, the caspase recruitment domain-containing protein 9 (CARD9)S12N risk allele has been linked to increased abundance of the yeast M. restricta in IBD patients.123 In contrast, Sokol et al. showed that the dectin-1 Clec7a single nucleotide polymorphism (SNP) rs2078178 (also associated with medically refractory UC) was inversely associated with a different Malassezia species, M. sympodialis during IBD flares.120 Similarly, the IBD-associated CARD9 SNP rs10781499 trended toward negative correlation (p = 0.072) with the abundance of S. cerevisiae,120 potentially supporting a protective role of some fungal species in preventing intestinal inflammation. Altogether, these studies portray the important contributions of mycobiome–immune interactions to the development and persistence of IBD.
Accordingly, several of the immune functions implicated in GWAS have been evaluated in animal models. Dectin-1 deficiency resulted in increased severity of dextran sulfate sodium (DSS)-colitis and increased circulating anti-S. cerevisiae antibodies (ASCA) IgG and IgM,122 supporting a loss of tolerance against intestinal fungi. Similarly, mice lacking intestinal CX3CR1+ MNPs exhibit increased susceptibility to DSS-induced colitis due to a failure to mount protective antifungal immune responses.24 M. restricta was found highly abundant in the inflamed intestinal mucosa of CD patients and exacerbated the disease through CARD9-mediated activation of macrophages and DCs.123 Likewise, expansion of Debaryomyces hansenii has been identified in mucosal wounds in colitis models and CD patients, directly contributing to impaired wound healing through induction of the type-1 IFN-CCL5 axis in mucosal macrophages.124 However, activation of spleen tyrosine kinase (Syk)-CARD9 signaling in myeloid cells, downstream of CLR recognition of the gut mycobiome, protected against colitis and colon cancer in an azoxymethane-DSS model.125 This effect was mediated by inflammasome activation and subsequent maturation of IL-18, and reduced hyperplasia in colonic epithelia.125
Overall, these findings underline the delicate relationship between the mycobiome and intestinal inflammation, whereby the outcomes of immune interactions with these fungi is highly dependent on context. Research to date has unveiled innate detection of intestinal fungi as critical in maintaining appropriate mucosal interactions and preventing the development of fungi-associated inflammation. However, given the diversity of fungal species associated with dysbiosis of the mycobiome,46 further research is needed to understand the mechanisms by which other fungal taxa contribute to intestinal immunopathology. In addition, given the ecological effects of fungi on the bacterial microbiome,10,126 future studies should also incorporate bacterial microbiome analysis, as this may help explain divergent results from animal studies colonized with fungal species.
Mycobiome–immune interactions beyond the gut
Intestinal mycobiome in the gut-lung axis
The mycobiome can prime the immune system and modulate inflammatory outcomes at extraintestinal mucosal sites, especially in the airways (Fig. 2a). Using gnotobiotic mice, we determined a causal contribution of fungal colonization on early-life immune development and allergic airway disease, independently and in combination with bacteria.10 Exclusive yeast colonization augmented monocyte/macrophage lung infiltration in the airways in response to allergic ovalbumin challenge.10 Similarly, antibiotic treatment induced gut fungal overgrowth and alternative macrophage (M2) polarization in the airways, supporting allergic airway inflammation.127 Further, P. kudriavzevii, a yeast found increased in 3-month-old infants who developed atopic wheeze at 5 years, exacerbated house dust mite (HDM)-induced airway inflammation in mice.28,32 These mice displayed increased type-2 and type-17 airway inflammation and elevated circulating IgE, highlighting the ability of this yeast to prime adaptive immunity, despite only transient colonization of young mice.32 Beyond the classical Th2-induced allergic response, Th17 responses induced by C. albicans can have off-target effects by increasing airway inflammation in mice and humans.8,128 Intestinal colonization with C. albicans led to immune cross reactivity with other fungal antigens via Th17 cells.8 Expansion of these Th17 cells resulted in enhanced chronic immune responses against Aspergillus fumigatus in the airways of patients with inflammatory respiratory diseases, including asthma, chronic obstructive pulmonary disease, and cystic fibrosis.8 Ultimately, intestinal fungi have a role in priming immune responses and increasing susceptibility to allergic airway inflammation via Th2 and Th17 mechanisms.

