
Abstract
Different psychiatric disorders and symptoms are highly correlated in the general population. A general psychopathology factor (or “P-factor”) has been proposed to efficiently describe this covariance of psychopathology. Recently, genetic and neuroimaging studies also derived general dimensions that reflect densely correlated genomic and neural effects on behaviour and psychopathology. While these three types of general dimensions show striking parallels, it is unknown how they are conceptually related. Here, we provide an overview of these three general dimensions, and suggest a unified interpretation of their nature and underlying mechanisms. We propose that the general dimensions reflect, in part, a combination of heritable ‘environmental’ factors, driven by a dense web of gene-environment correlations. This perspective calls for an update of the traditional endophenotype framework, and encourages methodological innovations to improve models of gene-brain-environment relationships in all their complexity. We propose concrete approaches, which by taking advantage of the richness of current large databases will help to better disentangle the complex nature of causal factors underlying psychopathology.
Similar content being viewed by others
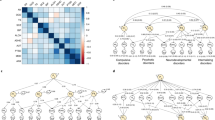

Introduction
A trend has emerged that shows striking parallels between clinical psychology, psychiatric genetics and neuroimaging. The dense covariation between a range of psychopathology-related traits is described in terms of new constructs. These take the form of three general dimensions or factors: a general psychopathology factor or “P-factor” [1, 2], a general dimension of genetic liability for psychopathology [3, 4], and a general dimension of brain structure and function [5, 6]. Here we refer to the general dimension derived from behavioural data as “phenotypic P-factor”, the general dimension derived from genomic data as “genomic P-factor”, and general dimensions derived from neuroimaging as “neural P-factors”. Whilst these general dimensions are receiving ample attention within each discipline, little has been done to interpret them together.
This Perspective Article has three main aims: (1) to integrate knowledge from the three types of P-factors into a new unified theory; (2) to evaluate implications of this theory for widely held assumptions behind endophenotypes; (3) to translate the theory into concrete future directions for psychiatric genetics and imaging genetics. In the first section, we introduce each of the general dimensions individually. This section is on purpose brief and not meant as an exhaustive literature review. The second section is the core of this paper, and where its novelty lies. There, we advance previous interpretations of genetic and phenotypic P-factors [7, 8] by integrating the neural P-factor into a unified theory of all three P-factors (aim 1), and by discussing important implications of this theory for the endophenotype concept in psychiatry (aim 2). In the third section, we translate our new theoretical perspective on gene-brain-behaviour interplay into concrete strategies for future research (aim 3).
General dimensions describing inter-individual variation in psychopathology, genetic liability for psychopathology, and brain structure and function
A phenotypic P-factor of psychopathology
Psychiatric disorders have fuzzy boundaries in their clinical definitions and phenomenological presentations. Few symptoms are unique to any diagnosis. A symptom can be a contributing criterion to multiple disorders, and a common comorbidity across disorders. Moreover, disorders may develop into other disorders through maturation and ageing (i.e., “network theory”) [8,9,10]. Using structural equation modelling (SEM), several studies have identified a single common psychopathology dimension, or “P-factor” [1, 2, 11,12,13,14,15,16], which captures a significant portion of inter-individual variation (e.g., 23% [16]) in the presence and severity of psychiatric symptoms in the population. This phenotypic P-factor is a reflection of the correlation structure across symptom scales and the comorbidity of psychiatric disorders [8]. Whilst further interpretation of the P-factor itself has already received much attention in the clinical and behavioural domain [8, 17,18,19], its relationship to emerging general constructs in genetics and neuroimaging has not been addressed in detail.
A genomic P-factor
The phenotypic P-factor is heritable [20,21,22], and phenotypic overlap may be partly driven by shared genetic liability [12]. Twin and family studies have long shown that genetic risk for psychiatric disorders is indeed shared across diagnostic categories [23, 24]. More recently, this shared genetic risk has also been observed from single nucleotide polymorphisms (SNPs) in DNA [25,26,27]. For example, attention-deficit/hyperactivity disorder (ADHD), anxiety disorders, major depressive disorder, bipolar disorder and schizophrenia all have pairwise genetic correlations exceeding ~0.20, ranging up to ~0.75 [26].
To identify a genomic dimension that captures this shared genetic liability, SNP-based genetic correlations have been further analysed using genomic SEM [3] and principal component analysis (PCA) [4]. A single genomic factor (or “polygenic P-factor” [4]) containing genome-wide factor-loadings representing each SNP’s contribution to cross-disorder liability, was derived. This genomic P-factor fitted well to the data [3], explained 20%-43% of the SNP-effects across disorders [4], and improved power for genome-wide association study (GWAS) [3].
A neural P-factor
The phenotypic P-factor is associated with brain structure and function [28,29,30,31,32,33,34,35,36]. A meta-analysis of structural magnetic resonance imaging (MRI) studies showed that case-control differences of six psychiatric disorders co-localized to the same brain regions [37], while very few diagnosis-specific associations were seen. Recent meta-analyses from the ENIGMA Consortium confirm this neuroanatomical overlap across adult psychiatric disorders [38, 39], but less so for neurodevelopmental disorders [39, 40]. Regarding functional MRI, a meta-analysis of case-control studies disorders also revealed highly significant spatial overlap across five psychiatric disorders, in the absence of diagnosis-specific effects [41].
