
Abstract
Lysinuric protein intolerance (LPI) is caused by dysfunction of the dibasic amino acid membrane transport owing to the functional abnormality of y+L amino acid transporter-1 (y+ LAT-1). LPI is associated with autosomal recessive inheritance and pathological variants in the responsible gene SLC7A7 are also observed. The pathophysiology of this disease had earlier been understood as a transport defect in polarized cells (e.g., intestinal or renal tubular epithelium); however, in recent years, transport defects in non-polarized cells such as lymphocytes and macrophages have also been recognized as important. Although the former can cause death, malnutrition, and urea cycle dysfunction (hyperammonemia), the latter can induce renal, pulmonary, and immune disorders. Furthermore, although therapeutic interventions can prevent hyperammonemic episodes to some extent, progression of pulmonary and renal complications cannot be prevented, thereby influencing prognosis. Such pathological conditions are currently being explored and further investigation would prove beneficial. In this study, we have summarized the basic pathology as revealed in recent years, along with the clinical aspects and genetic features.
Similar content being viewed by others

Outline
Lysinuric protein intolerance (LPI) is caused by dysfunction resulting from functional abnormality of y+L amino acid transporter-1 (y+LAT-1), a transport protein of dibasic amino acids (lysine, arginine, and ornithine). Impaired absorption of these amino acids in the intestinal epithelium and impaired resorption in the kidneys causes disruption in the amino acid balance and a decrease in protein synthesis, as well as various clinical symptoms primarily caused by hyperammonemia and growth disorders. LPI exhibits autosomal recessive inheritance and pathological variants in the responsible gene SLC7A7 are observed [1,2,3,4].
Metabolic pathway
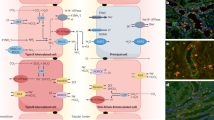
Human y+LAT-1 is a light subunit of heteromeric amino acid transporters. It has a protein structure with 12 transmembrane regions and its molecular weight is ~40 kDa. It induces system y+L activity when co-expressed with a heavy chain of cell surface antigen 4F2 heavy chain (4F2hc, also known as CD98). This transporter exists mainly on the basolateral membrane of polarized cells, such as those of the kidney and small intestine (Fig. 1). And it is responsible for transport of cationic amino acids (CAAs) such as lysine, arginine, and ornithine.

Membranous transport of cationic amino acids(CAA). y+LAT and b0+AT transport both CAA and neutral amino acids. In addition, y+LAT is Na dependent. a CAA transport at an polarized cell. b CAA transport at a non-polarized cell. CAT1/2 (cationic transporter-1/2) are Na+ independent
In y+LAT-1 mutants, the expression rate of y+LAT-1 protein was significantly lower and cellular mortality was markedly increased compared with the wild type, but heteromerization of y+LAT-1 and 4F2hc within the cell was not disrupted [7]. Impairment of intestinal malabsorption and renal tubular reabsorption of CAAs by y+LAT-1 dysfunction causes a reduction of these amino acid pools in the body, resulting in various symptoms. Arginine and ornithine, a part of the dibasic amino acids, are also substrates for the urea cycle. These deficiencies may lead to dysfunction of the urea cycle and cause hyperammonemia, but details are unknown.
On the other hand, l-arginine is also a precursor substance of endogenous nitric oxide (NO) synthesis. In this disease, impaired regulation of CAA intra-extracellular transport due to dysfunction of y+LAT-1, which may result in an increase in intracellular arginine, inducing intracellular NO accumulation. It is also estimated to be involved in the complex pathology such as the dysfunction of the immune, bone metabolism, kidney, and lung systems. Below are some studies related to NO in LPI.
Kayanoki et al. [8] reported that serum NO metabolites decreased in Japanese male patient with LPI. They showed that intravenous administration of arginine increased serum NO derivatives and platelet count, and decreased serum thrombin antithrombin III complex and fibrin/fibrinogen degradation products in patients with LPI [8].