a The intestinal mycobiome has been associated with increased susceptibility to airway inflammation. Th2 airway inflammation has been shown to be increased by various intestinal fungi, including P. kudriavzevii, A. amstelodami, E. nigrum, and W. mellicola.27,32,46 C. albicans colonization leads to an expansion of C. albicans-specific and cross-reactive Th17 cells, which promotes pathogenic inflammatory responses to A. fumigatus in the airways.8,128 Yeast colonization from early life has been demonstrated to increase monocyte infiltration in the lungs during ovalbumin-induced inflammation in adulthood.10 Antibiotic perturbation of the mycobiome, leads to C. albicans overgrowth and consequent increases in prostaglandin E2 (PGE2), leading to the polarization and activation of pulmonary M2 macrophages.127 Fungal colonization and antibiotic-induced fungal expansion also enhanced allergic responses and eosinophilia in the airways.10,26,27,81 b Enhanced systemic immunity by the intestinal mycobiome provides protection from infection. C. albicans colonization primes Th17 cells and enhances IL-17 responsive neutrophils, significantly contributing to anti-fungal defenses.128 The wild mycobiome is associated with increased circulating CD44hi T cells and neutrophils.138 Through trained immunity, intestinal fungi may epigenetically reprogram monocytes, leading to an increase in the secretion of canonical proinflammatory cytokines which are protective against systemic infections.72,134,135,136 In addition, fungal colonization can increase IgG, with C. albicans colonization being integral to the production of protective IgG responses against infection.9 Immune-responses elicited by C. albicans intestinal colonization protects the host from extraintestinal dissemination, besides inducing cross-kingdom protection from infection (e.g., S. aureus and C. difficile).128,131
Other less abundant members of the mycobiome have also been implicated in intestinal immune priming of airway inflammation. Wheeler et al. showed that colonization with A. amstelodami, E. nigrum, and W. sebi was sufficient to recapitulate the HDM-induced airway inflammation seen with antifungal-induced expansion of these fungal species.46 It was later determined that Syk signaling in CX3CR1+ MNPs in the intestine is necessary to induce the Th2 phenotype underlying the exacerbated HDM-induced airway inflammation in this model.81 Expanded intestinal Wallemia mellicola was also associated with exacerbated airway disease in an HDM-induced airway inflammation model.27 HDM restimulation of mediastinal lymph nodes from these mice resulted in elevated IL-13 expression by CD4+ T cells, but no differences in IFN-γ or IL-17.27 This work suggests that fungi-induced allergic airway inflammation can occur through several immune mechanisms beyond the potent Th17 responses elicited by C. albicans, and likely involves different fungal colonizers, many of which remain to be studied.
Influence of mycobiome–immune interactions on systemic defense against pathogens
Intestinal fungal colonization can increase protection against infection through immune priming. C. albicans intestinal colonization induces protective immune responses against invasive candidiasis, mediated by elevated systemic anti-C. albicans Th17 cells and IL-17 responsive neutrophils128 (Fig. 2b). However, it remains unclear whether these responses would be protective against infections by other fungal colonizers. C. albicans intestinal colonization is also critical for the development of protective IgG responses against disseminated infection of Candida auris to the brain but not the kidneys.9 In contrast, common environmental, skin, and food derived fungi, such as Cladosporium cladosporioides, M. restricta, and S. cerevisiae, respectively, failed to elicit IgG responses in the host.9 This reveals less potent immunogenicity in these colonizers and suggests that cross protection against other fungi implicates phylogenetically similar fungal taxa.
As with mucosal responses, innate immune detection is critical for immune control of systemic infections. CARD9 signaling in intestinal CX3CR1+ MNPs is required for the development of anti-C. albicans IgG.9 Individuals with mutant CARD9 alleles suffering from systemic candidiasis displayed reduced circulating anti-C. albicans IgG relative to wild-type individuals.9 However, the relative risk of systemic infections in individuals carrying mutant CARD9 alleles remains unknown. In addition, C. albicans colonization resulted in more robust production and improved reactivity of anti-Candida systemic IgG, than systemic C. albicans infection,85 emphasizing the importance of mucosal immune interactions in driving these systemic responses.
Aside from enhancing protection against fungal infections, the mycobiome can also promote protective immune responses against heterologous pathogens (Fig. 2b). For example, C. albicans colonization primed Teff responses that provided cross-kingdom protection against systemic Staphylococcus aureus infection.128 Consistent with these findings, a vaccine against the N-terminus of Candida adhesin Aslp3 was reported to provide T cell mediated cross protection against both systemic candidiasis and S. aureus infection in mice.129,130 Additionally, C. albicans colonization prior to Clostridium difficile infection mediated increased expression of IL-17A in the colon in a protective manner.131 Therefore, expansion of C. albicans in the gut, which is commonly associated with mycobiome dysbiosis, may result in collateral beneficial consequences for host responses to infection, and may provide a framework for effective cross-kingdom preventative strategies against invasive pathogens.
The intestinal mycobiome may also influence host defenses by eliciting trained immunity (Fig. 2b). While immunological memory is a property usually associated with adaptive immunity, epigenetic changes in innate immune cells have emerged as another mechanism of immune memory induction, allowing for faster responses to previously exposed pathogens.132,133 It was first discovered that trained immunity applied to antifungal defense when low-dose C. albicans infection in Rag1−/− mice improved survival to subsequent lethal C. albicans infection.134 In this study, dectin-1 recognition of C. albicans β-glucan elicited epigenetic changes in monocytes, mediated by rapidly accelerated fibrosarcoma protein 1 (Raf-1) signaling, ultimately leading to enhanced IL-6 and TNF-ɑ secretion and candidacidal activity.134 This same pathway was also found to induce trained immunity in human peripheral blood mononuclear cells (PBMC) to TLR ligands and commensal bacteria in vitro,135 supporting cross kingdom priming of innate immunity by this fungal colonizer. Similarly, S. cerevisiae has also been reported to induce trained immunity mediated by chitin recognition in monocytes.72 Innate immune training with chitin improved survival and fungal clearance in systemic C. albicans infection, accompanied by increased IL-6, TNF-ɑ, IL-1β, and IL-10 in kidneys.72 This highlights an important role in host defense by what is typically considered a benign and minimally immunogenic member of the mycobiome. Interestingly, gut evolved strains of C. albicans that lack hyphal formation have an enhanced ability to induce trained immunity against systemic C. albicans, A. fumigatus, Pseudomonas aeruginosa, and S. aureus infections.53 These results were obtained following systemic immunization with an evolved C. albicans strain, suggesting that evolutionary pressures that permit intestinal colonization may also provide signals for both intra- and interkingdom protection against infection. Notably, this protective immune training can also be passed on transgenerationally, with non-lethal systemic C. albicans infection or zymosan stimulation of parental mice enhancing protection in offspring against systemic infection with E. coli or Listeria monocytogenes, respectively.136 This protection was sustained up to the second (F2) generation and was attributed to functional, transcriptional, and epigenetic changes that primed myeloid cells to respond to future infections.136 Overall, fungi-induced trained immunity has important implications for the impact of intestinal colonization on host immunological fitness, displaying true co-evolutionary fitness between the mycobiome and host. At least in principle, uptake of intestinal fungi or mycobiome-secreted soluble factors (e.g., β-glucans and chitin) at mucosal sites could lead to regional and systemic trained immunity in myeloid cells. However, evidence for trained immunity in intestinal immune cells in response to the commensal mycobiome has not been illustrated, thus the exact role of intestinal fungi in this process, and their implications in early-life immune training warrants further exploration.
