
Abstract
Myeloproliferative neoplasms (MPNs) are frequently associated with classic driver mutations involving JAK2, MPL or CALR. SRSF2 is among the most frequently mutated splicing genes in myeloid neoplasms and SRSF2 mutations are known to confer a poor prognosis in patients with MPNs. In this study, we sought to evaluate the clinicopathologic spectrum of myeloid neoplasms harboring concurrent MPN-driver mutations and SRSF2 mutations. The study cohort included 27 patients, 22 (82%) men and five (19%) women, with a median age of 71 years (range, 51–84). These patients presented commonly with organomegaly (n = 15; 56%), monocytosis (n = 13; 48%), morphologic dysplasia (n = 11; 41%), megakaryocytic hyperplasia and/or clustering (n = 10; 37%) and bone marrow fibrosis >MF-1 (17/22; 77%). About one third of patients either initially presented with acute myeloid leukemia (AML) or eventually progressed to AML. Eighteen (68%) patients had a dominant clone with SRSF2 mutation and nine (33%) patients had a dominant clone with a classic MPN-associated driver mutation. Our data suggest that the presence of an SRSF2 mutation preceding the acquisition of a MPN driver mutations is not a disease-defining alteration nor is it restricted to any specific disease entity within the spectrum of myeloid neoplasms. In summary, patients with myeloid neoplasms associated with concurrent SRSF2 and classic MPN driver mutations have clinical and morphologic features close to that of classic MPNs often with frequent dysplasia and monocytosis.
Similar content being viewed by others

Introduction
Myeloproliferative neoplasms (MPNs) are a heterogeneous group of hematopoietic stem cell disorders characterized by a clonal proliferation of erythroid, myeloid and/or megakaryocytic cells. Philadelphia (Ph)-negative MPNs are further subclassified into polycythemia vera, characterized by overproduction of red blood cells; essential thrombocythemia, defined by overproduction of platelets; and primary myelofibrosis, characterized by proliferation of abnormal megakaryocytes and granulocytes and stepwise evolution of bone marrow fibrosis depending on the stage of disease. However, distinguishing between these entities based solely on clinical and morphologic findings can be challenging; a subset of cases have overlapping features between the aforementioned entities or with myelodysplastic syndromes and are therefore classified as MPN-unclassifiable or myelodysplastic/myeloproliferative neoplasm (MDS/MPN)-unclassifiable, respectively1,2.
The advent of massive parallel sequencing and identification of recurrent somatic gene mutations has had an impact on the classification and prognostication of myeloid neoplasms including MPNs. Mutations of JAK2, MPL, and CALR have been recognized as driver mutations in MPNs, and these mutations have been incorporated into the diagnostic criteria for MPNs. Yet, these mutations do not occur in isolation and are often seen in concert with other mutations and chromosomal alterations, many of which also influence disease phenotype and prognosis3. Mutations of spliceosome regulator genes can occur in MPNs and have been shown to be enriched in cases classified as primary myelofibrosis and MDS/MPN; these mutations are associated with an increased risk for progression to myelofibrosis3.
SRSF2 (Serine/arginine-rich Splicing Factor 2) is located on chromosome 17q25.2 and is among the most frequently mutated spliceosome regulator genes in myeloid neoplasms4,5. The most common SRSF2 mutations involve the proline 95 (P95) residue and alter the RNA binding specificity of SRSF2. These mutations lead to aberrant splicing of hematopoietic regulators, eventually resulting in impaired hematopoiesis. Murine models have shown that Srsf2 P95H results in myeloid dysplasia in an EZH2-dependent manner6. Numerous clinical studies have shown that SRSF2 mutations occur in various myeloid neoplasms including MDS, MDS/MPN (with particular enrichment in chronic myelomonocytic leukemia), MPNs, and acute myeloid leukemia (AML)7,8,9,10. Moreover, SRSF2 mutations confer a poorer prognosis in patients with myeloid neoplasms3,4,11,12.