Kamada et al. [9] reported the functional impairment of vascular endothelial cells in patients with LPI associated with arginine deficiency. In detail, myocardial ischemic change in positron emission tomography was improved by arginine administration and presumed to be due to a decrease in serum NO [9]. Furthermore, Takeda et al. [10] evaluated portal flow in seven patients and reported a decline in portal circulation as a result of l-arginine deficiency.
The above-mentioned reports describe the pathological conditions caused by a deficiency of l-arginine, i.e., a deficiency of NO.
On the other hand, there are experimental studies that intracellular arginine (NO substrate) is increased in in vitro patient cells, which forms part of the pathological condition in LPI, as cited below.
Mannucci et al. [11] measured plasma and urinary nitrite/nitrate (NO2−/ NO3−) concentration in three patients and in vitro NO2− production in cultured skin fibroblasts, and found that both plasma NO3− levels and NO2− release in cultured skin fibroblasts increased. Kurbo et al. [12] showed LPI macrophages secreted significantly less NO than control macrophages.
From the above, there are likely two pathophysiological states in this disease. One is a transport defect in polarized cells (intestinal or renal tubular epithelium), another is a transport defect in non-polarized cells (lymphocytes or macrophages). In non-polarized cells, there is an intracellular imbalance between CAA accumulation and hyperproduction of NO synthase 2 (NOS2 or inducible NO synthase (iNOS)).
The former can cause failure to thrive, malnutrition, and dysfunction of the urea cycle (hyperammonemia), and the latter can induce renal, pulmonary, and immune disorders [13]. Such bipolar pathological conditions are currently being elucidated and further investigation would prove beneficial.
Clinical findings
Clinical symptoms and severity of LPI are diverse [14, 15]. Symptoms are not generally recognized at birth and are only noticed after weaning, when protein intake increases.
Physical features
After weaning, short stature (limb/trunk equilibrium type) and low body weight become gradually apparent. Infants are often referred to hospitals for consultation related to poor weight gain, hepatosplenomegaly, and short stature. Occasionally, hepatosplenomegaly can be identified in the neonatal period. Some patients with short stature also have complicated growth hormone (GH) deficiencies [15, 16]. Recurrent fractures occur frequently and may indicate bone disease [17]. The rate of osteoporosis is high and severe from childhood to adulthood [18, 19]. Sometimes, a delay in bone maturation, bone deformity, and osteoarthritis are also recognized. In addition, sparse hair growth, loose skin, and excessive extension of joints may be observed.
Hyperammonemia/neurological symptoms
After excessive protein intake, discomfort, behavioral changes, and loss of consciousness occur due to hyperammonemia. There has also been a reported infant case with recurrent episodic mild encephalopathy [20]. Starvation, infection, and stress can trigger hyperammonemia in many cases. However, some patients do not display consistent symptoms of hyperammonemia. Instead, they often show transient hyperammonemia only after meals (after protein loading), resulting in difficult diagnosis of this disorder. Therefore, diagnosis may be delayed into adulthood.
Gastrointestinal symptoms
After excessive intake of protein-rich foods, patients display nausea, vomiting, abdominal pain, and diarrhea, in addition to the neurological symptoms listed above. Generally, gastrointestinal symptoms occur after weaning at ~1 year and hence patients develop an extreme dislike of these foods. Protein aversion is a characteristic feature of this disease and is present in about 80% of patients.
Immune abnormality, autoimmune disease, and blood cell phagocytosis syndrome
Aggravated viral infections are frequently shown in LPI patients. It has been reported that patients with LPI had severe varicella infection combined with severe interstitial pneumonitis, hepatitis, decreased platelet count, bleeding, and elevated serum lactate dehydrogenase (LDH)/ferritin levels [21]. Other viral infections (measles encephalitis, and Epstein-Barr virus infection) also tend to be deteriorated. In an immunological study, low levels of leukocyte phagocytic, cytotoxic, and natural killer (NK) cell activity were reported, along with increased spontaneous proliferation of lymphocytes [22]. In these cases, although a vaccine is useful, it can be difficult to acquire antibodies [23].