Finally, recent wild microbiome studies have highlighted a pivotal role of indigenous fungi normally absent in laboratory mice. Derived through C57BL/6 embryo transfer to wild dams, wildling mice displayed increased cecal fungal load and a distinct immunological landscape in the spleen and peripheral circulation compared to lab mice.137 Additionally, wildling mice more accurately recapitulated the immunological outcomes of clinical trials for the CD28-superagonist monoclonal antibody and anti-TNF-ɑ treatment in sepsis.137 In another study, lab mice released into an outdoor enclosure, termed rewilded mice, displayed elevated intestinal filamentous fungi.138 Rewilded mice demonstrated increased activation of the systemic immune system, characterized by an increased proportion of circulating neutrophils and activated (CD44hi) CD4+ and CD8+ T cells.138 These findings suggest that laboratory microbial environments may be masking fungi-derived immune functions with essential roles in disease outcomes and responses to treatment, which may be key to improved translatability of immunological research.
Results from wild microbiome studies further highlight the double-edged immune outcomes from intestinal colonization with fungi. On one hand, their immunogenic nature stimulates anti-pathogen immunity at mucosal and systemic sites, enhancing protection against a spectrum of pathogens. Simultaneously, these more robust mechanisms of anti-pathogen immunity may elicit off-target effects yielding collateral damage to host cells and tissues, contributing to the pathogenesis of sepsis. In this context, wildling mice may be a more suitable model of the immunological balance between systemic host defense and collateral immune-mediated organ injury, however, further experimentation must be performed to test this hypothesis.
Concluding remarks
With our emerging appreciation of the importance of intestinal fungi towards mucosal and systemic immune regulation, and in maintaining the balance between health and disease, a critical outstanding question remains in the field: how should we define mycobiome dysbiosis? Studies have clearly demonstrated that the mechanisms of dysregulated fungi–immune interactions in disease go far beyond simply observing shifts in fungal communities, and include important contributions of mycobiome function (e.g., cellular morphogenesis, fungal products, and metabolites), transkingdom interactions with other commensal microbes, and how these contribute to bi-directional interactions with the host.120,139,140,141 Therefore, we propose a holistic framework for studying fungal dysbiosis in disease that integrates taxonomic community shifts, functional mycobiology, morphological shifts and transkingdom interactions, together with the cellular and molecular mechanisms of immune dysregulation that collectively mediate disease. This comprehensive framework for dysbiosis will not only enable a deeper understanding of the role of colonizing fungi in disease, but will be crucial to designing precision-guided mycobiome-modifying therapies. Currently, much of our understanding of fungi–immune interactions in the gut have been derived from studies focused on a single organism, C. albicans. While this model organism has uncovered groundbreaking discoveries, much remains to be revealed about the role of other fungi, as well as the cumulative functional impact of the multikingdom interactions that contribute to fungal dysbiosis and its impact on intestinal and extraintestinal diseases.
Change history
11 May 2022
A Correction to this paper has been published: https://doi.org/10.1038/s41385-022-00523-w
References
Sender, R., Fuchs, S. & Milo, R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 14, e1002533 (2016).
Huttenhower, C. et al. Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214 (2012).
Iliev, I. D. & Leonardi, I. Fungal dysbiosis: immunity and interactions at mucosal barriers. Nat. Rev. Immunol. 17, 635–646 (2017).
Ward T. L., Dominguez-Bello M. G., Heisel T., Al-Ghalith G., Knights D., Gale C. A. Development of the human mycobiome over the first month of life and across body sites. mSystems 3, e00140–17 (2018).
Nash, A. K. et al. The gut mycobiome of the Human. Microbiome Proj. Healthy Cohort. Microbiome 5, 153 (2017).
Raimondi, S. et al. Longitudinal survey of fungi in the human gut: ITS profiling, phenotyping, and colonization. Front. Microbiol. 10, 1575 (2019).
Lim, E. S. et al. Early life dynamics of the human gut virome and bacterial microbiome in infants. Nat. Med. 21, 1228–1234 (2015).
Bacher, P. et al. Human anti-fungal Th17 immunity and pathology rely on cross-reactivity against candida albicans. Cell 176, 1340–1355 (2019).
Doron, I. et al. Human gut mycobiota tune immunity via CARD9-dependent induction of anti-fungal IgG antibodies. Cell 184, 1017–1031 (2021).
van Tilburg Bernardes, E. et al. Intestinal fungi are causally implicated in microbiome assembly and immune development in mice. Nat. Commun. 11, 2577 (2020).