Akin to the phenotypic and the genetic domain, these cross-diagnostic similarities have led to the construction of general dimensions describing inter-individual variation in neuroimaging traits. Such “neural P-factors” have been identified from functional connectivity [6], diffusion MRI [42], neuroanatomical data [5], and multi-modal data encompassing all the above [43]. In white matter, a single whole-brain factor of diffusion anisotropy was associated with the phenotypic P-factor in a community sample of 10-year old children [42]. In the resting-state connectivity study, a single dimension capturing a combination of functional connectivity, with environmental and behavioural variables was identified, which explained up to 17% of variance in input variables. The traits in this combined phenotypic/neural P-factor showed an interesting pattern with traits generally regarded as positive (intelligence, life satisfaction, educational attainment) at one end of the spectrum, and traits generally regarded as negative and associated with psychiatric liability (substance abuse, poor sleep quality, aggression, stress) on the other end [6]. An analogous neural P-factor based on structural MRI was recently described in the same sample, and correlated with the same behavioural, demographic and environmental measures [5]. A similar multimodal neural P-factor explained 10–40% of variance in environmental and demographic variables in an independent sample of children and adolescents [43].
In summary, recent neuroimaging and genetic research across psychiatric traits mirrors observations at the phenotypic level. Psychiatric symptoms and their biological correlates co-vary highly in the population. Within each discipline, this covariation can be captured by single phenotypic, genomic and neural P-factors.
An integrative interpretation of the general dimensions
Explaining variance versus understanding mechanisms
Together, the scientific developments described above mark a perhaps unsurprising, yet new development in our understanding of biological underpinnings of psychiatric disorders. Decades of work on pairwise associations between genome, brain, and single diagnoses generally revealed only modest effects. In comparison, the diagnosis-general phenotypic, genomic and neural P-factors explain undeniably large amounts of inter-individual variation. Whilst this quality is immediately appealing from a statistical perspective, it does not directly lead to mechanistic insights or clinical impact [44]. As the P-factors are built solely from covariance structures, they are arguably ultimate cases of “correlation without causation.” Yet, we know from basic and experimental neuroscience that the associations between the genome, brain and behaviour are governed by tightly regulated mechanisms of molecular pathways and protein interactions. To arrive at a new integrated perspective on potential mechanisms underlying the different P-factors, we first discuss some technical and conceptual differences between the three P-factors.
Methodological differences between the P-factors
Tables 1 and 2 summarize key methods used to extract P-factors in each discipline. Generally, the phenotypic P-factor has been derived from continuous symptom ratings in population-based samples [1, 2, 14]. Whilst different designs and approaches have led to inconsistent results for specific cross-diagnostic factors, such as “thought disorder” or “fear and distress” factors, the general P-factor has been largely consistent, regardless of demographics or diagnostic instrument [1, 2, 12,13,14, 17]. The finding that binary diagnostic phenotypes and continuous dimensional traits yield similar results is in line with taxometric properties of phenotypic variability in the population [45] and the “liability threshold model”, under which diagnoses are the extremes of latent continuous traits [46].
Similar to the phenotypic P-factor, neural P-factors tend to be derived from continuous traits in population-based cohorts [5, 6, 42]. In contrast, genomic P-factors are generally based on case-control comparisons [3, 4]. The degree to which the clinician-based case-control genomic P-factor can be generalised to population-based traits has been questioned [47]. This design-specific effect may be particularly strong for psychotic disorders, whereas case-control GWAS of depression [48], ADHD [49], and autism spectrum disorders [50, 51] show high overlap with continuous traits in community samples.
We ought to keep in mind these methodological differences until this has been tested directly (see Section 3). Nevertheless, we hypothesise that the three types of P-factors reflect to some degree the same inter-individual variation, on the basis of their similarities.
Similarities between the P-factors
The three P-factors share several key properties:
-
1.
They are based on the high degree of covariance between many variables.
-
2.
They describe substantial amounts of inter-individual variation.
-
3.
They are diffuse, comprised of subtle, widespread effects throughout the phenotypic, genomic, and neural domains, as opposed to restricted to a few brain regions, genomic loci, or behavioural domains.
-
4.
They do not infer directions of causality (except for DNA, which due to its stability is more likely cause than effect).
-
5.
They are associated with heritable variables that are rather “environmental” in nature. The phenotypic P-factor and the neural P-factors are strongly associated with household income, years of education, and welfare benefit use [1, 5, 6]. While the genomic P-factor has not yet been directly associated with environmental variables, many environmental factors are genetically correlated with psychiatric disorders [7, 52, 53].
The shared properties 1-3 and 5 suggest that the three types of P-factors, despite their differences, explain at least some similar aspects of inter-individual variation. The 4th shared property—the lack of causal information—is a critical limitation that impedes translation from correlations to mechanisms, and ultimately to clinical impact. We propose that the 5th property—the association with the heritable environment—may be particularly important in disentangling these potential causal mechanisms.
The heritable environment and gene-environment correlations
In the context of quantitative genetics, the “heritable environment” may seem paradoxical, because in genetics “environment” tends to equal everything that is not explained by genetic variation. Here, we mean by “environment” the common sense conceptual notion, i.e., variables mainly located outside the body. Environmental variables like recreation, educational attainment and socio-economic status have heritability estimates around 40–50% in twin studies and 5–27% in SNP-based estimation [52, 54,55,56,57,58].
The heritability of environmental measures revives seminal papers of Plomin et al. [59], Kendler et al. [60], and Scarr and McCartney [61], who first described the importance of possible gene-environment correlations (rGE) in this context. RGE occurs when exposure to an environmental risk factor is influenced by the same genetic variation as a (psychiatric) trait of interest [59, 62]. As a consequence, rGE can give rise to “environmentally mediated pleiotropy” [7, 63, 64]. For example, hypothetically genetic variation influencing one’s degree of openness to new experiences in turn may influence a person’s attitude toward substance use, which is a risk factor for several psychiatric disorders. Although scarcely considered in the GWAS era so far [62], rGE is abundant in psychiatric genetics. For example, household income and educational attainment are genetically correlated with multiple psychiatric disorders [52, 53]. In line with the possibility of “environmentally mediated pleiotropy” [4, 7, 63], statistically removing the variance of socio-economic status significantly alters genetic correlations between psychiatric disorders [65].
