
Abstract
Gastric mixed adenoneuroendocrine carcinoma (MANEC) is a clinically aggressive subtype of mixed neuroendocrine–nonneuroendocrine neoplasm (MiNEN) with unclear clonal origin. In this study, we analyzed high-resolution copy number (CN) profiling data using the OncoScan CNV Assay in the neuroendocrine carcinoma (NEC) and adenocarcinoma components of eight MANECs. Some common CNVs, including the gain of CCNE1 (19q12) and the loss of FAT1 (4q35.2), were frequently detected in both components; these CNVs were verified by FISH, qPCR and immunohistochemistry staining assays in samples with sufficient material. The identification of common CNVs in both components supports the likelihood of single clonal origin of morphologically heterogeneous tumor cells and suggests several novel genetic events potentially involved in the development of gastric MANEC. We also detected and validated some CNVs and alterations specific for the NEC component, such as MAPK1 loss and MAPK signaling pathway alterations, which could contribute to the neuroendocrine differentiation of gastric MANEC. In addition, we found that the NEC component presented more CNVs and greater CN loss than the adenocarcinoma component (P = 0.007 and P = 0.004, respectively); the NEC components from different cases were not clustered in the hierarchical clustering analysis, indicating the marked genetic heterogenicity of the NEC component in gastric MANEC. In summary, this study describes the cytogenetic characteristics of each component of gastric MANEC, providing some clues for further studies on the development and progression of gastric MANEC as well as providing some potential therapeutic targets.
Similar content being viewed by others

Introduction
Mixed neuroendocrine–nonneuroendocrine neoplasms (MiNENs) were described in the fifth edition of the WHO classification of digestive system tumors in 2019; MiNEN occurs when both components (neuroendocrine and nonneuroendocrine) are substantial (accounting for ≥30% of each neoplasm) [1]. In the stomach, mixed adenoneuroendocrine carcinoma (MANEC) is the most frequent subtype of MiNEN, in which the nonneuroendocrine component is adenocarcinoma. Gastric MANECs, accounting for ~20% of all digestive MANECs [2], are morphologically heterogeneous and clinically aggressive. Gastric MANECs present a worse prognosis than gastric adenocarcinoma; however, it remains unclear whether the prognosis of gastric MANECs is better or worse than that of gastric neuroendocrine carcinoma (NEC) [2,3,4,5]. In the clinic, when feasible, surgery is performed on patients with gastric MANEC. Limited adjuvant chemotherapy and target therapy regimens have been used [6,7,8], but for the most part, there have not been effective uniform treatments for gastric MANECs.
The mechanism underlying the tumorigenesis and differentiation of gastric MiNENs is also controversial [4,5,6, 9,10,11,12,13,14]. There were two main hypotheses regarding the origin of MANEC [6]: some researchers have proposed that adenocarcinoma cells originate from multipotent stem cells, whereas NEC cells originate from embryonic neural cells; the second view is that both adenocarcinoma and NEC cells originate from a single endoderm multipotent stem cell. Recently, the same loci of loss of heterozygosity (LOH) were detected in both tumor components of gastric MANECs via microallelotyping analyses; these loci included the TP53 locus and loci on chromosomes 3p, 11q17q, and 18q [14, 15]. The next-generation sequencing and expression data obtained from two gastric MANECs also detected the same TP53 mutation and level of p53 protein expression in both components [10], supporting a monoclonal origin of gastric MANECs. On the other hand, LOHs and mutations of some cancer-related genes were only restricted in the neuroendocrine component [10, 14], suggesting a multistep progression model of MANEC from initial homogeneous adenocarcinoma to heterogeneous adenocarcinoma and NEC and proposing a model of dual differentiation occurring in parallel after the initial mitosis of a precursor cell based on the results of widespread reciprocal LOHs from one gastric MANEC case [14].
To gain a better understanding of gastric MANECs, we pathologically verified the gastric MANECs in our center and analyzed their clinicopathological features. We extracted DNA from each tumor component and normal tissue of gastric MANECs and performed genome-wide copy number variation (CNV) analysis for each component. The common CNVs of both components and special CNVs of the NEC component were identified and validated by FISH and qPCR, followed by immunohistochemistry staining of related proteins. Based on the CNV status and protein expression in this study, we found some novel candidate genes involved in both tumor components, which supports the probable single clonal origin of morphologically heterogeneous tumor cells and supplies some possible driver genes in tumor development and implies some potential therapeutic targets for gastric MANEC. Our study also revealed a high level of heterogeneity in NEC components and provided insight into some potential genes and signaling pathways specific for NEC components, revealing a mechanism of neuroendocrine differentiation in gastric MANEC.
Materials and methods
Patients selection
All of the gastric neuroendocrine neoplasm cases in this study were patients treated at the Tianjin Medical University Cancer Institute and Hospital from March 2011 to December 2017. The diagnosis of gastric MANEC was agreed upon by two pathologists according to the WHO classification criteria [1] after they reviewed all of the hematoxylin and eosin (H&E)-stained sections and immunohistochemistry-stained sections for Syn (DAK-SYNAP, Dako, Denmark), CgA (DAK-A3, Dako, Denmark), CD56 (123C3, Dako, Denmark), and Ki-67 (MIB-1, Dako, Denmark). The clinicopathological data and survival information were collected for all cases of gastric MANEC. This study was approved by the Institutional Review Board of Tianjin Medical University Cancer Institute and Hospital in compliance with the Helsinki Declaration.