Concurrent classic MPN driver mutations (JAK2, MPL, CALR) with SRSF2 mutations can give rise to myeloid neoplasms with features overlapping between classic Ph-negative MPNs and those with a phenotype more akin to MDS/MPNs or MDS. Recognition of the clinicopathologic spectrum of these entities is helpful for the most appropriate classification of these entities, given that patient management and clinical trial enrollment criteria may differ based on pathologic classification13,14,15. Although SRSF2 mutations play a substantial role in the phenotypic manifestations of myeloid neoplasms as well as patient outcomes, these mutations are thus far considered to be non-driver mutations.
In this study, our aim was to focus on SRSF2 mutations in the context of myeloid neoplasms with MPN-associated driver mutations. We correlated the presence of SRSF2 mutations with disease classification and sought to explore the phenotypic features of myeloid neoplasms harboring classic MPN-associated driver mutations and concurrent SRSF2 mutation.
Materials and methods
We searched our sequencing archives for myeloid neoplasms harboring concurrent classic MPN driver mutations (JAK2, MPL, CALR) and SRSF2 mutations resulted between 09/28/2016 (assay go-live) and 07/01/2019. In order to capture the entire spectrum of disease presentation, we included de novo and untreated myeloid neoplasms as well as cases that were previously treated with this combination of mutations. Using this selection criteria, we identified a total of 27 patients among 4740 patients (0.5%) that had undergone sequencing for suspected myeloid neoplasms during the time interval. Clinical and laboratory data were obtained from the electronic medical records. This study was approved by the Institutional Review Board (IRB) at MD Anderson Cancer Center (MDACC) and conducted in accord with the Declaration of Helsinki.
Bone marrow morphology
Hematoxylin and eosin-stained sections of bone marrow (BM) trephine biopsy specimens and Wright–Giemsa-stained BM aspirate smears were used for morphologic classification according to current World Health Organization diagnostic criteria16. A 200- or 500-nucleated cell differential cell count was performed when possible. Special attention was paid to megakaryocytic morphology (number, loose vs tight clustering, atypia vs dysplasia including small forms or nuclear hypolobation). Furthermore, erythroid and/or granulocytic lineages were also examined for dysplasia. Dysplasia was assessed using the criteria proposed by Della Porta et al.17. Reticulin and Masson trichrome stains were performed using tissue sections prepared from paraffin-embedded trephine biopsy specimens and an automated stainer (Leica Biosystems, Buffalo Grove, IL). We evaluated the degree of reticulin and collagen fibrosis and BM fibrosis was graded according to the criteria proposed by the European Bone Marrow Fibrosis Consensus Group18.
Conventional karyotyping
Bone marrow aspirate cells were cultured and harvested after 24 and 48 h, and chromosome slides were prepared according to standard protocol for G banding as described previously19. A minimum of 20 metaphases were analyzed when possible and karyotypes were reported using the 2016 International System for Human Cytogenetic Nomenclature20.
Molecular analysis
Next-generation sequencing (NGS) analysis was performed interrogating all exons or hotspot regions of 81 genes mutated frequently in myeloid malignancies (Supplementary Table 1) validated at the CLIA-certified molecular diagnostic laboratory at MDACC as described previously21. A sequencing library was prepared using 250 ng of genomic DNA and respective sequencing libraries were subjected to the Illumina MiSeq (Illumina, Inc., San Diego, CA, USA) sequencer. Variant calling was performed using the SureCall application (Agilent HaloPlex Target Enrichment System). The Integrative Genomics Viewer (IGV, Broad Institute) was used for data visualization. A minimum sequencing coverage of x250 (bidirectional true paired-end sequencing) was required. The analytical sensitivity was established at 1–2% mutant reads in a background of wild type reads.