Importantly, many of these environmental variables also have spatially overlapping associations with brain traits [5, 6, 43], which is captured by the neural P-factor (see section 1.3). Therefore, the consideration of gene-environment correlations has important consequences for the identification of brain mechanisms on the causal pathway from genes to behaviour.
Reconsidering the brain as mediator between genes and behaviour
Brain structure and function are heritable [64, 66,67,68,69], and theoretically, the brain is the mediator in the causal chain from genetic effect to behaviour and environment. Therefore, studying brain traits - as “endophenotypes” - in relation to genetic risk factors is generally considered to help unravelling causal mechanisms from genome to behaviour [70,71,72,73]. However, the complexity of environmentally mediated pleiotropy [4, 7, 63] also holds for genetic influences on the brain, and challenges the bottom-up causality from genome to brain to behaviour (Fig. 1). Consequently, the causal chain from genes to behaviour includes a brain-environment loop. For example, the SNPs that confer risk for major depression may influence the brain directly throughout development, but many of these SNPs also contribute to environmental exposures such as household income [52], social deprivation [52], traumatic experiences [74], poor sleep [75], socio-economic status [76], to name a few. Some of these factors will affect the brain as well. These indirect, environmentally mediated effects accumulate over time. Thus, in theory, the brain is the mediator between the genome and behaviour, but in practice the genetic effects on the brain we measure can be anywhere on the causal brain-environment loop that perpetuates and broadens from the moment of conception to the moment of MRI measurement. Considering the number of causal routes to psychopathology this permits, it should be no surprise that psychiatric disorders and their associated brain traits are highly polygenic [77,78,79], and non-specific to brain regions, tissues or circuits [50, 80, 81]. In line with Avinun et al. (2020) [7], we argue that this convolution of many causal routes induces not only highly correlated and subtle effects in the genome, but also in the brain, and these are now captured by general dimensions in the form of P-factors.

Intuitive theories and concepts, like endophenotypes, logically assume that genetic effects on behaviour (and on environment) pass through the brain. However, the heritable environment induces rGEs, which propagate over time, through brain, environment and behaviour. Hence, the model is updated with a causal brain-behaviour-environment loop. This loop permits a multitude of causal mechanisms between the genome and behaviour, inside and outside the brain, and we propose that this is reflected in the cross-trait covariance captured by the P-factor and its genetic and neural equivalents. Note that the exact mechanisms by which the environment influences the brain and behaviour are many, and may potentially involve tissues outside the brain. For the sake of clarity, we here do not include routes from genome to behaviour that are outside of the brain.
Summary: an integrated interpretation of three P-factors
To summarise, the phenotypic, genomic and neural P-factors all capture inter-individual variation extremely efficiently in a single variable. However, this efficiency does not readily translate to mechanistic insights. We note an important contribution of heritable environmental risk factors as a common feature to all three P-factors. Insofar as we can speculate on causal relationships, it seems likely that variables within and across dimensions have bidirectional causal relationships, at least when considered throughout the human lifespan. In this context, we emphasise that heritable environmental risk factors can, just like neural processes, be mediators on the causal pathway from genes to behaviour. Thus, as a general conclusion, general dimensions of psychopathology and genetic liability for psychopathology reflect a dense web of mutually reinforcing traits that propagate throughout development, via the environment and the brain, in conferring risk for psychiatric disorders. While the P-factors in the brain and the genome may not be the only explanation for all patterns at the phenotypic level, our interpretation also fits with “network theory”:[9, 10] the web of mutually reinforcing traits may, at different stages of development and under different circumstances, clinically manifest as a sequence of different symptoms or diagnoses.
Onwards: quantifying and using gene-environment correlations
Our conclusion may appear daunting: if all risk factors are correlated and their causal effects are bidirectional, it is difficult to dissect their relationships into mechanistic insights that can be therapeutically interfered with. However, it is only by acknowledging this complexity that we can make headway in biological psychiatry, for example using imaging genetics. Both neuroimaging and genetics studies have been troubled by lack of replication and limited clinical impact. This is largely due to subtle and widespread effects in the genome and the brain, which call for new approaches that can more effectively model these patterns of effects. We finally have large sample sizes to work with (e.g. UK Biobank [64, 82], ENIGMA [83, 84], PGC [27]), and a range of new methods to apply to them.
Concretely, we consider the following strategies to move toward understanding biological mechanisms underpinning the P-factors:
-
1.
To test directly to what extent the three P-factors reflect the same inter-individual variation.
-
2.
To quantify rGEs using GWAS data and extract multiple dimensions, which may reflect more distinct mechanisms.
-
3.
To extend multivariate genetic models to include data types at multiple molecular and neurophysiological scales of investigation.
-
4.
To apply multivariate models to longitudinal cohorts, to get a glimpse of sensitive age-windows and possibly causal dynamics of gene-brain-environment interplay.
Testing to what extent the three P-factors reflect the same variation in the population
Different designs and statistical approaches were used for deriving the three P-factors (Table 2 and Section 2.2). Using the same statistical method across phenotypic and brain traits and applying it to the same sample, would be a first step to understand the shared biological mechanisms behind these new constructs and help identify any potential biases and confounders in their derivation. In addition, the validity and generalisability of the P-factors needs to be examined across healthy and clinical populations of different ancestries and demographics (e.g., age, sex).