Copy number variation analysis
CNV analysis was performed in eight gastric MANECs. On the basis of the H&E and immunohistochemistry-stained slides, we labeled the NEC and adenocarcinoma components on formalin-fixed and paraffin-embedded (FFPE) tissue. When we scraped each tumor component from the FFPE sections for DNA extraction, we remade an H&E slide to ensure that the percentage of neoplastic cells was ≥85% for each component. DNA was then extracted with the QIAamp DNA FFPE Tissue Kit according to the manufacturer’s protocol (Qiagen, CA, USA) and quantified using a NanoDrop2000 (Thermo Scientific, MA, USA). High-resolution copy number (CN) analysis was processed with OncoScan CNV Assay (Affymetrix, CA, USA) according to the manufacturer’s protocol. The data were analyzed with Nexus Express Software (BioDiscovery, CA, USA). All of the altered chromosomal regions and the genes located at these regions were selected. To define the significant chromosome regions and candidate genes, we selected 0.25 and −0.25 as thresholds for the CN gain and loss, respectively.
Hierarchical clustering was performed, and JAVA TreeView was used to draw heat maps to visualize the clustering results. By matching the two components of single cases, common focal genes were collected. The genes that only showed alterations in the NEC component but not in the adenocarcinoma component were also collected. Gene Ontology enrichment analysis and functional annotation of the gene gain or loss were performed using the Search Tool for the Retrieval of Interacting Genes (STRING) database (https://string-db.org/). Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis was performed, and the results were visualized with the ClueGo plug-in of Cytoscape software (version 3.4.0). A P value < 0.05 was set as the cutoff criterion. The protein–protein interaction (PPI) networks of genes with CNV were identified using the STRING database.
Fluorescence in situ hybridization
To confirm the CNV results, interphase fluorescence in situ hybridization (FISH) was performed on 4-μm sections from FFPE tissue using commercial dual-color probes made by Shaoxing Jinlu Biotechnology (Shaoxing City, Zhejiang Province, China). The candidate genes (CCNE1, FAT1, and MAPK1) are labeled orange, and their corresponding centromeres are labeled green. The information of these probes is summarized in Supplementary Table S1. Hybridization was performed in an automated hybridizer overnight according to the standard protocol. Fluorescence signals were captured by a Zeiss Axio Imager 2 Research Microscope with an Axiocam 506 color camera and processed by ZEN Imaging Software (ZEISS, Germany). The FISH images were evaluated simultaneously by two professionals. For each tumor component, the numbers of orange and green signals per nucleus were counted in 50 nonoverlapping tumor cells, and the ratio of the orange signals to green signals was calculated. We used thresholds of ≥1.3 for gain and ≤0.75 for the loss of DNA as previously described [16].
Quantitative real-time PCR
Quantitative real-time PCR (qPCR) for CCNE1, FAT1, and MAPK1 was separately performed for NEC and adenocarcinoma components as well as normal tissue using a StepOnePlus Real-Time PCR System (Applied Biosystems, CA, USA). The primer information is listed in Supplementary Table S2. The 20 μL reaction mixture included 10 μL of 2 × Fast SYBR Green Master Mix from Applied Biosystems, 400 nM of each primer, and 20 ng of genomic DNA. Each sample was analyzed in triplicate with β-actin as the internal control. The results were analyzed using StepOne Software v2.2 (Applied Biosystems, CA, USA). Further analysis was performed using the ΔΔCt method and then normalized by setting a normal control to a value of 1 [17].
Immunohistochemical analysis
Immunohistochemical (IHC) staining was performed on 4-μm sections from FFPE tumor tissues and normal control tissues with cyclin E (HE12, dilution 1:100, Invitrogen), FAT1 (FAT/FAT1, dilution 1:50, Abcam), and ERK2 (E460, dilution 1:100, Abcam) antibodies according to the protocol. A positive control was used based on the manufacturer’s recommendations, and a negative control was included with the omission of the primary antibody step. Brown staining in the nucleus was defined as positive expression.
Statistical analysis
Statistical analysis was performed with SPSS 22.0 (IBM SPSS, IL, USA). The analysis for progression-free survival (PFS) and overall survival (OS) was performed using the Kaplan–Meier method with the log-rank test. Multivariate analyses based on the stepwise Cox proportional hazards model were used to identify the most significant factors. The CN of each component and normal tissue were compared with paired t-tests. A significant difference was considered when the P value < 0.05.