Results
Patients
The study group included 22 (82%) men and five (19%) women with a median age of 71 years (range, 51–84). At the time of molecular characterization the cases were classified as follows: primary myelofibrosis (PMF, 14 cases, 52%), acute myeloid leukemia (AML, 4 cases, 15%), chronic myelomonocytic leukemia (CMML, three cases, 11%), polycythemia vera (PV, 2 cases, 7.4%), post-ET/PV myelofibrosis (two cases, 7.4%), MPN-unclassifiable (MPN-U, one case, 3.6%), and MDS/MPN-unclassifiable (MDS/MPN-U, one case, 3.6%) (Fig. 1A).

A Spectrum of diseases in the study group, (B) Spectrum of mutations in SRSF2, (C) Spectrum of MPN-related mutations.
All patients except two with the diagnosis of PV had molecular characterization months after the initial diagnosis. The median interval between the time of initial diagnosis and molecular characterization was 18.9 months (range, 0–293) and the breakdown for each category was as follows: PMF, 18.7 months (range, 0.6–81.6); post-ET/PV myelofibrosis, 37.2 months (range, 18.8–55.5); CMML, 36.2 months (range, 12.6–66.4); AML, 34.9 months (range, 7.1–292.6), MPN-U, 1.4 months, and MDS/MPN-U, 14.7 months. Seventeen of 27 (63%) patients had received therapy for their myeloid neoplasm (other than cytoreductive agents alone) prior to molecular characterization(Table 1). Therapeutic agents included ruxolitinib (n = 15); azacitidine (n = 3); thalidomide (n = 2); cladribine+ low dose cytarabine (n = 1). These agents were administered alone or in combination with one another.
Peripheral blood indices at the time of molecular characterization were variable among these patients (Table 1). The median absolute monocyte count (AMC) was as follows: PMF, 0.945 × 109/L (range, 0–7.59); PV, 0.90 × 109/L (range, 0.31–1.48); post-ET/PV myelofibrosis, 0.06 × 109/L (range, 0–0.12); CMML, 1.58 × 109/L (range, 0.97–17.56); AML, 0.26 × 109/L (range, 0.02–7.4); MPN-U, 6.48 × 109/L; and MDS/MPN-U, 0.15 × 109/L. Overall, 13 (48%) patients had absolute monocytosis (>1 × 109/L), predominantly in patients with PMF and CMML (Fig. 2A, E).

PMF with monocytosis, patient 22 (top panel, (A) peripheral blood smear, (B) bone marrow aspirate smear, (C) H&E stained trephine core biopsy section, (D) reticulin stained trephine core biopsy section) and CMML with MF-3, patient 29 (bottom panel, (E) peripheral blood smear, (F) bone marrow aspirate smear, (G) H&E stained trephine core biopsy section, (H) reticulin stained trephine core biopsy section).
Fifteen of 27 (55.5%) patients had splenomegaly at the time of molecular characterization. (Table 1). Transfusion-dependency was present in four of 14 (28.6%) patients with PMF, one of two (50%) patients with AML patients and one patient with MDS/MPN-U (Table 1).
Morphologic dysplasia was present in 11 (40.7%) patients including 3/8 (38%) previously untreated patients (all three with disease manifesting as PMF); seven had dysplasia in ≥2 lineages (six granulocytic, six erythroid, and four megakaryocytic) and four had single lineage dysplasia in granulocytes (n = 2) and megakaryocytes (n = 2). The morphologic classification of these 11 neoplasms included six PMF, two CMML, two AML and one MDS/MPN-U. The PMF group included three with dysplasia in granulocytic and erythroid lineages, one with dysplasia in granulocytic and megakaryocytic lineages, one with granulocytic dysplasia, and one with dysplasia in megakaryocytes. One patient with CMML had trilineage dysplasia and the other had granulocytic dysplasia. AML patients included one with dysplasia in the erythroid and megakaryocytic lineages and one with erythroid dysplasia. The patient with MDS/MPN-U had trilineage dysplasia (Table 1).