Quantifying genetic correlations and gene-environment correlations using GWAS data
Classic twin models can be extended to multivariate models to quantify the shared genetic influence on multiple variables, including environmental measures [85, 86]. This way, rGEs can be quantified. For example, the effect of exposure to aggressive media on aggressive behaviour in children may be partly explained by genetically driven media preferences [87]. Since multivariate models can also be applied to GWAS output [25], their scope and feasibility has increased. For example, using LD-score regression [25], genetic overlap between socio-economic status and several major psychiatric disorders and risk factors was quantified, and accounting for genetic variation of socio-economic status changed the genetic correlations between those psychopathology traits [76]. In addition, multivariate models like genomic SEM can generate not only a single general factor, but also more specific factors [27]. Similarly, at the phenotypic level, two or three more specific factors are often derived [1, 2, 13, 14, 33, 45, 88, 89]. The high dimensionality of neuroimaging data already tends to be described by dozens of dimensions rather than a single one [5, 36, 43]. Combining data more systematically across disciplines and drastically increasing the number of phenotypes to hundreds or more, would allow better estimation of many more genomic factors as well. Table 2 describes data-driven methods suitable for such high-dimensional multivariate analyses. Multiple independent factors may expose more specific patterns of gene-brain-behaviour associations, thereby aiding interpretation in terms of concrete mechanisms. In the genome, for example, bioinformatics [90, 91] may reveal that multiple independent factors map to molecular pathways or cell types with higher sensitivity and specificity than a single general factor. This could ultimately help to disentangle multiple specific mechanisms from the web of correlated risk factors.
Including multi-level neurobiological data at multiple scales of investigation
We focused mostly on integration of neuroimaging, genomic and phenotypic research. However, the underlying mechanisms involve every step from alleles, through molecular interactions, cell morphology, neural circuits to behaviour [92], and even non-brain related mechanisms that contribute to behaviour. The examples of multivariate models above can be extended with multi-layer biological information, including transcriptomics, epigenetics, and proteomics. Doing so could yield new P-factors and more specific factors (as described above) across biological levels of investigation. Just like in genomic SEM, which is based on the covariance of SNP-phenotype associations, covariance matrices of methylation-brain or transcriptome-brain associations be decomposed into novel constructs whose factor loadings indicate which variables across levels work together in influencing phenotypes of interest. For example, N-dimensional PCA of transcriptome-neuroimaging associations returns N factors containing loadings of brain measures, plus N respective vectors of transcript loadings. Transcripts and brain measures loading highly on the same component jointly describe a distinct portion of brain-transcript covariance, and therefore may point to a distinct mechanism of how gene expression influences brain structure and function, or vice versa. Neural and transcript-dimensions can be further linked to molecular pathways [90, 91] and atlases of brain function [93], which while allowing no direct causal inference, help generate new testable hypotheses for experimental studies to test causality. To model many variables with complex relations and different distributions, new methods such as MiXeR [94] could be considered to quantify genetic overlap more flexibly, while Mendelian Randomization [95] could give first glimpses of causal inference.
Modelling the dynamics of gene-environment correlations over time
Longitudinal twin research indicates that the phenotypic P-factor and its genetic underpinnings are largely stable over time [96]. Extensions of longitudinal genetic models can give insights into which gene-brain-environment associations and interactions are most relevant for psychiatric symptoms at different age-windows, and as a first test of causality. For example, a longitudinal twin study suggests that the genetic influence on temperament at age 3 years partly determines peer problems at a later age [97]. Currently, SNP-based analyses have several advantages over twin studies: GWAS data are currently more widely available than twin samples; and SNP-based results can be further investigated to understand the molecular basis of the general and specific factors. Established methods from twin research provide a ready-made framework for further extending SNP-based approaches [25, 98] to better understand mechanisms underlying rGEs. Although longitudinal modelling of single variables has been performed [99, 100], further extensions of these models including environmental variables and/or multivariate genomic, neural and psychopathology factors reviewed above, are now feasible with the increased availability of summary statistics of large consortia [27, 64, 82, 83, 101].
General conclusion
Pioneers of P-factor concept wrote: “Correlations [between different symptom dimensions] are not a problem, but a profoundly important source of information about the nature of psychopathology.” [17]. The same is true for the highly correlated biological risk factors of psychopathology we reviewed here. In the last few decades, the limited clinical impact of psychiatric genetics and neuroimaging in psychiatry has been largely attributed to two issues: (1) tiny effect sizes in any specific (genomic or neuroanatomical) location, and (2) a lack of specificity to diagnoses or symptoms. Following our integrated interpretation of the recently identified general dimensions in psychopathology, psychiatric genetics, and neuroimaging, our perspective is that the solution to these problems lies in accepting the complexity of the nature of the causal mechanisms we aim to find, and in modelling them more accurately. We suggest that the neural P-factor, like its genetic and phenotypic equivalents [7], reflects an abundance of rGEs underlying multifactorial neuropsychiatric traits. The expansion of available data and new multivariate methods provide promising new ways to account for and quantify the nature of the covariance of psychopathology with a multitude of biological and environmental risk factors. In the near future, we anticipate exciting new research that will take our understanding of gene-brain-behaviour-environment relationships from a web of associations to new hypotheses of causal relationships.
References
Caspi A, Houts RM, Belsky DW, Goldman-Mellor SJ, Harrington H, Israel S, et al. The p factor: one general psychopathology factor in the structure of psychiatric disorders? Clin Psychol Sci. 2014;2:119–37.
Lahey BB, Applegate B, Hakes JK, Zald DH, Hariri AR, Rathouz PJ. Is there a general factor of prevalent psychopathology during adulthood? J Abnorm Psychol. 2012;121:971–7.
Grotzinger AD, Rhemtulla M, de Vlaming R, Ritchie SJ, Mallard TT, Hill WD, et al. Genomic structural equation modelling provides insights into the multivariate genetic architecture of complex traits. Nat Hum Behav. 2019;3:513–25.
Selzam S, Coleman JRI, Caspi A, Moffitt TE, Plomin R. A polygenic p factor for major psychiatric disorders. Transl Psychiatry. 2018;8:205.