Results
Clinicopathological characteristics and survival analysis
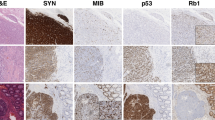
A total of 35 gastric MANECs were identified with typical morphology and IHC phenotype (Fig. 1a). These cases showed heterogeneous morphology. The NEC components presented the pattern of large cell NEC in 33 cases and small cell NEC in only 2 cases, while the adenocarcinoma components showed a wide spectrum of histology, including tubular, papillary, mucinous adenocarcinoma, or signet ring cell carcinoma. The ratio of the two components varied in different cases, from 3:7 to 7:3. The median Ki-67 index was 60% (30–90%) in the NEC component and 60% (25–90%) in the adenocarcinoma component. This cohort of patients had a male-to-female ratio of 29:6 and a median age of 60 years. The tumors were most frequently found in the cardia–fundus (40%), followed by the gastric body (28.6%). All patients received surgery. The median tumor size was 5 cm (0.8–10 cm). Intravascular carcinoma thrombus and nerve invasion were detected in 12 (34.3%) and 13 (37.1%) gastric MANECs, respectively. Lymphatic metastasis was found at diagnosis in 23 (65.7%) patients, and the metastatic cells were adenocarcinoma cells in 7 cases, neuroendocrine cells in 12 cases, and both tumor components in 4 cases. Five patients presented with distant metastasis at diagnosis, with sites including the liver (two cases), transverse colon (one case), lung (one case), and kidney (one case). The liver metastatic tumors were resected synchronously, and only the NEC component was detected in both metastatic tumors (Supplementary Fig. S1). According to the 8th version, American Joint Committee on Cancer staging system, 7 (20.0%), 5 (14.3%), 18 (51.4%), and 5 (14.3%) patients were classified as stage I, II, III, and IV, respectively. Twenty-five patients, including 6 stage I/II and 19 stage III/IV patients, received adjuvant chemotherapy (etoposide + cisplatin, EP) after surgery, of which patients received combined with dendritic cell-cytokine-induced killer cell immunotherapy and two patients received combined with local radiotherapy. Information on the tumors and patients is listed in Table 1.

a H&E staining (100×) showed the separated NEC (arrow) and adenocarcinoma (cross-shape) components. Immunohistochemistry staining (100×) showed positive Syn expression only in the NEC component and positive CK8/18 expression in both the NEC and adenocarcinoma components. Ki-67 indicated the cell proliferation activity of both tumor components. b Univariate survival analyses of PFS and OS in 34 cases of gastric MANEC. MANEC mixed adenoneuroendocrine carcinoma, H&E hematoxylin and eosin, NEC neuroendocrine carcinoma, PFS progression-free survival, OS overall survival, LN lymph node.
The follow-up time for 34 patients (1 missing from the database) ranged from 1 to 89 months. During the follow-up period, 19 patients (55.9%) presented with recurrence and/or metastasis to the liver, retroperitoneum, bone, lung, or brain. The median PFS time was 13.5 months. Univariate analysis showed that the PFS was significantly shorter in patients with lymph node metastasis (P = 0.039), distant metastasis (P = 0.025), and advanced stage (P = 0.048) (Fig. 1b). The patients with Ki-67 index of NEC component ≥45 and ≥50% tended to have a poorer PFS than those with NEC components <45% (P = 0.060) and <50% (P = 0.079); however, the differences were not significant (Supplementary Table S3). The patients’ age, sex, tumor location, tumor size, intravascular carcinoma thrombus, nerve invasion, or postoperative adjuvant therapy were not significant prognostic factors for PFS (Table 1). Cox regression analysis indicated that lymph node metastasis (P = 0.015, HR = 0.221, 95% CI: 0.065–0.749) and distant metastasis (P = 0.006, HR = 0.192, 95% CI: 0.059–0.621) were independent poor prognostic factors for PFS in gastric MANECs. At the end of follow-up, 14 patients had (41.2%) died. The 1-year and 3-year survival rates were 88.1% and 56.7%, respectively. The univariate analyses for the Ki-67 index of the NEC component showed that the OS curves were significantly different when the cutoff value was 35% (P = 0.048), 40% (P = 0.048), 45% (P = 0.025), or 50% (P = 0.018) (Supplementary Table S3, Fig. 1b). In addition, lymph node metastasis at diagnosis (P = 0.012), distant metastasis (P = 0.014), advanced stage (P = 0.007), and recurrence and/or metastasis (P < 0.001) during the follow-up period were significantly associated with poor OS (Fig. 1b). OS was not significantly correlated with other clinicopathological parameters (Table 1). Cox regression analysis showed that recurrence and/or metastasis (P = 0.057, HR = 0.013, 95% CI: 0.000–1.140) was an independent poor prognostic factor for OS in gastric MANECs.
Copy number variation (CNV) analysis
CNV analysis was performed on both the adenocarcinoma and NEC components in the eight gastric MANECs using the OncoScan CNV Assay. The chromosome segments of CNV identified in NEC and adenocarcinoma components are shown in Fig. 2. As shown, the merged CN pattern of the eight patients showed arm-level and focal CNVs across all chromosomes. Taking 0.25 or −0.25 as the threshold values of significant gain or loss, all chromosome segments and genes identified on these segments were collected. Gains of chromosome regions 1p36.22, 1q21.1-q21.3, 1q44, 5p15.33-p12, 6q23.3, 8q21.11-q23.3, 11q13.4, 14q32.33, 18q12.1-q12.2, 19q12, 19q13.32-q13.41, 20p12.1-p11.21, 20p13-p12.3, 20p11.21-q13.33 and losses of chromosome regions 1p36.33, 2q33.2, 4q12-q13.1, 4q28.1-q28.3, 4q32.2-35.2, 5q12.1, 5q31.3, 5q35.1-q35.3, 7q36.3, 8p23.3, 10p14-p13, 10p12.1-p11.1, 11p15.5, 15q11.1-q13.3, 15q14-q15.1, 15q21.3-q26.3, 16q12.1-q22.1, 16q23.1, 17p13.3-p11.2, 19q13.2,22q11.1-q11.23, 22q12.1-q12.2 were frequently identified in the NEC components (in at least three cases). Gains of chromosome regions 19q12, 20q11.21-q13.12, and 20q13.2 and losses of chromosome regions 4q35.1-q35.2, 9p13.2-p13.3, and 22q11.23 were frequently identified in adenocarcinoma components (in at least three cases).