Wampach L., Heintz-Buschart A., Hogan A., Muller E. E. L., Narayanasamy S., Laczny C. C. et al. Colonization and Succession within the Human Gut Microbiome by Archaea, Bacteria, and Microeukaryotes during the First Year of Life. Front. Microbiol. 8, 738 (2017).
Bliss, J. M., Basavegowda, K. P., Watson, W. J., Sheikh, A. U. & Ryan, R. M. Vertical and horizontal transmission of Candida albicans in very low birth weight infants using DNA fingerprinting techniques. Pediatr. Infect. Dis. J. 27, 231–235 (2008).
Moossavi, S. et al. Human milk fungi: environmental determinants and inter-kingdom associations with milk bacteria in the CHILD Cohort Study. BMC Microbiol. 20, 146 (2020).
Boix-Amorós, A. et al. Mycobiome profiles in breast milk from healthy women depend on mode of delivery, geographic location, and interaction with bacteria. Appl. Environ. Microbiol. 85, e02994–18 (2019).
Heisel, T., Nyaribo, L., Sadowsky, M. J. & Gale, C. A. Breastmilk and NICU surfaces are potential sources of fungi for infant mycobiomes. Fungal Genet. Biol. 128, 29–35 (2019).
Chow, B. D. et al. Expressed breast milk as a predictor of neonatal yeast colonization in an intensive care setting. J. Pediatr. Infect. Dis. Soc. 3, 213–220 (2014).
Cui, L., Morris, A. & Ghedin, E. The human mycobiome in health and disease. Genome Med. 5, 63 (2013).
Fiers, W. D., Leonardi, I. & Iliev, I. D. From birth and throughout life: fungal microbiota in nutrition and metabolic health. Annu. Rev. Nutr. 40, 323–343 (2020).
Schei, K. et al. Early gut mycobiota and mother-offspring transfer. Microbiome 5, 107 (2017).
Fujimura, K. E. et al. Neonatal gut microbiota associates with childhood multisensitized atopy and T cell differentiation. Nat. Med. 22, 1187–1191 (2016).
Hoffmann, C. et al. Archaea and fungi of the human gut microbiome: correlations with diet and bacterial residents. PLoS One 8, e66019 (2013).
LaTuga, M. S. et al. Beyond bacteria: a study of the enteric microbial consortium in extremely low birth weight infants. PLoS One 6, e27858 (2011).
Strati, F. et al. Age and gender affect the composition of fungal population of the human gastrointestinal tract. Front. Microbiol. 7, 1227 (2016).
Leonardi, I. et al. CX3CR1(+) mononuclear phagocytes control immunity to intestinal fungi. Science 359, 232–236 (2018).
Zhang, Z. et al. Peripheral lymphoid volume expansion and maintenance are controlled by gut microbiota via RALDH+ Dendritic Cells. Immunity 44, 330–342 (2016).
Noverr, M. C., Noggle, R. M., Toews, G. B. & Huffnagle, G. B. Role of antibiotics and fungal microbiota in driving pulmonary allergic responses. Infect. Immun. 72, 4996–5003 (2004).
Skalski, J. H. et al. Expansion of commensal fungus Wallemia mellicola in the gastrointestinal mycobiota enhances the severity of allergic airway disease in mice. PLoS Pathog. 14, e1007260 (2018).
Arrieta, M.-C. et al. Associations between infant fungal and bacterial dysbiosis and childhood atopic wheeze in a nonindustrialized setting. J. Allergy Clin. Immunol. 142, 424–434.e410 (2018).
Noverr, M. C. & Huffnagle, G. B. Regulation of Candida albicans morphogenesis by fatty acid metabolites. Infect. Immun. 72, 6206–6210 (2004).
Dalle, F. et al. Cellular interactions of Candida albicans with human oral epithelial cells and enterocytes. Cell Microbiol. 12, 248–271 (2010).
Guinan, J., Wang, S., Hazbun, T. R., Yadav, H. & Thangamani, S. Antibiotic-induced decreases in the levels of microbial-derived short-chain fatty acids correlate with increased gastrointestinal colonization of Candida albicans. Sci. Rep. 9, 8872 (2019).
Boutin, R. C. et al. Bacterial-fungal interactions in the neonatal gut influence asthma outcomes later in life. Elife 10, e67740 (2021).
Yamaguchi, N. et al. Gastric colonization of Candida albicans differs in mice fed commercial and purified diets. J. Nutr. 135, 109–115 (2005).
Ribeiro, F. C., de Barros, P. P., Rossoni, R. D., Junqueira, J. C. & Jorge, A. O. Lactobacillus rhamnosus inhibits Candida albicans virulence factors in vitro and modulates immune system in Galleria mellonella. J. Appl. Microbiol. 122, 201–211 (2017).
MacAlpine, J. et al. A small molecule produced by Lactobacillus species blocks Candida albicans filamentation by inhibiting a DYRK1-family kinase. Nat. Commun. 12, 6151 (2021).
Xu, X. L. et al. Bacterial peptidoglycan triggers Candida albicans hyphal growth by directly activating the adenylyl cyclase Cyr1p. Cell Host Microbe 4, 28–39 (2008).
Tan, C. T., Xu, X., Qiao, Y. & Wang, Y. A peptidoglycan storm caused by β-lactam antibiotic’s action on host microbiota drives Candida albicans infection. Nat. Commun. 12, 2560 (2021).