Megakaryocytic hyperplasia, defined as >6 megakaryocytes per high power field on average, and/or clustering was observed in 10 (37%) cases: six PMF, two CMML, and two PV (Fig. 2C, G). We further evaluated the morphology of megakaryocytes in each group: PMF patients had predominantly hybrid MDS/MPN-like megakaryocytic morphology (nine of 14, 64.3%), followed by MPN-like (two of 14, 14.3%), and MDS-like (one of 14, 7.1%). Most of the remaining groups had at least one case with hybrid morphologic features.
Bone marrow fibrosis was present in all cases assessed (n = 22, including 7/7 evaluable untreated cases): five (23%) MF-1, 11 (50%) MF-2, and six (27%) MF-3. The 17 patients with MF-2 and MF-3 had PMF (n = 12), post-ET/PV myelofibrosis (n = 2), CMML (n = 2) and PV (n = 1) (Table 1) (Fig. 2D, H). Ten (37%) patients had osteosclerosis; among these, eight had PMF.
Conventional cytogenetic analysis at the time of molecular studies revealed 16 (59.3%) cases had a diploid karyotype and 11 (40.7%) cases had an abnormal karyotype. Among eight previously untreated cases, seven had a diploid karyotype and one case had del(13)(q12q22). Five of 11 patients had a complex karyotype or monosomy 5: four patients with PMF and one patient with AML-MRC (Table 1). The most prevalent aberrations were del(20q) (n = 3) and del(13q) (n = 3) and all of these patients had PMF.
Spectrum of genetic changes
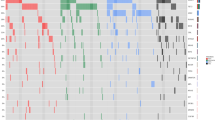
SRSF2 mutations included P95H (n = 17, 63%), P95R (n = 6, 22.2%), P95L (n = 2, 7.4%), p.P95_R102del (n = 2, 7.4%) with a median variant allelic frequency (VAF) of 44.5% (range, 2–65%) (Fig. 1B). MPN driver mutations involved JAK2 (n = 19, 70.3%), MPL (n = 7, 26%) and CALR (n = 1, 3.7%); these mutations were mutually exclusive (Figs. 1C and 3). All patients with JAK2 mutations had the canonical JAK2 V617F variant with a median VAF of 36% (range, 2–73%). MPL mutations had a median VAF of 24.2% (range, 6–88%) and included the W515L (n = 5), L629Q (n = 1), V501M (n = 1), and S493A (n = 1) variants; one patient had two concurrent MPL mutations: W515L and S493A. The one patient with CALR mutation had a CALR K385fs variant with VAF of 58%.

Patients 1, 4, 6, 11, 14, 17, 22, 25 were previously untreated (de novo).
Other recurrent mutations were present. Eighteen (66.6%) patients had ASXL1 mutations; one patient had two concurrent ASXL1 mutations (S1028* and G658*). The median VAF for ASXL1 mutations was 22% (range, 5–46%) and the most frequent mutation was ASXL1 G646fs (n = 5). Twelve (44%) patients had TET2 mutations; eight had two different TET2 mutations and one patient had three distinct mutations. The median VAF for TET2 mutations was 37% (range, 4–94%). Eight (29.6%) patients had mutations involving the RAS pathway: six NRAS and two KRAS. The median VAF of the NRAS mutations was 23% (range, 2–46%) and the mutations included G12D/A/C/S (n = 5) and Y64D (n = 1). The KRAS mutations were G12R with VAF of 15% and A59T with VAF of 45%. Six (22%) patients had IDH2 R140Q mutations with a median VAF of 45% (range, 3–48%); of note these patients had chronic myeloid neoplasms, mostly PMF. Other mutations were present with a lower frequency (Fig. 3).