The chromosomes (1–22, X and Y) are shown in turn. For each chromosome, the CNVs of eight cases are shown on the right (from left to right: case 1 to case 8 in turn). Blue bars indicate copy number gains, and red bars indicate copy number losses. Copy number gains (a) and losses (b) in the NEC component. Copy number gains (c) and losses (d) in the ADC component.
Identification and validation of the common genes for both tumor components
First, we selected the genes that were embodied in the CNV simultaneously in the matching NEC and adenocarcinoma components. The average number of these common gene gains was much greater than that of common gene losses (727.5 vs 209.5, P = 0.038). CN gain of CCNE1 (located at 19q12) identified in four cases and loss of FAT1 (located at 4q35.2) identified in three cases were the most frequent alterations in both components. The incidence rates of CCNE1 gain and FAT1 loss in our gastric MANEC cases (50% and 37.5%, respectively) were higher than those in TCGA stomach adenocarcinoma (11.2% and 3.5%, respectively) and TCGA PanCancer Atlas Studies (ranging from 0.4–19.15% and 0.29–15.38%, respectively) (http://www.cbioportal.org/).
We next performed interphase FISH analysis to confirm the CN status of CCNE1 or FAT1 on the eight gastric MANECs using custom-designed dual-color fluorescent probes. The gain of CCNE1 was detected in both components in the four cases with CN gain in CNV analysis, and in the other four cases, CCNE1 showed a normal copy in accordance with the CNV results. Similarly, the loss of FAT1 was detected in both components in the three cases with CN loss in CN analysis, while in the other cases/components, FAT1 showed a normal copy. Representative images of CNV and FISH for CCNE1 and FAT1 in each component of the same cases are shown in Fig. 3a, b, respectively.

a The gain of 19q12 (CCNE1, down arrow) and loss of 4q35.2 (FAT1, up arrow) were observed in both components in the whole genome view. b FISH analysis detected multiple copies of CCNE1 (orange) in both components (top panels) and the loss of the FAT1 gene (orange) in both components (lower panels). c qPCR validation of CCNE1 and FAT1 in 18 gastric MANECs. The CN of CCNE1 was significantly higher in NEC and ADC components than in normal tissue, and the CN of FAT1 was significantly lower in NEC and ADC components than in normal tissue. There was no difference in the CN of CCNE1 and FAT1 between NEC and ADC components. d Immunohistochemistry staining (200×) showed diffusely positive expression of cyclin E (the protein product of CCNE1) in both components but negative expression in normal mucosa. FAT1 was negative in both tumor components but positive in normal mucosa.
We further validated the CN of CCNE1 and FAT1 using qPCR in 18 gastric MANECs (including the above 8 cases with OncoScan and FISH results), which contained enough good quality DNA from separate NEC and adenocarcinoma components for the analysis. In the eight cases with CNV results (Supplementary Fig. S2), the CN of CCNE1 was significantly higher in the NEC component than in normal tissue (P = 0.039) and tended to be higher in the adenocarcinoma component than in normal tissue (P = 0.131). Similarly, the CN of FAT1 was significantly lower in the NEC component than in normal tissue (P = 0.028) and tended to be lower in the adenocarcinoma component than in normal tissue (P = 0.089). Among all 18 gastric MANECs, the CN of CCNE1 in NEC and adenocarcinoma components was higher than that in normal tissue (P = 0.011 and P = 0.018, respectively); FAT1 showed a significantly lower CN in NEC and adenocarcinoma components than in normal tissue (P < 0.001 and P = 0.009, respectively); and there was no difference in CCNE1 and FAT1 between NEC and adenocarcinoma components (Fig. 3c).
To further investigate the association between CN and protein expression, we analyzed the expression of cyclin E (the protein product of CCNE1) and FAT1 in 30 gastric MANECs in which both components were available for IHC analysis. Coinciding with the CNV and FISH results, diffusely strong positive staining of cyclin E was detected in both tumor components of the four cases with gain of CCNE1 (Fig. 3d), while in the other four cases, cyclin E was negative in both tumor components. FAT1 was negative in both tumor components in the three cases with loss of FAT1 (Fig. 3d) but positive in the other cases/components. Among the 30 gastric MANECs, diffusely strong positive staining of cyclin E in both NEC and adenocarcinoma components was detected in 60.0% (18/30) of cases, and FAT1 was negative in both NEC and adenocarcinoma components in 33.3% (10/30) of cases.
The comparison of CNVs between NEC and adenocarcinoma components
Overall, the average CNV of chromosome regions in the NEC components was much greater than that in the adenocarcinoma components (67.5 vs 26.6, P = 0.007). The CN losses in the NEC components were significantly greater than those in the adenocarcinoma components (28.3 vs 8.5, P = 0.004), while the difference in CN gain was not significant (39.3 for NEC vs 18.9 for adenocarcinoma, P = 0.07). Hierarchical clustering analysis, based on all genes related to the significant gain or loss regions, showed that adenocarcinoma components from different cases were clustered together, while the two components from each case, except for case 1 and case 3, did not cluster together; NEC components from different cases were not clustered (Fig. 4a). We carefully reviewed the H&E staining and immunohistochemically stained slides for all cases to explain the results in cases 1 and 3. We found that although NEC cells and adenocarcinoma cells separated in most areas of the tumor, a few NEC cells were scattered in the adenocarcinoma area in cases 1 and 3 (Fig. 4b). On the other hand, the NEC component and adenocarcinoma component were separated in almost all areas in other cases (Fig. 4c).