Furthermore, several complications such as hemophagocytic syndrome [24, 25], autoimmune disease (systemic lupus erythematosus with antinuclear antibodies) [26, 27], autoimmune hepatitis, and rheumatoid arthritis are reported. A 9-month-old boy was diagnosed as a result of atypical hemophagocytic lymphohistiocytosis findings [28]. Several immunological studies in vitro have also been reported in recent years [29]. Barilli et al. [30] reported that system y+L activity is markedly lowered in monocytes and alveolar macrophages from LPI patients, and demonstrated that arginine influx and efflux through the y+L system is significantly lower in LPI macrophages than in those of normal subjects, indicating that the phagocytic activity of LPI macrophages is severely impaired [31]. In addition, granulocyte-macrophage colony-stimulating factor (GM-CSF) induces expression of SLC7A7 in both normal and LPI monocytes, suggesting that the gene is a cytokine target [30]. On the other hand, a study of LPI patient gene expression profiles using genome-wide microarray technology showed a significant accumulation of reactions related to inflammatory processes, immunity, and apoptosis observed in a functional annotation analysis [32]. In metabolome analysis of molecular lipids and polar metabolites derived from patients, it was suggested that amino acid imbalance affected the Tricarboxylic acid (TCA) cycle, β-oxidation, lipid metabolism, oxidative stress, and apoptosis [33].
Rotoli et al. [34] examined the effects of macrophages and airway epithelial cells on the lung and immune systems using a gene silencing approach. Significant induction of the expression and release of inflammatory mediators interleukin-1β and tumor necrosis factor-α was observed in cells by inhibition of SLC7A7. This effect was regulated mainly at the transcriptional level through activation of the nuclear factor-κB signaling pathway. The results indicated that downregulation of y+LAT-1 enhances the stimulatory effect of cytokines on CCL5/RANTES (regulated on activation, normal T-cell expressed and secreted) expression and release [34]. Kurko et al. [12] have also observed cytokine fluctuation and signaling abnormality mediated by Toll-like receptor. These results show that y+LAT-1 contributes to immune system function and inhibition of inflammation, in addition to CAA transport.
Renal involvement
Renal disease is a frequent and progressive complication in LPI [35,36,37]. In many patients, mild proteinuria and microhematuria are observed. Sometimes the initial symptom observed may be only mild proteinuria [36, 37]. Tanner et al. [37] described that in a cohort of 39 patients in Finland (mean age 30 years, 1–62), 74% had proteinuria, 38% had hematuria, 59% showed a serum cystatin C increase, and 38% showed a serum creatinine increase. Glomerulonephritis has also been often reported [36,37,38].
These findings are frequently observed in adulthood (from childhood in some patients) and are slowly progressive [37,38,39]. Renal histological findings are heterogeneous from tubulointestinal disorder to distinct glomerulonephritis, often showing membranous or mesangial proliferative glomerulonephritis [36,37,38, 40, 41]. There is a necropsy report of glomerulonephritis with IgA deposition [42], as well as reports of French patients showing membranous proliferative glomerulonephritis with polyclonal immunoglobulin deposition [38]. Furthermore, renal tubular acidosis [38] and Fanconi syndrome [43, 44] may also accompany LPI and require appropriate treatment. Urinary β-2 microglobulin was elevated in 90% of LPI cases, suggesting an early marker of renal involvement and indicating that regular monitoring of this marker is beneficial [39]. Some cases ultimately lead to renal failure [37]; therefore, caution is needed for treatment and observation of renal function.
In end-stage renal failure associated with LPI, awareness of potential osteoporosis and carnitine deficiency should also be considered [37].