We inferred clonal dominance based on VAF with the mutation with the highest VAF considered as being present in a dominant clone and found that when all mutations were considered, SRSF2 was present in the dominant clone in 13 (48.1%) patients with a median VAF of 47% (range, 24–65%). Other co-dominant mutations included JAK2 [n = 3; median VAF of 60% (range, 23–63 %), IDH2 (n = 2; VAFs: 44 and 43%), TET2 (n = 2; one case with two mutations with VAFs of 48 and 49% and the other case with VAF of 95%), MPL (n = 2; VAFs: 88 and 32%), ASXL1 (n = 2; VAFs: 45% each), CBL (n = 1, VAF: 82%), RUNX1 (n = 1, VAF: 88%), SETBP1 (n = 1, VAF: 49%) and CALR (n = 1, VAF: 58%).
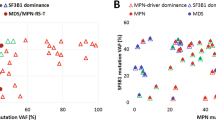
We then subclassified the patients based on the dominance of the SRSF2 mutant clone or classic MPN-associated mutations, only considering these two mutation groups. Eighteen (66.7%) patients had a dominant SRSF2 clone and these neoplasms were PMF (n = 9, 50%), AML (n = 4, 22.2%), CMML (n = 2, 11.1%), PV (n = 2, 11.1%) and MDS/MPN-U (n = 1, 5.6%) (Fig. 4A). Nine (33.3%) patients had classic dominant MPN-associated mutations and these neoplasms were PMF (n = 5, 55.6%), post-PV/ET MF (n = 2, 22.2%), CMML (n = 1, 11.1%), and MPN-U (n = 1, 11.1%) (Fig. 4B). We further examined the clonal dominance of SRSF2 and MPN-associated mutations in each disease group (Fig. 5).

A Spectrum of diseases with SRSF2 dominant clones, (B) Spectrum of diseases with MPN-related dominant clones.

Violin plot of VAF for SRSF2 and MPN-related mutations in each disease group.
Although concurrent splicing factor mutations are commonly mutually exclusive in myeloid neoplasms5, in this cohort three patients had 2 splicing factor mutations. Two patients had PMF with concurrent SRSF2 and U2AF1 mutations; one with SRSF2 P95H (VAF: 33.5%) and U2AF1 Q157P (VAF: 15%) and the other with SRSF2 P95H (VAF: 2%) and U2AF1 Q157P (VAF: 37%). One patient had AML-MRC with a SRSF2 P95R (VAF: 52%) and ZRSR2 R169* (VAF: 37%) (Fig. 3). Notably, no splicing factor gene had more than one mutation and, expectedly, no patients had SF3B1 mutations22.
Clinical outcomes
Four patients with chronic neoplasms progressed to AML during the course of their disease. The median time to AML transformation was 35.5 months (range, 20.5–297.6). The initial diagnosis in these patients was PMF (n = 2), PV (n = 1), and CMML (n = 1) (Table 1). Among the four patients who progressed to AML, two were diagnosed with AML at the time of molecular characterization (index case in this cohort) and two cases (initially PV and PMF) progressed later. Both of the two patients that progressed later had SRSF2 mutations in their chronic phase of disease. One patient initially diagnosed with PV, retained their SRSF2 mutation but lost their JAK2 mutation with acquisition of additional mutations involving BRAF, KRAS and RAD21 at the time of AML transformation; the other patient, initially diagnosed with PMF, transformed with extramedullary disease (myeloid sarcoma of skin); the skin sample was not sequenced but the concurrent bone marrow had lost the SRSF2 mutation and retained the JAK2 mutation at this time, along with additional mutations involving NRAS and NF1. Among 27 patients, 24 patients were treated with agents other than supportive therapy as follows: 10 received a JAK2 inhibitor, five were treated with hypomethylating agents, and seven received both a JAK2 inhibitor and a hypomethylating agent. Four patients underwent allogeneic hematopoietic stem cell transplant (HSCT). The median overall survival of patients in this study was 41 months (range, 9.7–313.5). Among different disease groups, the median overall survival was relatively comparable, with patients in the post-PV/ET MF subgroup having the longest median overall survival [52 months, (45.3 and 58.6)] (Table 1). Six (20.7%) patients had died by the time we conducted this study. All patients who received an allogeneic HSCT were alive with no disease recurrence at last follow up with a median survival of 42.7 months (range, 17.3–49.3).