a Clustering analysis showed that adenocarcinoma components in different cases tended to be grouped, except in cases 1 and 3, while the NEC components in different cases were not clustered. b Histological images of case 3 with the two tumor components clustered. Although H&E staining (100×) showed the separated NEC (arrow) and adenocarcinoma (cross-shape) components, the immunohistochemistry staining of CgA (200×) demonstrated that neuroendocrine carcinoma cells (triangle) were scattered in the adenocarcinoma area. c Histological images of case 4 with the two tumor components not clustered. H&E staining (100×) showed the separated NEC (arrow) and adenocarcinoma (cross-shape) components, and immunohistochemistry staining of CD56 (100×) indicated that CD56-positive cells were only located in the NEC area.
Identification and validation of the specific genes only changed in the NEC component
To identify the candidate genes that might be related to NEC differentiation, we collected the genes located at CNV segments only detected in the NEC component. The enriched signaling pathways were detected by KEGG pathway analysis (Supplementary Fig. S3a), and the PPI network of the top 100 genes was constructed in Supplementary Fig. S3b. The MAPK signaling pathway and the loss of MAPK1 were highly ranked. For the highly reproducible genes, we performed an integrated analysis based on the P value of the KEGG pathway and combined it with the score of the PPI network. MAPK signaling, cell cycle, oxytocin signaling, Rap1 signaling, extracellular matrix-receptor interaction, tyrosine metabolism, and p53 signaling were highly ranked pathways (Fig. 5). Next, from the eight cases, we collected all the genes with CNV that are involved in the MAPK signaling pathway and performed an integrative analysis (Supplementary Fig. S4). Both the upstream factors of MAPK1 (MAP3Ks and MAP2Ks) and their downstream transcription factors (MEF2A, MEF2C, FoxO3B, ATF4, etc.) showed CN loss in the NEC component.

The proteins related to the genes located in the CNV regions specific for the NEC component were enriched and were present in the top 100 results of the protein–protein interaction analysis. Pathways including MAPK signaling, cell cycle, oxytocin signaling, Rap1 signaling, extracellular matrix-receptor interaction, tyrosine metabolism, and p53 signaling are highlighted. Octagonal nodes represent the pathway, and circular nodes represent the genes. The magnitude of the octagonal nodes represents the extent to which the pathways are enriched. The circular nodes with black borders represent copy number loss, and those without borders represent copy number gain.
We next performed interphase FISH analysis to confirm the CN status of MAPK1 on the eight gastric MANECs. One copy deletion of MAPK1 was detected by FISH in the NEC component of the four cases with CN loss in the CNV analysis, while MAPK1 showed a normal CN in adenocarcinoma components of the same four cases. In the other four cases with normal CN status of MAPK1, FISH also detected a normal copy in both components. Representative images of CNV and FISH for MAPK1 in each component of the same cases are shown in Fig. 6a, b, respectively. The qPCR results also confirmed significantly lower CN of MAPK1 in the NEC component than in the adenocarcinoma component (P = 0.043) and normal tissue (P = 0.006) in the eight cases with CNV results (Supplementary Fig. S2). Among the 18 gastric MANECs, the CN of MAPK1 in the NEC component was significantly lower than that in adenocarcinoma components and normal tissue (P = 0.004 and P = 0.026, respectively), and there was no significant difference between the adenocarcinoma component and normal tissue (Fig. 6c). The immunohistochemistry staining showed negative ERK2 (the protein product of MAPK1) expression in NEC components but positive expression in adenocarcinoma components in the four cases with loss of MAPK1 only in NEC (Fig. 6d). ERK2 was positive in both components of the other four cases. Among the 30 gastric MANECs with immunohistochemistry results, 43.3% (13/30) of cases showed negative ERK2 expression in NEC components, while weakly or strongly positive ERK2 expression was observed in adenocarcinoma components and normal tissues of the same cases.

a The loss of 22q11.21 (including MAPK1, black arrow) was much more significant in the NEC component than in the ADC component in the whole genome view. b FISH analysis showed loss of the MAPK1 gene (one orange signal) in the NEC component and diploid signals (pairs of orange signals) in the ADC component. c qPCR validation of MAPK1 in 18 gastric MANECs. The CN of MAPK1 was significantly lower in the NEC component than in the ADC component and normal tissue. d Immunohistochemistry staining (200×) showed the negative expression of ERK2 (the protein product of MAPK1) in the NEC component and the positive expression of ERK2 in the ADC component and normal mucosa.
Discussion
Gastric MANEC, a subtype of MiNEN, is a morphologically heterogeneous and clinically aggressive stomach tumor subtype. In accordance with our study, both Chen [11] and Nie [13] reported that the adenocarcinoma component of gastric MANEC was characterized by a wide spectrum of histology, while the NEC component was mostly large cell NEC. In another study, however, the NEC component was mainly small cell NEC in gastric MANEC [12]. The NEC component was reported to decide the clinical behavior of MANEC in a great part [2, 11, 13]. In our cohort, only the NEC component was detected in the liver metastatic tumors of patients with gastric MANEC. In addition, a higher Ki-67 index of the NEC component was associated with shorter OS in our univariate survival analysis, which is consistent with a study on gastroenteropancreatic MANEC [2]. In other studies, the increasing proportion of NEC components and metastatic NEC components in lymph nodes were related to unfavorable prognosis [11, 13]. As gastric MANEC is rare, more multiple-center studies with large patient cohorts are needed to clarify its clinicopathological features and biological behaviors.