Llera A, Wolfers T, Mulders P, Beckmann CF. Inter-individual differences in human brain structure and morphology link to variation in demographics and behavior. Elife. 2019;8:e44443.
Smith SM, Nichols TE, Vidaurre D, Winkler AM, Behrens TE, Glasser MF, et al. A positive-negative mode of population covariation links brain connectivity, demographics and behavior. Nat Neurosci. 2015;18:1565–7.
Avinun R. The E is in the G: gene-environment-trait correlations and findings from genome-wide association studies. Perspect Psychol Sci. 2020;15:81–9.
Caspi A, Moffitt TE. All for one and one for all: mental disorders in one dimension. Am J Psychiatry. 2018;175:831–44.
Fried EI, Cramer AOJ. Moving forward: challenges and directions for psychopathological network theory and methodology. Perspect Psychol Sci. 2017;12:999–1020.
Borsboom D, Cramer AO. Network analysis: an integrative approach to the structure of psychopathology. Annu Rev Clin Psychol. 2013;9:91–121.
Carragher N, Teesson M, Sunderland M, Newton NC, Krueger RF, Conrod PJ, et al. The structure of adolescent psychopathology: a symptom-level analysis. Psychol Med. 2016;46:981–94.
Lahey BB, Krueger RF, Rathouz PJ, Waldman ID, Zald DH. Validity and utility of the general factor of psychopathology. World Psychiatry. 2017;16:142–4.
Martel MM, Pan PM, Hoffmann MS, Gadelha A, do Rosario MC, Mari JJ, et al. A general psychopathology factor (P factor) in children: structural model analysis and external validation through familial risk and child global executive function. J Abnorm Psychol. 2017;126:137–48.
Noordhof A, Krueger RF, Ormel J, Oldehinkel AJ, Hartman CA. Integrating autism-related symptoms into the dimensional internalizing and externalizing model of psychopathology. The TRAILS study. J Abnorm Child Psychol. 2015;43:577–87.
Patalay P, Fonagy P, Deighton J, Belsky J, Vostanis P, Wolpert M. A general psychopathology factor in early adolescence. Br J Psychiatry. 2015;207:15–22.
Michelini G, Barch DM, Tian Y, Watson D, Klein DN, Kotov R. Delineating and validating higher-order dimensions of psychopathology in the Adolescent Brain Cognitive Development (ABCD) study. Transl Psychiatry. 2019;9:261.
Lahey BB, Rathouz PJ, Keenan K, Stepp SD, Loeber R, Hipwell AE. Criterion validity of the general factor of psychopathology in a prospective study of girls. J Child Psychol Psychiatry. 2015;56:415–22.
Tackett JL, Lahey BB, van Hulle C, Waldman I, Krueger RF, Rathouz PJ. Common genetic influences on negative emotionality and a general psychopathology factor in childhood and adolescence. J Abnorm Psychol. 2013;122:1142–53.
Conway CC, Forbes MK, Forbush KT, Fried EI, Hallquist MN, Kotov R, et al. A hierarchical taxonomy of psychopathology can transform mental health research. Perspect Psychol Sci. 2019;14:419–36.
Neumann A, Pappa I, Lahey BB, Verhulst FC, Medina-Gomez C, Jaddoe VW, et al. Single nucleotide polymorphism heritability of a general psychopathology factor in children. J Am Acad Child Adolesc Psychiatry. 2016;55:1038–1045 e1034.
Franke B. Heritability of a general psychopathology factor in the population: potential implications for classification and treatment. J Am Acad Child Adolesc Psychiatry. 2016;55:1016–7.
Allegrini AG, Cheesman R, Rimfeld K, Selzam S, Pingault JB, Eley TC, et al. The p factor: genetic analyses support a general dimension of psychopathology in childhood and adolescence. J Child Psychol Psychiatry. 2020;61:30–9.
Kendler KS, Aggen SH, Knudsen GP, Roysamb E, Neale MC, Reichborn-Kjennerud T. The structure of genetic and environmental risk factors for syndromal and subsyndromal common DSM-IV axis I and all axis II disorders. Am J Psychiatry. 2011;168:29–39.
Lichtenstein P, Yip BH, Bjork C, Pawitan Y, Cannon TD, Sullivan PF, et al. Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: a population-based study. Lancet. 2009;373:234–9.
Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh PR, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet. 2015;47:1236–41.
Brainstorm Consortium, Anttila V, Bulik-Sullivan B, Finucane HK, Walters RK, Bras J, et al. Analysis of shared heritability in common disorders of the brain. Science. 2018;360:eaap8757.
Cross-Disorder Group of the Psychiatric Genomics Consortium. Genomic relationships, novel loci, and pleiotropic mechanisms across eight psychiatric disorders. Cell. 2019;179:1469–82 e1411.
Romer AL, Knodt AR, Houts R, Brigidi BD, Moffitt TE, Caspi A, et al. Structural alterations within cerebellar circuitry are associated with general liability for common mental disorders. Mol Psychiatry. 2018;23:1084–90.
Romer AL, Knodt AR, Sison ML, Ireland D, Houts R, Ramrakha S, et al. Replicability of structural brain alterations associated with general psychopathology: evidence from a population-representative birth cohort. Mol Psychiatry. 2019;10.1038/s41380-019-0621-z (Epub ahead of print).
Romer AL, Elliott ML, Knodt AR, Sison ML, Ireland D, Houts R et al. Pervasively thinner neocortex as a transdiagnostic feature of general psychopathology. Am J Psychiatry. 2020: appiajp202019090934.
Elliott ML, Romer A, Knodt AR, Hariri AR. A connectome-wide functional signature of transdiagnostic risk for mental illness. Biol Psychiatry. 2018;84:452–9.