Lung involvement
Respiratory disease is a serious complication affecting prognosis. Pulmonary complications include interstitial pneumonia and pulmonary alveolar proteinosis (PAP) [45]. Although infrequent in childhood, the onset and severity of respiratory disease varies among individuals. Although asymptomatic in early stages, interstitial lesions are seen in chest X-rays and can also be observed by chest high-resolution computed tomography (HRCT) [46, 47]. Diffuse reticular nodular stromal shadows are characteristically seen in the chest X-ray or HRCT over time.
Valimehamed-Mitha et al. [45] observed pulmonary involvement in 10 out of 14 children with LPI. Five of them had acute symptoms. HRCT was conducted in seven cases and interstitial lesions were observed in all cases, and fibrosis in four cases [45]. Mauhin et al. [35] also observed PAP in 10 out of 16 patients during 11 years of follow-up in France, and half of the patients had pulmonary fibrosis.
In bronchoalveolar lavage with PAP, an increase in the number of cells and foamy macrophages are observed. Lung biopsy of LPI patients may show cholesterol granulomas and/or alveolar proteinosis. Alveolar proteinosis can progress rapidly and become life-threatening, which is considered typical of terminal-stage pulmonary complications [40, 46, 48]. Respiratory symptoms may occur at any age and be the initial symptoms in new patients. Even if standard treatment has been introduced, symptoms may proceed on a monthly or yearly basis [40, 41, 47, 49]. The exact cause of PAP is unknown; however, intracellular accumulation of NO may be related to the occurrence of PAP [35].
Liver involvement
Hepatomegaly in infancy is recognized in about 70% of patients. However, elevation of serum transaminase is mild and often aspartate aminotransferase (AST) dominant. Jaundice and cholestasis are rarely observed, except in progressive liver cirrhosis. Steatosis is observed in many cases, although there are not many findings on LPI liver pathology. It is speculated that symptoms are caused by protein malnutrition. Generally, the liver of Kwashiorkor patients shows increased steatogenesis and apolipoprotein synthesis, as well as inhibition of lipoprotein lipase activity. In Kwashiorkor, lipid droplet deposition first appears in the hepatic portal area, not around the central vein, a process similarly observed in LPI, suggesting that low protein intake by the patient will result in similar symptoms to Kwashiorkor. Steatosis and fibrosis of portal areas are also seen in urea cycle disorders such as ornithine transcarbamylase deficiency, carbamyl phosphate synthase deficiency, and hyperornithinemia-hyperammonemia-homocitrullinuria syndrome. These diseases are common in metabolic dysfunctions related to arginine and citrulline; protein intake is as low as in LPI, but the correlation with pathological findings is unknown. In an LPI case where a liver biopsy was performed in early childhood, only mild portal tract chronic inflammation existed at 19 months of age, but a repeat biopsy at 4 years of age showed portal fibrous septal extension and bridging fibrosis by thin portal-to-portal septa, with focal regenerative nodules, micro steatosis, and focal glycogenosis [50]. In addition, focal glycogenosis in a 3-year-old [51, 52] showed an increase in collagenous fibers around the portal vein, and bridging fibrosis and diffuse cirrhosis in autopsies of another patient [41] have been observed. These findings represent various stages of slowly progressive pathological results. Pathology is assumed to begin with the deposition of lipid droplets and inflammation of the portal area, but gradually bridging fibrosis becomes apparent, and finally progresses to diffuse cirrhosis. In LPI, impairment of the urea cycle, portal circulation disorder due to NO decline due to arginine deficiency [10], and immune abnormality [13] are also considered to contribute to liver damage.
Other symptoms
There are few reported cases of cardiovascular symptoms. Myocardial ischemic change after exercise [9], myocardial infarction, and sinus arrest requiring a pace-maker [37] are reported. Vascular lesions (cerebral infarction) thought to be based on vascular endothelial dysfunction have been observed, in addition to a patient case displaying Moyamoya disease [53]. Acute pancreatitis is also occasionally observed, although the association with hyperlipidemia is unknown.