In addition to the clinicopathological analyses, we performed a genome-wide CNV analysis for each component of gastric MANECs and identified novel common molecular alterations in both components and specific alterations in the NEC component in this study. By comparing the NEC and adenocarcinoma components from the same cases, we identified some frequent CNVs that exist in both components, supporting the probable single clonal origin of morphologically heterogeneous tumor cells in gastric MANECs. Among these common CNVs, CCNE1 gain (located at 19q12) and FAT1 loss (located at 4q35.2) were most frequent, which were further demonstrated by FISH and qPCR. Cyclin E, the protein product of CCNE1, is a nuclear protein critically involved in the regulation of the G1/S cell cycle transition [18]. CCNE1 gain and cyclin E overexpression have been observed in many malignancies, resulting in cellular proliferation, centrosome hypergains, or chromosome instability and tumorigenesis accordingly [19]. Cyclin E overexpression was detected in 35.3% of gastroenteropancreatic neuroendocrine tumors, and high expression of cyclin E indicated a worse prognosis [20]. Our study revealed for the first time that CCNE1 gain is a key CNV in gastric MANEC. Gain of CCNE1 may contribute to the tumorigenesis of gastric MANEC by regulating cell cycle G1/S checkpoint signaling pathways. FAT1, a member of the cadherin superfamily, was first identified as a tumor suppressor that was essential for controlling cell proliferation during drosophila development [21]. The functions of FAT1 in human cancer development and progression appear to be dependent on the tumor context. Knockdown of FAT1 expression promoted tumor growth and epithelial–mesenchymal transition in esophageal squamous cell carcinoma [22]. FAT1 was also detected as homozygous deletion, LOH, epigenetic silencing, or downregulated in several human tumors [23,24,25]. On the other hand, suppression of FAT1 inhibited the cell proliferative, migratory, and antiapoptotic potential of hepatocellular carcinoma cells in vitro and led to a delayed onset and increased apoptosis in the animal model [26]. FAT1 was also detected to be upregulated in leukemia and cholangiocarcinoma [27, 28]. The loss of FAT1 in our study suggested a tumor suppression function of FAT1 in MANECs, but this will need to be tested in future laboratory investigations. The gain of 5p15.33-p12, containing PTGER4, which was the most frequent chromosome alteration in colonic MANEC [29], was also found in both components of two cases in this study. Other common CNVs, such as the gain of 19q12, 20q11.21-q13.12, and 20q13.2 and the loss of 4q35.1-q35.2, were also identified in a pan-cancer study [30, 31]. Further studies on the common CNVs in both components will shed light on the development of gastric MANEC.
Our analysis also highlighted CNVs and signaling pathways specific for the NEC component of gastric MANEC, such as MAPK1 loss and marked alterations in the MAPK signaling pathway. MAPK1 belongs to the mitogen-activated protein kinase family, which participates in regulating many pathophysiological processes, including cell proliferation, differentiation, apoptosis, metabolism, invasion, inflammation, and angiogenesis though the MAPK signaling pathway [32]. The heterodimer abnormal activation of MAPK1 and MAPK3 (also called extracellular regulated protein kinase 2 and 1) has been reported in a variety of malignancies [33]. MAPK1/3 activity was reported to promote neuroendocrine differentiation in the human prostate cancer cell line LNCaP [34]. The loss of MAPK1 has been identified in some neoplastic and nonneoplastic lesions, leading to developmental disorders, developmental dysplasia, and differentiation abnormalities [35,36,37,38]. Furthermore, MAPK family members have been reported to be major factors in deciding the cell fate in response to cisplatin depending on the types of activated MAPK [39]. In the cisplatin-induced human cervical carcinoma HeLa cell line, the level of MAPK1 was decreased, which led to a lower level of apoptosis and accordingly resistance to cisplatin [40]. MEK inhibitor attenuated the activity of MAPK1/3 and enhanced the sensitivity to cisplatin in the human ovarian carcinoma SKOV3 cell line. Considering the remarkable difference in the CNV frequency between NEC and adenocarcinoma components, we speculate that MAPK1 loss and abnormalities in the MAPK signaling pathway might play an important role in the neuroendocrine differentiation of gastric MANEC and probably influence cisplatin sensitivity, which is worth studying in the future. Furthermore, MYC amplification and RB1 deletion, which were reported in a single case of colorectal MiNEN [41], were also identified in the NEC component in our cohort, albeit not presenting with high reproducibility. In addition, the NEC component presented more CNVs and CN losses than the adenocarcinoma component, and the NEC components from different cases were not clustered in this study, which suggests that the NEC component of gastric MANEC probably has more genetic instability. Exploring the possible mechanism underlying the increased CNV in the NEC component and the key molecular alterations in neuroendocrine differentiation will be helpful to improve treatments for gastric MANEC.