Hinton KE, Lahey BB, Villalta-Gil V, Meyer FAC, Burgess LL, Chodes LK, et al. White matter microstructure correlates of general and specific second-order factors of psychopathology. Neuroimage Clin. 2019;22:101705.
Alnaes D, Kaufmann T, Doan NT, Cordova-Palomera A, Wang Y, Bettella F, et al. Association of heritable cognitive ability and psychopathology with white matter properties in children and adolescents. JAMA Psychiatry. 2018;75:287–95.
Moberget T, Alnaes D, Kaufmann T, Doan NT, Cordova-Palomera A, Norbom LB, et al. Cerebellar gray matter volume is associated with cognitive function and psychopathology in adolescence. Biol Psychiatry. 2019;86:65–75.
Kaczkurkin AN, Moore TM, Calkins ME, Ciric R, Detre JA, Elliott MA, et al. Common and dissociable regional cerebral blood flow differences associate with dimensions of psychopathology across categorical diagnoses. Mol Psychiatry. 2018;23:1981–9.
Kaczkurkin AN, Park SS, Sotiras A, Moore TM, Calkins ME, Cieslak M, et al. Evidence for dissociable linkage of dimensions of psychopathology to brain structure in youths. Am J Psychiatry. 2019;176:1000–9.
Goodkind M, Eickhoff SB, Oathes DJ, Jiang Y, Chang A, Jones-Hagata LB, et al. Identification of a common neurobiological substrate for mental illness. JAMA Psychiatry. 2015;72:305–15.
Kochunov P, Hong LE, Dennis EL, Morey RA, Tate DF, Wilde EA, et al. ENIGMA-DTI: Translating reproducible white matter deficits into personalized vulnerability metrics in cross-diagnostic psychiatric research. Hum Brain Mapp. 2020. https://doi.org/10.1002/hbm.24998 (Epub ahead of print).
Opel N, Goltermann J, Hermesdorf M, Berger K, Baune BT, Dannlowski U. Cross-disorder analysis of brain structural abnormalities in six major psychiatric disorders: a secondary analysis of mega- and meta-analytical findings from the ENIGMA consortium. Biol Psychiatry. 2020;88:678–86.
Boedhoe PSW, van Rooij D, Hoogman M, Twisk JWR, Schmaal L, Abe Y, et al. Subcortical brain volume, regional cortical thickness, and cortical surface area across disorders: findings from the ENIGMA ADHD, ASD, and OCD Working Groups. Am J Psychiatry. 2020;177:834–43.
Sprooten E, Rasgon A, Goodman M, Carlin A, Leibu E, Lee WH, et al. Addressing reverse inference in psychiatric neuroimaging: Meta-analyses of task-related brain activation in common mental disorders. Hum Brain Mapp. 2017;38:1846–64.
Neumann A, Muetzel RL, Lahey BB, Bakermans-Kranenburg MJ, van IMH, Jaddoe VW, et al. White matter microstructure and the general psychopathology factor in children. J Am Acad Child Adolesc Psychiatry. 2020;59:1285–96.
Alnaes D, Kaufmann T, Marquand AF, Smith SM, Westlye LT. Patterns of socio-cognitive stratification and perinatal risk in the child brain. bioRxiv 2020;839969.
Turkheimer E. Three laws of behavior genetics and what they mean. Curr Directions Psychological Sci. 2000;9:160–4.
Haslam N, McGrath MJ, Viechtbauer W, Kuppens P. Dimensions over categories: a meta-analysis of taxometric research. Psychol Med. 2020;50:1418–32.
Falconer DS. The inheritance of liability to certain diseases, estimated from the incidence among relatives. Ann Hum Genet. 1965;29:51–76.
Mallard TT, Linner RK, Okbay A, Grotzinger AD, de Vlaming R, Meddens SFW, et al. Not just one p: Multivariate GWAS of psychiatric disorders and their cardinal symptoms reveal two dimensions of cross-cutting genetic liabilities. bioRxiv 2019;603134.
Wray NR, Ripke S, Mattheisen M, Trzaskowski M, Byrne EM, Abdellaoui A, et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat Genet. 2018;50:668–81.
Demontis D, Walters RK, Martin J, Mattheisen M, Als TD, Agerbo E, et al. Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat Genet. 2019;51:63–75.
Grasby KL, The ENIGMA Consortium, Jahanshad N, Painter JN, Colodro-Conde L, Bralten DP, et al. The genetic architecture of the human cerebral cortex. BioRXiv 2018; bioRxiv 399402.
Glasser MF, Smith SM, Marcus DS, Andersson JL, Auerbach EJ, Behrens TE, et al. The human connectome project’s neuroimaging approach. Nat Neurosci. 2016;19:1175–87.
Hill WD, Hagenaars SP, Marioni RE, Harris SE, Liewald DCM, Davies G, et al. Molecular genetic contributions to social deprivation and household income in UK biobank. Curr Biol. 2016;26:3083–9.
Hill WD, Davies NM, Ritchie SJ, Skene NG, Bryois J, Bell S, et al. Genome-wide analysis identifies molecular systems and 149 genetic loci associated with income. Nat Commun. 2019;10:5741.
Polderman TJ, Benyamin B, de Leeuw CA, Sullivan PF, van Bochoven A, Visscher PM, et al. Meta-analysis of the heritability of human traits based on fifty years of twin studies. Nat Genet. 2015;47:702–9.
Trzaskowski M, Harlaar N, Arden R, Krapohl E, Rimfeld K, McMillan A, et al. Genetic influence on family socioeconomic status and children’s intelligence. Intelligence. 2014;42:83–88.
Davies G, Marioni RE, Liewald DC, Hill WD, Hagenaars SP, Harris SE, et al. Genome-wide association study of cognitive functions and educational attainment in UK Biobank (N=112 151). Mol Psychiatry. 2016;21:758–67.