Laboratory findings
General blood examinations
Serum LDH usually increases in all fractions (especially LDH 4 and LDH 5) and is often increased to 600–1000 IU/L; however, although serum ferritin is recognized in many patients, its value varies. Most cases have episodes of hyperammonemia. The maximum blood ammonia level in adults usually ranges from 180 to 240 μmol/L (300–400 μg/dL), occasionally rising to ~600 μmol/L (1000 μg/dL). In newborns and infants, values of >120 μmol/L (200 μg/dL) and >60 μmol/L (100 μg/dL) have been reported, respectively. In some cases, transient hyperammonemia is detected by examining blood ammonia only after meals, which may lead to diagnosis of this disease. Approximately one-third of patients have some hematological abnormalities (leukocytopenia, thrombocytopenia, and anemia). Peripheral blood counts include normochromic or hypochromic anemia, leukopenia, thrombocytopenia, and latent intravascular coagulopathy. Bone marrow aspiration shows megakaryocyte reduction and erythroblast phagocytosis. In a study of coagulability, defective primary hemostasis, fibrin abnormality, and other deficiencies are detected [54]. High levels of serum total cholesterol/triglyceride and low levels of high-density lipoprotein have also been reported. Observation of 39 patients in Finland showed the average total cholesterol of patients was 7.6 mmol/L in adults and 5.4 mmol/L in children; patient triglyceride was 4.5 mmol/L in adults and 2.5 mmol/L in children, showing values higher than those in healthy subjects 56). The precise causes of hyperlipidemia are not yet known. In a study by Tanner, lipid intake of LPI patients does not significantly differ from that of the general population in Finnish patients, suggesting that hyperlipidemia does not occur because of excessive lipid intake [55]. Insulin-like growth factor-1 (IGF-1) values of LPI are low (5–13 nmol/L) [38]. Only some individuals exhibit GH deficiency [38]. The exact pathophysiology of GH deficiency is unknown, although specific nutritional factors have been identified. For example, amino acids such as arginine and lysine can stimulate GH secretion. Some patients respond to GH treatment for short stature due to GH deficiency, suggesting the GH/IGF-1 axis should be investigated in LPI [15]. In addition, slight increases in AST/alanine aminotransferase (ALT) (AST > ALT), thyroid-binding protein increases, deficient B-cell functions, low concentrations of immunoglobulin G subclasses [21], hypergammaglobulinemia, decreased leukocyte phagocytosis, decreased fungicidal activity, decreased NK cell activity, hypocomplementemia, and decreases in the CD4/CD8 ratio may be observed.
Amino acid analysis in blood/urine
Blood concentrations of dibasic amino acids (lysine, arginine, and ornithine) are distributed from about one-third of normal to the lower limit of the normal range [14]. As a secondary change, blood glutamine, alanine, glycine, serine, and proline levels tend to be elevated. Glutamine levels reflects hyperammonemia and are often elevated 800–1200 nmol / mL (11.7–17.5 mg / dL). Urine concentration of dibasic amino acids usually increases (lysine shows the largest increase, arginine and ornithine increases are moderate, and cysteine increases are mild), in almost all cases. Blood and urine amino acid analyses are important during diagnosis, but under malnutritive conditions the overall blood amino acid level is low and urinary excretion may also be reduced. In newborns and premature infants, excretion of amino acids in urine is excessive. Furthermore, under administration of amino acids and in Fanconi syndrome, excessive excretion of urinary amino acids occurs. Clinicians should carefully evaluate levels of amino acids in neonatal urine.
In some cases, it may be necessary to calculate the renal clearance of these amino acids [14]. In urinary organic acid analysis, urinary orotic acid is increased, accompanied by hyperammonemia.
Evaluation of the transport ability of dibasic amino acids is difficult in cultured skin fibroblasts and lymphocytes, as transport capacity is compensated by y+ LAT-2 expression, which is the isoform of the dibasic amino acid transporter [30, 56].