Discussion
In this study, we evaluated the clinicopathologic spectrum of myeloid neoplasms harboring concurrent MPN-driver mutations and SRSF2 mutations. We show that the co-occurrence of these mutations is seen predominantly in elderly men who present commonly with organomegaly, monocytosis, morphologic dysplasia, megakaryocytic hyperplasia and/or clustering and bone marrow fibrosis. Mostly, these patients have classic MPN-like phenotypes. About one third of these patients either presented with AML or eventually progressed to AML. A limitation of our study is the inclusion of patients that had undergone therapy prior to molecular characterization (17/27), however, most of these patients were treated with ruxolitinib or hypomethylating agents or a combination of the two, which typically do not substantially alter disease morphology or phenotype; only one patient had received cytotoxic chemotherapy prior to molecular characterization.
Given the different management and therapeutic options for patients with MPN versus those with MDS/MPN cases, more specifically PMF versus CMML, the definitive diagnosis of myeloid neoplasms with intermediate features between these two entities is of clinical interest13,14,15 (Fig. 2). Monocytosis is frequently seen in myeloid neoplasms with SRSF2 mutations which was also common in our study cohort13. In our cohort, eight of 19 MPN cases had absolute and relative monocytosis. Moreover, all three CMML cases had fibrosis to a variable degree. In this cohort, 22 patients already had an established diagnosis prior to molecular studies; thus, the monocytosis or fibrosis did not lead to disease reclassification. However, it is important to recognize that monocytosis and fibrosis can occur in the course of disease and lead to manifestations of MDS/MPN-like features.
SRSF2 is a member of the serine/arginine-rich (SR) protein family that plays a role in pre-mRNA splicing through its RNA recognition motif (RRM) domain. SRSF2 mutations occur predominantly at proline 95. P95H/L/R mutants enhance binding affinity of the RRM domain via conformational changes up to 5 fold greater than wild type SRSF26,23. In contrast, the P95A mutant does not influence binding affinity. In our cohort, 27 neoplasms had missense mutations, including P95H in 19 cases, P95R in six, and P95L in two. Two patients had P95_R102del (c.284_307del).
Kim et al.6 showed in a detailed mouse study that Srsf2P95H/wild type mice develop macrocytic anemia and leukopenia with preserved BM cellularity and myeloid and erythroid dysplasia, recapitulating the clinicopathologic features of MDS. Numerous studies also have shown that SRSF2 mutations are frequent in MDS and MDS/MPN patients4,9,12. In our cohort, slightly more than half of patients had dysplasia in single or multiple lineages. Moreover, Papaemmanuil et al.4 suggested that SRSF2 mutations occur as early genetic events in MDS pathogenesis and predestine neoplastic clones to acquire specific genetic and genomic alterations. Mutant SRSF2 was a dominant clone in most cases in our cohort; yet these neoplasms mostly showed MPN-type features. Our observations suggest that SRSF2 mutations could be early genetic events in a subset of MPN patients.
Lee et al.22 showed that a minority (~2%) of patients with myeloid neoplasms have >1 splicing factor mutation and that concurrent SRSF2 and SF3B1 mutations cannot be tolerated during hematopoiesis due to impaired HSPC self-renewal, differentiation, and survival. In our cohort, co-mutant SRSF2 and SF3B1 did not exist. However, we identified four cases with coexistent splicing factor mutations including ZRSR2 and U2AF1. Using bulk and single cell analyses, Tyler and colleagues23 reported rare myeloid neoplasms with escape of such mutational epistasis demonstrating a selection for less common mutants in the presence of concomitant splicing factor mutations. We also observed the same pattern in two patients: concomitant SRSF2 P95H and a rare missense mutation in ZRSR2 with similar VAFs (45 and 50%, respectively) and SRSF2 P95R with a truncating ZRSR2 R169* mutation with slightly different VAFs (53 and 37%, respectively). The other two patients had concurrent common mutations, SRSF2 P95H and U2AF1 Q157P; however, the VAFs of the mutants were substantially different: one with 2% and 37%, respectively, and the other with 34% and 15%, respectively, suggesting that these mutations exist in distinct clones. This observation could be of importance in designing future targeted therapies for patients with myeloid neoplasms with mutated splicing factor genes; however, further characterization is limited in our study due lack of single cell genomics data.