Gao J, Davis LK, Hart AB, Sanchez-Roige S, Han L, Cacioppo JT, et al. Genome-wide association study of loneliness demonstrates a role for common variation. Neuropsychopharmacology. 2017;42:811–21.
Dalvie S, Maihofer AX, Coleman JRI, Bradley B, Breen G, Brick LA, et al. Genomic influences on self-reported childhood maltreatment. Transl Psychiatry. 2020;10:38.
Plomin R, DeFries JC, Loehlin JC. Genotype-environment interaction and correlation in the analysis of human behavior. Psychol Bull. 1977;84:309–22.
Kendler KS, Eaves LJ. Models for the joint effect of genotype and environment on liability to psychiatric illness. Am J Psychiatry. 1986;143:279–89.
Scarr S, McCartney K. How people make their own environments: a theory of genotype greater than environment effects. Child Dev. 1983;54:424–35.
Plomin R. Genotype-environment correlation in the era of DNA. Behav Genet. 2014;44:629–38.
Gage SH, Davey Smith G, Ware JJ, Flint J, Munafo MR. G = E: what GWAS Can Tell Us about the Environment. PLoS Genet. 2016;12:e1005765.
Elliott LT, Sharp K, Alfaro-Almagro F, Shi S, Miller KL, Douaud G, et al. Genome-wide association studies of brain imaging phenotypes in UK Biobank. Nature. 2018;562:210–6.
Marees AT, Smit DJA, Abdellaoui A, Nivard MG, van den Brink W, Denys D et al. Genetic correlates of socio-economic status influence the patterns of shared heritability across mental health traits. medRXiv. 2020;2020.02.26.20028092.
Grasby KL, Jahanshad N, Painter JN, Colodro-Conde L, Bralten J, Hibar DP, et al. The genetic architecture of the human cerebral cortex. Science. 2020;367:eaay6690.
Hibar DP, Stein JL, Renteria ME, Arias-Vasquez A, Desrivieres S, Jahanshad N, et al. Common genetic variants influence human subcortical brain structures. Nature. 2015;520:224–9.
McKay DR, Knowles EE, Winkler AA, Sprooten E, Kochunov P, Olvera RL, et al. Influence of age, sex and genetic factors on the human brain. Brain Imaging Behav. 2014;8:143–52.
Jahanshad N, Kochunov PV, Sprooten E, Mandl RC, Nichols TE, Almasy L, et al. Multi-site genetic analysis of diffusion images and voxelwise heritability analysis: a pilot project of the ENIGMA-DTI working group. Neuroimage. 2013;81:455–69.
Gottesman II, Gould TD. The endophenotype concept in psychiatry: etymology and strategic intentions. Am J Psychiatry. 2003;160:636–45.
Glahn DC, Knowles EE, McKay DR, Sprooten E, Raventos H, Blangero J, et al. Arguments for the sake of endophenotypes: examining common misconceptions about the use of endophenotypes in psychiatric genetics. Am J Med Genet B Neuropsychiatr Genet. 2014;165B:122–30.
Greenwood TA, Shutes-David A, Tsuang DW. Endophenotypes in schizophrenia: digging deeper to identify genetic mechanisms. J Psychiatr Brain Sci. 2019;4:e190005.
Walters JT, Owen MJ. Endophenotypes in psychiatric genetics. Mol Psychiatry. 2007;12:886–90.
Clarke TK, Zeng Y, Navrady L, Xia C, Haley C, Campbell A, et al. Genetic and environmental determinants of stressful life events and their overlap with depression and neuroticism. Wellcome Open Res. 2018;3:11.
Gasperi M, Herbert M, Schur E, Buchwald D, Afari N. Genetic and environmental influences on sleep, pain, and depression symptoms in a community sample of twins. Psychosom Med. 2017;79:646–54.
Marees AT, Smit DJA, Abdellaoui A, Nivard MG, van den Brink W, Denys D et al. Genetic correlates of socio-economic status influence the pattern of shared heritability across mental health traits. medRXiv 2020;2020.02.26.20028092.
Sullivan PF, Agrawal A, Bulik CM, Andreassen OA, Borglum AD, Breen G, et al. Psychiatric genomics: an update and an agenda. Am J Psychiatry. 2018;175:15–27.
Shi H, Kichaev G, Pasaniuc B. Contrasting the genetic architecture of 30 complex traits from summary association data. Am J Hum Genet. 2016;99:139–53.
Loh PR, Bhatia G, Gusev A, Finucane HK, Bulik-Sullivan BK, Pollack SJ, et al. Contrasting genetic architectures of schizophrenia and other complex diseases using fast variance-components analysis. Nat Genet. 2015;47:1385–92.
Van Erp TGM, Walton E, Hibar DP, Schmaal L, Jiang W, Glahn DC, et al. Cortical brain abnormalities in 4474 individuals with schizophrenia and 5098 control subjects via the enhancing neuro imaging genetics through meta analysis (ENIGMA) consortium. Biol Psychiatry. 2018;84:644–54.
Schmaal L, Hibar DP, Samann PG, Hall GB, Baune BT, Jahanshad N, et al. Cortical abnormalities in adults and adolescents with major depression based on brain scans from 20 cohorts worldwide in the ENIGMA Major Depressive Disorder Working Group. Mol Psychiatry. 2017;22:900–9.
Miller KL, Alfaro-Almagro F, Bangerter NK, Thomas DL, Yacoub E, Xu J, et al. Multimodal population brain imaging in the UK Biobank prospective epidemiological study. Nat Neurosci. 2016;19:1523–36.