Genetic analysis
Biallelic pathogenic variants are detected in SLC7A7 (gene encoding y+ LAT-1) [1,2,3,4,5,6]. SLC7A7 is located on chromosome 14q11.2 and the full genomic length is about 46.5 kB, composed of 11 exons, and encodes 512 amino acids. There have been more than 60 types of genetic pathological variants reported, including small insertions/deletions, large insertions/deletions, missense, nonsense, and splice site variants [12, 56,57,58].
Finland has the largest number of patients [5] in whom c.895-2A>T exists as a high frequency mutation. In Italy, there are several families with c.1381_1384dupATCA, and c.726G>A [2, 4]. In Japan, nine types of pathological variants have been identified [9, 55, 59]. One of these variants, p.R410* (c. 1228C>T), is accumulated in the northern part of Japan [6, 60, 61]. Notably, many of the mutations in Japan have not been reported in other countries (except some in East Asia), indicating the genetic heterogeneity of Japanese patients. In addition, several novel patients have been recently reported worldwide: a Korean patient (with the same variant reported in Japan (IVS4+ 1G>A) [62], four novel types of variants in Turkish patients [63], c.147delCTTT as a common mutation in Tunisia [64], and others in Malaysia [65] and China [66].
During diagnosis, 92–95 % of patients are detected by two pathogenic mutations using the usual sequencing analysis, but the remainder cannot be diagnosed genetically. For example, the 3′-region Alu Y repeats within this gene [9, 55, 67]. In some patients, large deletions caused by the recombination of Alu repeats in introns 3 and 5 have been confirmed. MLPA (Multiplex ligation-dependent probe amplification) analysis is useful for detection of such large deletions during LPI molecular diagnosis. It is said that this genomic rearrangement is a well-known site of recombination, and that the site reaches 38% of the rearrangement reported in the past [67]. Genotype–phenotype correlations have not been found and there are few reports of prenatal diagnosis [68].
Differential diagnosis
Urea cycle disorders: all patients exhibit hyperammonemia. They can be differentiated by blood and urine amino acid analysis to some extent, but genetic analysis is useful for a definite diagnosis. In some cases, measurements of hepatic enzyme activity in the urea cycle are required. In cases of lysosomal disease, Gaucher or Niemann-Pick disease may be suspected from indications of hepatomegaly, interstitial lung disease, and blood abnormality. Gastrointestinal disorders such as cyclic vomiting, food allergies, chronic abdominal pain, and malabsorption syndrome are observed. These diseases can be judged based on digestive symptoms induced by protein intake [69]. Epilepsy and psychomotor developmental delay are secondary findings resulting from hyperammonemia, but when recurrent hyperammonemia goes unnoticed, they may be regarded as a developmental delay of unknown origin. Immunodeficiency, macrophage activation syndrome, and interstitial pneumonia are initial symptoms observed in childhood in rare cases.
Treatment
Acute phase treatment
To distinguish each urea cycle disorder that causes hyperammonemia, blood/urine amino acid analysis and urinary orotic acid analysis should be submitted before administration of therapeutic drugs. In addition, the possibility of this disease could be indicated by elevated serum LDH and ferritin. For definitive diagnosis, genetic analysis should be considered.
Acute phase
In the acute phase of hyperammonemia with clinical symptoms such as nausea, vomiting, and disturbance of consciousness, protein (nitrogen load) is removed and sufficient calories with parenteral nutrition are supplemented for preventing protein catabolism by the following methods.
1) Glucose infusion
: An infusion of 10% glucose or high concentration infusion (60–100 kcal/kg/day) should be started via a central venous catheter. In cases of hyperglycemia, concurrent use of insulin is also taken into consideration.