Cabral, D. J., Penumutchu, S., Norris, C., Morones-Ramirez, J. R. & Belenky, P. Microbial competition between Escherichia coli and Candida albicans reveals a soluble fungicidal factor. Micro. Cell 5, 249–255 (2018).
Mathieu, L. G., Dube, D. & Lebrun, M. Increased in vitro sensitivity of Candida albicans to amphotericin B when grown in mixed culture with Escherichia coli. Can. J. Microbiol. 24, 1482–1489 (1978).
Bandara, H., Yau, J. Y. Y., Watt, R. M., Jin, L. J. & Samaranayake, L. P. Escherichia coli and its lipopolysaccharide modulate in vitro Candida biofilm formation. J. Med. Microbiol. 58, 1623–1631 (2009).
Hummel, R. P., Oestreicher, E. J., Maley, M. P. & Macmillan, B. G. Inhibition of Candida albicans by Escherichia coli in vitro and in the germfree mouse. J. Surg. Res. 15, 53–58 (1973).
Pérez-Cobas, A. E. et al. Gut microbiota disturbance during antibiotic therapy: a multi-omic approach. Gut 62, 1591–1601 (2013).
Dollive, S. et al. Fungi of the murine gut: episodic variation and proliferation during antibiotic treatment. PLoS One 8, e71806 (2013).
Seelbinder, B. et al. Antibiotics create a shift from mutualism to competition in human gut communities with a longer-lasting impact on fungi than bacteria. Microbiome 8, 133 (2020).
Fan, D. et al. Activation of HIF-1α and LL-37 by commensal bacteria inhibits Candida albicans colonization. Nat. Med. 21, 808–814 (2015).
Wheeler, M. L. et al. Immunological consequences of intestinal fungal dysbiosis. Cell Host Microbe 19, 865–873 (2016).
Zhai, B. et al. High-resolution mycobiota analysis reveals dynamic intestinal translocation preceding invasive candidiasis. Nat. Med. 26, 59–64 (2020).
Heisel, T. et al. High-fat diet changes fungal microbiomes and interkingdom relationships in the murine gut. mSphere 2, e00351–00317 (2017).
Mar Rodríguez, M. et al. Obesity changes the human gut mycobiome. Sci. Rep. 5, 14600 (2015).
Borgo, F. et al. Relative abundance in bacterial and fungal gut microbes in obese children: a case control study. Child Obes. 13, 78–84 (2017).
Yilmaz, B. et al. Long-term evolution and short-term adaptation of microbiota strains and sub-strains in mice. Cell Host Microbe 29, 650–663.e659 (2021).
Pande, K., Chen, C. & Noble, S. M. Passage through the mammalian gut triggers a phenotypic switch that promotes Candida albicans commensalism. Nat. Genet. 45, 1088–1091 (2013).
Tso, G. H. W. et al. Experimental evolution of a fungal pathogen into a gut symbiont. Science 362, 589–595 (2018).
Sudbery, P. E. Growth of Candida albicans hyphae. Nat. Rev. Microbiol. 9, 737–748 (2011).
Mayer, F. L., Wilson, D. & Hube, B. Candida albicans pathogenicity mechanisms. Virulence 4, 119–128 (2013).
Gow, N. A., van de Veerdonk, F. L., Brown, A. J. & Netea, M. G. Candida albicans morphogenesis and host defence: discriminating invasion from colonization. Nat. Rev. Microbiol. 10, 112–122 (2011).
Vautier, S. et al. Candida albicans colonization and dissemination from the murine gastrointestinal tract: the influence of morphology and Th17 immunity. Cell Microbiol. 17, 445–450 (2015).
Böhm, L. et al. The yeast form of the fungus Candida albicans promotes persistence in the gut of gnotobiotic mice. PLoS Pathog. 13, e1006699 (2017).
Ost, K. S. et al. Adaptive immunity induces mutualism between commensal eukaryotes. Nature 596, 114–118 (2021).
Doron, I. et al. Mycobiota-induced IgA antibodies regulate fungal commensalism in the gut and are dysregulated in Crohn’s disease. Nat. Microbiol. 6, 1493–1504 (2021).
McCusker, J. H., Clemons, K. V., Stevens, D. A. & Davis, R. W. Saccharomyces cerevisiae virulence phenotype as determined with CD-1 mice is associated with the ability to grow at 42 degrees C and form pseudohyphae. Infect. Immun. 62, 5447–5455 (1994).
Kim, S. H. et al. Global analysis of the fungal microbiome in cystic fibrosis patients reveals loss of function of the transcriptional repressor Nrg1 as a mechanism of pathogen adaptation. PLoS Pathog. 11, e1005308 (2015).
Kornitzer D. Regulation of Candida albicans Hyphal Morphogenesis by Endogenous Signals. J. Fungi (Basel). 5, 21 (2019).
Bernier, V. et al. Skin colonization by Malassezia species in neonates: a prospective study and relationship with neonatal cephalic pustulosis. Arch. Dermatol. 138, 215–218 (2002).
Saunte, D. M. L., Gaitanis, G. & Hay, R. J. Malassezia-associated skin diseases, the use of diagnostics and treatment. Front Cell Infect. Microbiol. 10, 112 (2020).
Prohic, A., Jovovic Sadikovic, T., Krupalija-Fazlic, M. & Kuskunovic-Vlahovljak, S. Malassezia species in healthy skin and in dermatological conditions. Int J. Dermatol. 55, 494–504 (2016).