Thompson PM, Stein JL, Medland SE, Hibar DP, Vasquez AA, Renteria ME, et al. The ENIGMA consortium: large-scale collaborative analyses of neuroimaging and genetic data. Brain Imaging Behav. 2014;8:153–82.
Thompson PM, Jahanshad N, Ching CRK, Salminen LE, Thomopoulos SI, Bright J, et al. ENIGMA and global neuroscience: a decade of large-scale studies of the brain in health and disease across more than 40 countries. Transl Psychiatry. 2020;10:100.
DiLalla LF, Jamnik MR. The Southern Illinois Twins/Triplets and Siblings Study (SITSS): a longitudinal study of early child development. Twin Res Hum Genet. 2019;22:779–82.
Vertsberger D, Abramson L, Knafo-Noam A. The longitudinal israeli study of twins (LIST) reaches adolescence: genetic and environmental pathways to social, personality and moral development. Twin Res Hum Genet. 2019;22:567–71.
Jamnik MR, DiLalla LF. A multimethodological study of preschoolers’ preferences for aggressive television and video games. J Genet Psychol. 2018;179:156–69.
Roelfs D, Alnaes D, Frei O, van der Meer D, Smeland OB, Andreassen OA, et al. Phenotypically independent mental health profiles are genetically related. MedRXiv 2020.03.30.20045591.
Waszczuk M, Miao J, Docherty A, Shabalin E, Michelini G, Jonas K, et al. General vs. specific vulnerabilities: polygenic risk scores and higher-order psychopathology dimensions in the adolescent brain cognitive development (ABCD) study. PsyArXiv 2020;10.31234/osf.io/km6v3.
Watanabe K, Taskesen E, van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun. 2017;8:1826.
Watanabe K, Umicevic Mirkov M, de Leeuw CA, van den Heuvel MP, Posthuma D. Genetic mapping of cell type specificity for complex traits. Nat Commun. 2019;10:3222.
Klein M, Onnink M, van Donkelaar M, Wolfers T, Harich B, Shi Y, et al. Brain imaging genetics in ADHD and beyond - Mapping pathways from gene to disorder at different levels of complexity. Neurosci Biobehav Rev. 2017;80:115–55.
Yarkoni T, Poldrack RA, Nichols TE, Van Essen DC, Wager TD. Large-scale automated synthesis of human functional neuroimaging data. Nat Methods. 2011;8:665–70.
Frei O, Holland D, Smeland OB, Shadrin AA, Fan CC, Maeland S, et al. Bivariate causal mixture model quantifies polygenic overlap between complex traits beyond genetic correlation. Nat Commun. 2019;10:2417.
Zhu Z, Zheng Z, Zhang F, Wu Y, Trzaskowski M, Maier R, et al. Causal associations between risk factors and common diseases inferred from GWAS summary data. Nat Commun. 2018;9:224.
Allegrini AG, Selzam S, Rimfeld K, von Stumm S, Pingault JB, Plomin R. Genomic prediction of cognitive traits in childhood and adolescence. Mol Psychiatry. 2019;24:819–27.
Hasenfratz L, Benish-Weisman M, Steinberg T, Knafo-Noam A. Temperament and peer problems from early to middle childhood: gene-environment correlations with negative emotionality and sociability. Dev Psychopathol. 2015;27:1089–109.
Lee SH, Yang J, Goddard ME, Visscher PM, Wray NR. Estimation of pleiotropy between complex diseases using single-nucleotide polymorphism-derived genomic relationships and restricted maximum likelihood. Bioinformatics. 2012;28:2540–2.
Deary IJ, Yang J, Davies G, Harris SE, Tenesa A, Liewald D, et al. Genetic contributions to stability and change in intelligence from childhood to old age. Nature. 2012;482:212–5.
Trzaskowski M, Yang J, Visscher PM, Plomin R. DNA evidence for strong genetic stability and increasing heritability of intelligence from age 7 to 12. Mol Psychiatry. 2014;19:380–4.
Jernigan TL, Brown SA, Coordinators AC. Introduction. Dev Cogn Neurosci. 2018;32:1–3.
Pettersson E, Larsson H, Lichtenstein P. Common psychiatric disorders share the same genetic origin: a multivariate sibling study of the Swedish population. Mol Psychiatry. 2016;21:717–21.
Acknowledgements
ES is supported by a Hypatia Tenure Track Grant (Radboudumc), Christine Mohrmann Fellowship (Radboud University), and a NARSAD Young Investigator Grant (Brain and Behavior Research Foundation, ID: 25034). BF is supported by the NWO through a personal Vici grant (grant number 016-130-669) and a pilot grant from the Dutch National Research Agenda for the NeuroLabNL project (grant 400 17 602). CUG is supported by the Netherlands Foundation of Mental Health (grant number 2016 7057).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
BF has received educational speaking fees from Medice, which had no influence on the present manuscript. The other authors (ES and CUG) have no conflicts of interest to disclose.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sprooten, E., Franke, B. & Greven, C.U. The P-factor and its genomic and neural equivalents: an integrated perspective. Mol Psychiatry 27, 38–48 (2022). https://doi.org/10.1038/s41380-021-01031-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-021-01031-2
This article is cited by
-
Perinatal and neonatal factors and mental disorders in children and adolescents: looking for the contributions of the early environment to common and dissociable aspects of psychopathology
European Child & Adolescent Psychiatry (2024)
-
Running in the FAMILY: understanding and predicting the intergenerational transmission of mental illness
European Child & Adolescent Psychiatry (2024)
-
A transdiagnostic network for psychiatric illness derived from atrophy and lesions
Nature Human Behaviour (2023)
-
Cortical profiles of numerous psychiatric disorders and normal development share a common pattern
Molecular Psychiatry (2023)
-
The harmonium model and its unified system view of psychopathology: a validation study by means of a convolutional neural network
Scientific Reports (2022)