In summary, our clinicopathological and genetic analyses revealed the clinicopathological features and CN characteristics of each tumor component in gastric MANECs. Analysis of the common CNVs and related molecular alterations frequently present in both components demonstrated that the morphologically heterogeneous tumor cells of gastric MANECs could originate from a single multipotent stem cell. Our study suggested that CCNE1 gain and FAT1 loss might promote the tumorigenesis of gastric MANEC partially through regulating cell cycle G1/S checkpoint signaling. Moreover, CNVs and molecular alterations, such as the loss of MAPK1 and alterations in the MAPK signaling pathway, specifically occurred in the NEC component, which might contribute to the neuroendocrine differentiation of gastric MANEC. However, the current results were based on a small number of samples and need to be validated in multiple-center studies with large patient cohorts. Furthermore, the precise function and mechanism of the genes that were suggested to be related to the tumorigenesis and neuroendocrine differentiation of gastric MANEC in this study deserve further investigation.
References
Klimstra DS, Klöppel G, La Rosa S, Rindi G. Classification of neuroendocrine neoplasms of the digestive system. In: WHO Classification of Tumors Editorial Board. Digestive System Tumors, WHO Classification of Tumors. 5th ed. Lyon: International Agency for Research on Cancer Press; 2019. p. 16–9.
Milione M, Maisonneuve P, Pellegrinelli A, Grillo F, Albarello L, Spaggiari P, et al. Ki67 proliferative index of the neuroendocrine component drives MANEC prognosis. Endocr Relat Cancer. 2018;25:583–93.
Levi Sandri GB, Carboni F, Valle M, Visca P, Garofalo A. Mixed adenoneuroendocrine gastric carcinoma: a case report and review of the literature. J Gastric Cancer. 2014;14:63–6.
Shen C, Chen H, Chen H, Yin Y, Han L, Chen J, et al. Surgical treatment and prognosis of gastric neuroendocrine neoplasms: a single-center experience. BMC Gastroenterol. 2016;16:111.
La Rosa S, Inzani F, Vanoli A, Klersy C, Dainese L, Rindi G, et al. Histologic characterization and improved prognostic evaluation of 209 gastric neuroendocrine neoplasms. Hum Pathol. 2011;42:1373–84.
Lin Z, Chen J, Guo Y. Efficacy of XELOX adjuvant chemotherapy for gastric mixed adenoneuroendocrine carcinoma: a case report. Medicine. 2019;98:e16000.
Strosberg JR, Coppola D, Klimstra DS, Phan AT, Kulke MH, Wiseman GA, et al. The NANETS consensus guidelines for the diagnosis and management of poorly differentiated (high-grade) extrapulmonary neuroendocrine carcinomas. Pancreas. 2010;39:799–800.
Kwok CM. Mixed adenoneuroendocrine carcinoma of the stomach. Case Rep Gastroenterol. 2015;9:241–5.
Makuuchi R, Terashima M, Kusuhara M, Nakajima T, Serizawa M, Hatakeyama K, et al. Comprehensive analysis of gene mutation and expression profiles in neuroendocrine carcinomas of the stomach. Biomed Res. 2017;38:19–27.
Scardoni M, Vittoria E, Volante M, Rusev B, Bersani S, Mafficini A, et al. Mixed adenoneuroendocrine carcinomas of the gastrointestinal tract: targeted next-generation sequencing suggests a monoclonal origin of the two components. Neuroendocrinology. 2014;100:310–6.
Chen H, Shu M, Chen S, Xue L, Lin Y. Clinicopathological features and lymph node metastatic patterns of gastric mixed adenoneuroendocrine carcinoma. Histol Histopathol. 2019;34:373–9.
Qiu W, Wang X, Zhang XH, Zhou XJ. Gastric mixed adenoneuroendocrine carcinoma: clinical and pathological analyses of 22 cases. Zhen Duan Bing Li Xue Za Zhi (Article in Chinese). 2015;22:1–7.
Nie L, Li M, He X, Feng A, Wu H, Fan X. Gastric mixed adenoneuroendocrine carcinoma: correlation of histologic characteristics with prognosis. Ann Diagn Pathol. 2016;25:48–53.
Kim KM, Kim MJ, Cho BK, Choi SW, Rhyu MG. Genetic evidence for the multi-step progression of mixed glandular-neuroendocrine gastric carcinomas. Virchows Arch. 2002;440:85–93.
Furlan D, Cerutti R, Genasetti A, Pelosi G, Uccella S, La Rosa S, et al. Microallelotyping defines the monoclonal or the polyclonal origin of mixed and collision endocrine-exocrine tumors of the gut. Lab Investig. 2003;83:963–71.
Kettunen E, Aavikko M, Nymark P, Ruosaari S, Wikman H, Vanhala E, et al. DNA copy number loss and allelic imbalance at 2p16 in lung cancer associated with asbestos exposure. Br J Cancer. 2009;100:1336–42.
Glessner JT, Bradfield JP, Wang K, Takahashi N, Zhang H, Sleiman PM, et al. A genome-wide study reveals copy number variants exclusive to childhood obesity cases. Am J Hum Genet. 2010;87:661–6.
Kuhn E, Bahadirli-Talbott A, Shih IeM. Frequent CCNE1 amplification in endometrial intraepithelial carcinoma and uterine serous carcinoma. Mod Pathol. 2014;27:1014–9.
Spruck CH, Won KA, Reed SI. Deregulated cyclin E induces chromosome instability. Nature. 1999;401:297–300.