Coelho, C. et al. Intranasal inoculation of cryptococcus neoformans in mice produces nasal infection with rapid brain dissemination. mSphere 4, e00483–00419 (2019).
Li, Q. et al. Dysbiosis of gut fungal microbiota is associated with mucosal inflammation in Crohn’s disease. J. Clin. Gastroenterol. 48, 513–523 (2014).
Sil, A. & Andrianopoulos, A. Thermally dimorphic human fungal pathogens-polyphyletic pathogens with a convergent pathogenicity trait. Cold Spring Harb. Perspect. Med 5, a019794 (2014).
Kozubowski, L. & Heitman, J. Profiling a killer, the development of Cryptococcus neoformans. FEMS Microbiol. Rev. 36, 78–94 (2012).
Jiang, T. T. et al. Commensal fungi recapitulate the protective benefits of intestinal bacteria. Cell Host Microbe 22, 809–816.e804 (2017).
Rizzetto, L. et al. Fungal chitin induces trained immunity in human monocytes during cross-talk of the host with saccharomyces cerevisiae. J. Biol. Chem. 291, 7961–7972 (2016).
Xin, L. et al. Commensal microbes drive intestinal inflammation by IL-17–producing CD4+ T cells through ICOSL and OX40L costimulation in the absence of B7-1 and B7-2. Proc. Natl Acad. Sci. 111, 10672–10677 (2014).
Sancho D., Sousa CRe. Signaling by myeloid C-type lectin receptors in immunity and homeostasis. Annual Rev. Immunol. 30, 491–529 (2012).
Plato, A., Hardison, S. E. & Brown, G. D. Pattern recognition receptors in antifungal immunity. Semin Immunopathol. 37, 97–106 (2015).
Ferwerda, B. et al. Human dectin-1 deficiency and mucocutaneous fungal infections. N. Engl. J. Med. 361, 1760–1767 (2009).
Taylor, P. R. et al. Dectin-1 is required for beta-glucan recognition and control of fungal infection. Nat. Immunol. 8, 31–38 (2007).
Cohen-Kedar, S. et al. Human intestinal epithelial cells respond to β-glucans via Dectin-1 and Syk. Eur. J. Immunol. 44, 3729–3740 (2014).
Wang, T. et al. Dectin-3 deficiency promotes colitis development due to impaired antifungal innate immune responses in the gut. PLoS Pathog. 12, e1005662 (2016).
Choteau, L. et al. Role of mannose-binding lectin in intestinal homeostasis and fungal elimination. Mucosal. Immunol. 9, 767–776 (2016).
Li, X. et al. Response to fungal dysbiosis by gut-resident CX3CR1(+) mononuclear phagocytes aggravates allergic airway disease. Cell Host Microbe 24, 847–856.e844 (2018).
Bonifazi, P. et al. Balancing inflammation and tolerance in vivo through dendritic cells by the commensal Candida albicans. Mucosal. Immunol. 2, 362–374 (2009).
Dillon, S. et al. Yeast zymosan, a stimulus for TLR2 and dectin-1, induces regulatory antigen-presenting cells and immunological tolerance. J. Clin. Invest 116, 916–928 (2006).
Choteau, L. et al. Role of TLR1, TLR2 and TLR6 in the modulation of intestinal inflammation and Candida albicans elimination. Gut Pathog. 9, 9 (2017).
Huertas, B. et al. Serum antibody profile during colonization of the mouse gut by candida albicans: relevance for protection during systemic infection. J. Proteome Res. 16, 335–345 (2017).
Prieto, D. et al. TLR2 modulates gut colonization and dissemination of Candida albicans in a murine model. Microbes Infect. 18, 656–660 (2016).
Saïd-Sadier, N., Padilla, E., Langsley, G. & Ojcius, D. M. Aspergillus fumigatus stimulates the NLRP3 inflammasome through a pathway requiring ROS production and the Syk tyrosine kinase. PLoS One 5, e10008 (2010).
Hise, A. G. et al. An essential role for the NLRP3 inflammasome in host defense against the human fungal pathogen Candida albicans. Cell Host Microbe 5, 487–497 (2009).
Gross, O. et al. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature 459, 433–436 (2009).
van der Graaf, C. A., Netea, M. G., Verschueren, I., van der Meer, J. W. & Kullberg, B. J. Differential cytokine production and Toll-like receptor signaling pathways by Candida albicans blastoconidia and hyphae. Infect. Immun. 73, 7458–7464 (2005).
Gasparoto, T. H. et al. Absence of functional TLR4 impairs response of macrophages after Candida albicans infection. Med. Mycol. 48, 1009–1017 (2010).
Netea, M. G. et al. The role of toll-like receptor (TLR) 2 and TLR4 in the host defense against disseminated candidiasis. J. Infect. Dis. 185, 1483–1489 (2002).
Napoli, J. L. Physiological insights into all-trans-retinoic acid biosynthesis. Biochim. Biophys. Acta 1821, 152–167 (2012).
Cassani, B., Villablanca, E. J., De Calisto, J., Wang, S. & Mora, J. R. Vitamin A and immune regulation: role of retinoic acid in gut-associated dendritic cell education, immune protection and tolerance. Mol. Asp. Med. 33, 63–76 (2012).
Ohnmacht, C. et al. MUCOSAL IMMUNOLOGY. The microbiota regulates type 2 immunity through RORγt+ T cells. Science 349, 989–993 (2015).