The results of this study highlight that concurrent MPN-driver mutations and SRSF2 mutations can occur in a variety of myeloid neoplasms with a predominance of a MPN-like phenotype and clinical features. SRSF2 mutation in myeloid neoplasms may precede the acquisition of MPN driver mutations and might affect the histologic features and clinical course of the disease; however, SRSF2 mutation is not a disease-defining alteration or restricted to any specific entity. Rather, SRSF2 mutation cooperates with other genomic alterations to exert its phenotypic effects.
Data availability
All data are available upon request.
References
Kvasnicka, H. M., Thiele, J., Orazi, A., Horny, H. P. & Bain, B. J. Myeloproliferative neoplasm, unclassifiable. In: S.H. Swerdlow et al. (eds). WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues 57–59 (IARC Lyon, 2017).
Orazi, A., Bennett, J. M., Bain, B. J., Baumann, I., Thiele, J., Bueso-Ramos, C. et al. Myelodysplastic I myeloproliferative neoplasm, unclassifiable. In: S.H. Swerdlow et al. (eds). WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues 95–96 (IARC: Lyon, 2017).
Grinfeld, J., Nangalia, J., Baxter, E. J., Wedge, D. C., Angelopoulos, N., Cantrill, R. et al. Classification and Personalized Prognosis in Myeloproliferative Neoplasms. New England Journal of Medicine 379, 1416–1430 (2018).
Papaemmanuil, E., Gerstung, M., Malcovati, L., Tauro, S., Gundem, G., Van Loo, P. et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 122, 3616–3627; quiz 3699 (2013).
Yoshida, K., Sanada, M., Shiraishi, Y., Nowak, D., Nagata, Y., Yamamoto, R. et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478, 64–69 (2011).
Kim, E., Ilagan, J. O., Liang, Y., Daubner, G. M., Lee, S. C., Ramakrishnan, A. et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 27, 617–630 (2015).
Patnaik, M. M., Lasho, T. L., Finke, C. M., Hanson, C. A., Hodnefield, J. M., Knudson, R. A. et al. Spliceosome mutations involving SRSF2, SF3B1, and U2AF35 in chronic myelomonocytic leukemia: prevalence, clinical correlates, and prognostic relevance. Am J Hematol 88, 201–206 (2013).
Makishima, H., Visconte, V., Sakaguchi, H., Jankowska, A. M., Abu Kar, S., Jerez, A. et al. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood 119, 3203–3210 (2012).
Wu, S. J., Kuo, Y. Y., Hou, H. A., Li, L. Y., Tseng, M. H., Huang, C. F. et al. The clinical implication of SRSF2 mutation in patients with myelodysplastic syndrome and its stability during disease evolution. Blood 120, 3106–3111 (2012).
Federmann, B., Abele, M., Rosero Cuesta, D. S., Vogel, W., Boiocchi, L., Kanz, L. et al. The detection of SRSF2 mutations in routinely processed bone marrow biopsies is useful in the diagnosis of chronic myelomonocytic leukemia. Hum Pathol 45, 2471–2479 (2014).
Vannucchi, A. M., Lasho, T. L., Guglielmelli, P., Biamonte, F., Pardanani, A., Pereira, A. et al. Mutations and prognosis in primary myelofibrosis. Leukemia 27, 1861–1869 (2013).
Zhang, S. J., Rampal, R., Manshouri, T., Patel, J., Mensah, N., Kayserian, A. et al. Genetic analysis of patients with leukemic transformation of myeloproliferative neoplasms shows recurrent SRSF2 mutations that are associated with adverse outcome. Blood 119, 4480–4485 (2012).