Liu SZ, Zhang F, Chang YX, Ma J, Li X, Li XH, et al. Prognostic impact of cyclin D1, cyclin E and P53 on gastroenteropancreatic neuroendocrine tumors. Asian Pac J Cancer Prev. 2013;14:419–22.
Bennett FC, Harvey KF. Fat cadherin modulates organ size in Drosophila via the Salvador/Warts/Hippo signaling pathway. Curr Biol. 2006;16:2101–10.
Hu X, Zhai Y, Kong P, Cui H, Yan T, Yang J, et al. FAT1 prevents epithelial mesenchymal transition (EMT) via MAPK/ERK signaling pathway in esophageal squamous cell cancer. Cancer Lett. 2017;397:83–93.
Nakaya K, Yamagata HD, Arita N, Nakashiro KI, Nose M, Miki T, et al. Identification of homozygous deletions of tumor suppressor gene FAT in oral cancer using CGH-array. Oncogene. 2007;26:5300–8.
Wang L, Lyu S, Wang S, Shen H, Niu F, Liu X, et al. Loss of FAT1 during the progression from DCIS to IDC and predict poor clinical outcome in breast cancer. Exp Mol Pathol. 2016;100:177–83.
Chosdol K, Misra A, Puri S, Srivastava T, Chattopadhyay P, Sarkar C, et al. Frequent loss of heterozygosity and altered expression of the candidate tumor suppressor gene ‘FAT’ in human astrocytic tumors. BMC Cancer. 2009;9:5.
Valletta D, Czech B, Spruss T, Ikenberg K, Wild P, Hartmann A, et al. Regulation and function of the atypical cadherin FAT1 in hepatocellular carcinoma. Carcinogenesis. 2014;35:1407–15.
de Bock CE, Ardjmand A, Molloy TJ, Bone SM, Johnstone D, Campbell DM, et al. The Fat1 cadherin is overexpressed and an independent prognostic factor for survival in paired diagnosis-relapse samples of precursor B-cell acute lymphoblastic leukemia. Leukemia. 2012;26:918–26.
Settakorn J, Kaewpila N, Burns GF, Leong AS. FAT, E-cadherin, beta catenin, HER 2/neu, Ki67 immuno-expression, and histological grade in intrahepatic cholangiocarcinoma. J Clin Pathol. 2005;58:1249–54.
Sinha N, Gaston D, Manders D, Goudie M, Matsuoka M, Xie T, et al. Characterization of genome-wide copy number aberrations in colonic mixed adenoneuroendocrine carcinoma and neuroendocrine carcinoma reveals recurrent amplification of PTGER4 and MYC genes. Hum Pathol. 2018;73:16–25.
Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, et al. Pan-cancer patterns of somatic copy number alteration. Nat Genet. 2013;45:1134–40.
Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905.
Igea A, Nebreda AR. The stress kinase p38alpha as a target for cancer therapy. Cancer Res. 2015;75:3997–4002.
Tartaglia M, Gelb BD. Disorders of dysregulated signal traffic through the RAS-MAPK pathway: phenotypic spectrum and molecular mechanisms. Ann N Y Acad Sci. 2010;1214:99–121.
Kim J, Jin H, Zhao JC, Yang YA, Li Y, Yang X, et al. FOXA1 inhibits prostate cancer neuroendocrine differentiation. Oncogene. 2017;36:4072–80.
Samuels IS, Karlo JC, Faruzzi AN, Pickering K, Herrup K, Sweatt JD, et al. Deletion of ERK2 mitogen-activated protein kinase identifies its key roles in cortical neurogenesis and cognitive function. J Neurosci. 2008;28:6983–95.
Yao Y, Li W, Wu J, Germann UA, Su MS, Kuida K, et al. Extracellular signal-regulated kinase 2 is necessary for mesoderm differentiation. Proc Natl Acad Sci USA. 2003;100:12759–64.
Imamura O, Satoh Y, Endo S, Takishima K. Analysis of extracellular signal-regulated kinase 2 function in neural stem/progenitor cells via nervous system-specific gene disruption. Stem Cells. 2008;26:3247–56.
Santos SD, Verveer PJ, Bastiaens PI. Growth factor-induced MAPK network topology shapes Erk response determining PC-12 cell fate. Nat Cell Biol. 2007;9:324–30.
Brozovic A, Osmak M. Activation of mitogen-activated protein kinases by cisplatin and their role in cisplatin-resistance. Cancer Lett. 2007;251:1–16.
Basu A, Tu H. Activation of ERK during DNA damage-induced apoptosis involves protein kinase Cdelta. Biochem Biophys Res Commun. 2005;334:1068–73.
Jesinghaus M, Konukiewitz B, Keller G, Kloor M, Steiger K, Reiche M, et al. Colorectal mixed adenoneuroendocrine carcinomas and neuroendocrine carcinomas are genetically closely related to colorectal adenocarcinomas. Mod Pathol. 2017;30:610–9.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81871990 and 81472263 to YS).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Sun, L., Zhang, J., Wang, C. et al. Chromosomal and molecular pathway alterations in the neuroendocrine carcinoma and adenocarcinoma components of gastric mixed neuroendocrine–nonneuroendocrine neoplasm. Mod Pathol 33, 2602–2613 (2020). https://doi.org/10.1038/s41379-020-0579-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-020-0579-z