Smith, P. M. & Garrett, W. S. The gut microbiota and mucosal T cells. Front. Microbiol. 2, 111 (2011).
van Wijk, F. & Cheroutre, H. Mucosal T cells in gut homeostasis and inflammation. Expert Rev. Clin. Immunol. 6, 559–566 (2010).
Atarashi, K. et al. Th17 cell induction by adhesion of microbes to intestinal epithelial cells. Cell 163, 367–380 (2015).
Becattini, S. et al. T cell immunity. Functional heterogeneity of human memory CD4+ T cell clones primed by pathogens or vaccines. Science 347, 400–406 (2015).
Lv, Q.-Z. et al. Priming with FLO8-deficient Candida albicans induces Th1-biased protective immunity against lethal polymicrobial sepsis. Cell. Mol. Immunol. 18, 2010–2023 (2021).
Fusco A., Savio V., Donniacuo M., Perfetto B., Donnarumma G. Antimicrobial Peptides Human Beta-Defensin-2 and -3 Protect the Gut During Candida albicans Infections Enhancing the Intestinal Barrier Integrity: In Vitro Study. Front. Cellular Infection Microbiol. 11, 666900 (2021).
Allert S. et al. Candida albicans-induced epithelial damage mediates translocation through intestinal barriers. mBio 9, e00915–18 (2018).
Moyes, D. L. et al. Candidalysin is a fungal peptide toxin critical for mucosal infection. Nature 532, 64–68 (2016).
Moyes, D. L. et al. A biphasic innate immune MAPK response discriminates between the yeast and hyphal forms of Candida albicans in epithelial cells. Cell Host Microbe 8, 225–235 (2010).
Yang, D. & Oppenheim, J. J. Alarmins and antimicrobial immunity. Med. Mycol. 47, S146–S153 (2009).
Moyes, D. L. et al. Protection against epithelial damage during Candida albicans infection is mediated by PI3K/Akt and mammalian target of rapamycin signaling. J. Infect. Dis. 209, 1816–1826 (2014).
Murzyn, A. et al. The effect of Saccharomyces boulardii on Candida albicans-infected human intestinal cell lines Caco-2 and Intestin 407. FEMS Microbiol. Lett. 310, 17–23 (2010).
Schirbel, A. et al. Intestinal epithelial cells and T cells differentially recognize and respond to Candida albicans yeast and hypha. Eur. J. Immunol. 48, 1826–1837 (2018).
Singer, M. & Sansonetti, P. J. IL-8 is a key chemokine regulating neutrophil recruitment in a new mouse model of Shigella-induced colitis. J. Immunol. 173, 4197–4206 (2004).
Richardson, J. P. et al. Candidalysin drives epithelial signaling, neutrophil recruitment, and immunopathology at the vaginal mucosa. Infect Immun. 86, e00645–17 (2018).
Swidergall, M. et al. Candidalysin Is Required for Neutrophil Recruitment and Virulence During Systemic Candida albicans Infection. J. Infect. Dis. 220, 1477–1488 (2019).
Kasper, L. et al. The fungal peptide toxin Candidalysin activates the NLRP3 inflammasome and causes cytolysis in mononuclear phagocytes. Nat. Commun. 9, 4260 (2018).
Cheng, S. C. et al. The dectin-1/inflammasome pathway is responsible for the induction of protective T-helper 17 responses that discriminate between yeasts and hyphae of Candida albicans. J. Leukoc. Biol. 90, 357–366 (2011).
Joly, S. et al. Cutting edge: Candida albicans hyphae formation triggers activation of the Nlrp3 inflammasome. J. Immunol. 183, 3578–3581 (2009).
van de Veerdonk, F. L. et al. The inflammasome drives protective Th1 and Th17 cellular responses in disseminated candidiasis. Eur. J. Immunol. 41, 2260–2268 (2011).
De Luca, A. et al. Functional yet balanced reactivity to Candida albicans requires TRIF, MyD88, and IDO-dependent inhibition of Rorc. J. Immunol. 179, 5999–6008 (2007).
Pandiyan, P. et al. CD4(+)CD25(+)Foxp3(+) regulatory T cells promote Th17 cells in vitro and enhance host resistance in mouse Candida albicans Th17 cell infection model. Immunity 34, 422–434 (2011).
De Luca, A. et al. IL-22 and IDO1 affect immunity and tolerance to murine and human vaginal candidiasis. PLoS Pathog. 9, e1003486 (2013).
Whibley, N. et al. Expansion of Foxp3(+) T-cell populations by Candida albicans enhances both Th17-cell responses and fungal dissemination after intravenous challenge. Eur. J. Immunol. 44, 1069–1083 (2014).
Sokol, H. et al. Fungal microbiota dysbiosis in IBD. Gut 66, 1039–1048 (2017).
Leonardi, I. et al. Fungal trans-kingdom dynamics linked to responsiveness to fecal microbiota transplantation (FMT) therapy in ulcerative colitis. Cell Host Microbe 27, 823–829.e823 (2020).
Iliev, I. D. et al. Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science 336, 1314–1317 (2012).
Limon, J. J. et al. Malassezia is associated with crohn’s disease and exacerbates colitis in mouse models. Cell Host Microbe 25, 377–388.e376 (2019).
Jain, U. et al. Debaryomyces is enriched in Crohn’s disease intestinal tissue and impairs healing in mice. Science 371, 1154–1159 (2021).