Chapman, J., Geyer, J. T., Khanlari, M., Moul, A., Casas, C., Connor, S. T. et al. Myeloid neoplasms with features intermediate between primary myelofibrosis and chronic myelomonocytic leukemia. Mod Pathol 31, 429–441 (2018).
Gur, H. D., Loghavi, S., Garcia-Manero, G., Routbort, M., Kanagal-Shamanna, R., Quesada, A. et al. Chronic Myelomonocytic Leukemia With Fibrosis Is a Distinct Disease Subset With Myeloproliferative Features and Frequent JAK2 p.V617F Mutations. Am J Surg Pathol 42, 799–806 (2018).
Hu, Z., Ramos, C. E. B., Medeiros, L. J., Zhao, C., Yin, C. C., Li, S. et al. Utility of JAK2 V617F allelic burden in distinguishing chronic myelomonocytic Leukemia from Primary myelofibrosis with monocytosis. Hum Pathol 85, 290–298 (2019).
WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. (IARC Lyon, 2017).
Della Porta, M. G., Travaglino, E., Boveri, E., Ponzoni, M., Malcovati, L., Papaemmanuil, E. et al. Minimal morphological criteria for defining bone marrow dysplasia: a basis for clinical implementation of WHO classification of myelodysplastic syndromes. Leukemia 29, 66–75 (2015).
Thiele, J., Kvasnicka, H. M., Facchetti, F., Franco, V., van Der Walt, J. & Orazi, A. European consensus on grading bone marrow fibrosis and assessment of cellularity. Haematologica 90, 1128–1132 (2005).
Khoury, J. D., Sen, F., Abruzzo, L. V., Hayes, K., Glassman, A. & Medeiros, L. J. Cytogenetic findings in blastoid mantle cell lymphoma. Hum Pathol 34, 1022–1029 (2003).
McGowan-Jordan J, S. A., Schmid M. ISCN 2016: An International System for Human Cytogenomic Nomenclature (2016). (Basel: S. Karger Publishing, 2016).
Ok, C. Y., Loghavi, S., Sui, D., Wei, P., Kanagal-Shamanna, R., Yin, C. C. et al. Persistent IDH1/2 mutations in remission can predict relapse in patients with acute myeloid leukemia. Haematologica 104, 305–311 (2019).
Lee, S. C., North, K., Kim, E., Jang, E., Obeng, E., Lu, S. X. et al. Synthetic Lethal and Convergent Biological Effects of Cancer-Associated Spliceosomal Gene Mutations. Cancer Cell 34, 225-241 e228 (2018).
Taylor, J., Mi, X., North, K. D., Binder, M., Penson, A., Lasho, T. L. et al. Single-cell genomics reveals the genetic and molecular bases for escape from mutational epistasis in myeloid neoplasms. Blood https://doi.org/10.1182/blood.2020006868 (2020).
Author information
Authors and Affiliations
Contributions
M.T. and S.L. designed the study, reviewed the pathology, collected and analyzed data. J.D.K., S.A.W., S.H., P.L. C.B.R., L.J.M. collected pathology data, M.J.R., R.L. K.P.P. C.Y.O., R.K.S. collected molecular data, SHE assisted in manuscript preparation, N.P., P.B. and S.V. manages patients and collected clinical data. All authors were involved in manuscript preparation and approved the final draft.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
This study was approved by the Institutional Review Board (IRB) at MD Anderson Cancer Center (MDACC) and conducted in accord with the Declaration of Helsinki. Consent is not applicable for this retrospective study.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Tashakori, M., Khoury, J.D., Routbort, M.J. et al. Clinicopathologic spectrum of myeloid neoplasms with concurrent myeloproliferative neoplasm driver mutations and SRSF2 mutations. Mod Pathol 35, 1677–1683 (2022). https://doi.org/10.1038/s41379-022-01118-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-022-01118-3