Malik, A. et al. SYK-CARD9 signaling axis promotes gut fungi-mediated inflammasome activation to restrict colitis and colon cancer. Immunity 49, 515–530.e515 (2018).
Erb Downward, J. R., Falkowski, N. R., Mason, K. L., Muraglia, R. & Huffnagle, G. B. Modulation of post-antibiotic bacterial community reassembly and host response by candida albicans. Sci. Rep. 3, 2191 (2013).
Kim, Y. G. et al. Gut dysbiosis promotes M2 macrophage polarization and allergic airway inflammation via fungi-induced PGE2. Cell Host Microbe 15, 95–102 (2014).
Shao, T. Y. et al. Commensal candida albicans positively calibrates systemic Th17 Immunological Responses. Cell Host Microbe 25, 404–417.e406 (2019).
Spellberg, B. J. et al. Efficacy of the anti-Candida rAls3p-N or rAls1p-N vaccines against disseminated and mucosal candidiasis. J. Infect. Dis. 194, 256–260 (2006).
Spellberg, B. et al. The antifungal vaccine derived from the recombinant N terminus of Als3p protects mice against the bacterium Staphylococcus aureus. Infect. Immun. 76, 4574–4580 (2008).
Markey, L. et al. Pre-colonization with the commensal fungus Candida albicans reduces murine susceptibility to Clostridium difficile infection. Gut Microbes 9, 497–509 (2018).
Netea, M. G., Quintin, J. & van der Meer, J. W. Trained immunity: a memory for innate host defense. Cell Host Microbe 9, 355–361 (2011).
Netea, M. G. et al. Trained immunity: A program of innate immune memory in health and disease. Science 352, aaf1098 (2016).
Quintin, J. et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe 12, 223–232 (2012).
Ifrim, D. C. et al. Candida albicans primes TLR cytokine responses through a Dectin-1/Raf-1-mediated pathway. J. Immunol. 190, 4129–4135 (2013).
Katzmarski, N. et al. Transmission of trained immunity and heterologous resistance to infections across generations. Nat. Immunol. 22, 1382–1390 (2021).
Rosshart, S. P. et al. Laboratory mice born to wild mice have natural microbiota and model human immune responses. Science 365, eaaw4361 (2019).
Yeung, F. et al. Altered immunity of laboratory mice in the natural environment is associated with fungal colonization. Cell Host Microbe 27, 809–822.e806 (2020).
Lemoinne, S. et al. Fungi participate in the dysbiosis of gut microbiota in patients with primary sclerosing cholangitis. Gut 69, 92–102 (2020).
Liguori, G. et al. Fungal dysbiosis in mucosa-associated microbiota of Crohn’s disease patients. J. Crohns Colitis 10, 296–305 (2016).
Coker, O. O. et al. Enteric fungal microbiota dysbiosis and ecological alterations in colorectal cancer. Gut 68, 654–662 (2019).
Leonardi, I. et al. Mucosal fungi promote gut barrier function and social behavior via Type 17 immunity. Cell 185, 831–846.e814 (2022).
Kolls, J. K. & Lindén, A. Interleukin-17 family members and inflammation. Immunity 21, 467–476 (2004).
Cha, H. R. et al. Downregulation of Th17 cells in the small intestine by disruption of gut flora in the absence of retinoic acid. J. Immunol. 184, 6799–6806 (2010).
Acknowledgements
MWG is funded by the Alberta Children’s Hospital Research Institute Graduate Scholarship, the Cumming School of Medicine Graduate Scholarship, the University of Calgary Faculty of Graduate Studies Master’s Research Scholarship, and the Canadian Institutes of Health Research (CIHR) Canada Graduate Scholarships Master’s Award. EvTB is funded by the Eye’s High Doctoral Recruitment Scholarship and the CIHR Frederick Banting and Charles Best Canada Graduate Scholarships Doctoral Award. BM is funded by the Cumming School of Medicine, the Snyder Institue for Chronic Diseases, the CIHR, Alberta Health Services and the Canadian Foundation for Innovation. M-CA is funded by the Cumming School of Medicine, the Alberta Children’s Hospital Research Institute, the Snyder Institute for Chronic Diseases, the W. Garfield Weston Foundation, the CIHR, the Natural Sciences and Engineering Research Council of Canada and Alberta Innovates. Figures created with BioRender.com (https://biorender.com/).
Author information
Authors and Affiliations
Contributions
M.W.G., E.v.T.B., D.C., B.M., and M.-C.A. conceptualized the manuscript. M.W.G., E.v.T.B., and D.C. wrote the original draft. B.M. and M.-C.A. reviewed and edited the manuscript. All authors approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: Due to an information missing in the acknowledgements section.
Rights and permissions
About this article

Cite this article
Gutierrez, M.W., van Tilburg Bernardes, E., Changirwa, D. et al. “Molding” immunity—modulation of mucosal and systemic immunity by the intestinal mycobiome in health and disease. Mucosal Immunol 15, 573–583 (2022). https://doi.org/10.1038/s41385-022-00515-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41385-022-00515-w
This article is cited by
-
Plastiphily is linked to generic virulence traits of important human pathogenic fungi
Communications Earth & Environment (2024)
-
The interactions between the host immunity and intestinal microorganisms in fish
Applied Microbiology and Biotechnology (2024)
-
Candida spp. in Human Intestinal Health and Disease: More than a Gut Feeling
Mycopathologia (2023)