2) Drug administration
: Administration of l-arginine (Argi U® 100–250 mg/kg/day, intravenously) is useful. If l-arginine is not sufficiently effective, sodium phenylbutyrate (Buphenyl®: 450–600 mg/kg/day in patients weighing <20 kg, or 9.9–13.0 g/m2/day in larger patients), and/or sodium benzoate (100–250 mg/kg/day, intravenously or orally) is used. These drugs can be used alone or in combination, depending on each situations.
3) Blood purification:
Although the aforementioned drug therapy can normalize blood ammonia levels in most cases, if ineffective, introduction of continuous hemodialysis (CHD) is recommended.
4) Other treatments
If necessary, antibiotics (kanamycin, neomycin), lactulose, and/or lactobacillus preparation (probiotics) are administered to reduce ammonia production and absorption in the intestinal tract.
Although an acute attack is described as hyperammonemia in this paragraph, interstitial pneumonia and hemophagocytosis syndrome can also have severe acute courses. After utilizing these treatments as a top priority, we begin diagnosis and treatment intervention for LPI.
Chronic phase treatment
Diet therapy
Sufficient caloric intake and appropriate protein restriction are essential. It is easy to develop an essential amino acid and energy shortage in LPI patients. According to a nutritional survey conducted for 28 cases (1.5–61 years old) by Tanner et al., the average caloric intake is 1499 kcal/day for adult females and 1976 kcal / day for males. In this study, only 6–7% of the total energy intake in children (<10 years) was derived from protein [70]. From the viewpoint of preventing hyperammonemia, 0.8 (1.0)−1.5 g/kg/day protein intake is recommended for children and 0.5–0.8 g/kg/day for adults [13, 29, 70]. However, actual patient daily protein intake was distributed between 39% and 244% of the recommended amount of protein given above and in some patients it was markedly low [70]. Many patients can increase their protein intake by pharmacological intervention such as citrulline administration. Protein intake is adjusted as appropriate by the patient. Calcium and vitamin D intake in particular tend to be deficient; in addition, iron and zinc are also likely to be low [70]. Inclusion of special medical foods (e.g., low protein foods, protein-free milk) should also be considered.
Pharmacotherapy
Use of l-citrulline (~100 mg/kg/day): as l-citrulline is a neutral amino acid, its absorption is not disturbed in LPI patients. It is converted to arginine and ornithine in the liver and its effectiveness has long been recognized for this disease [71]. In recent years, lower volume supplementation (~100 mg/kg/day) is recommended to avoid excessive NO production [13, 29]. By administering l-citrulline, decreases in blood ammonia level, increases in dietary intake, increases in daily life activity, and reduction of hepatomegaly are observed.
Use of l-Arginine (Argi U® 120–380 mg/kg/day): oral administration of l-Arginine is effective but limited, due to intestinal malabsorption causing osmotic diarrhea. In addition, although administration of l-Arginine is effective as a treatment of acute phase hyperammonemia, increases of arginine in the cell induce excess production of NO in this disease and administration is controversial [30, 31].
Use of l-carnitine (20–50 mg/kg/day): oral administration is used for secondary hypocarnitinemia [13, 29, 72, 73]. Particularly in patients with chronic renal diseases, the use of ammonia-scavenging drugs tends to cause hypocarnitinemia [73].
Use of l-lysine (20–50 mg/kg/day): Although the effect on oral administration is not clear due to intestinal malabsorption, there are reports of usefulness at low doses [74, 75]. Tanner et al. [75] administered 8–46 mg/kg/day (mean 22.7 mg /kg/day) of l-lysine orally to 27 patients for 12 months and reported improved blood lysine without exacerbation of digestive symptoms. However, the effect of l-lysine on long-term prognosis should be further evaluated.
Use of sodium phenylbutyrate (Buphenyl®: 450–600 mg/kg/day in patients weighing <20 kg or 9.9–13.0 g/m2/day in larger patients), sodium benzoate (100–250 mg/kg/day). In cases where blood ammonia level is unstable or persistently high, oral administration of these nitrogen scavengers must be considered [14, 15].
Use of other supplementation: administration of vitamin D and bisphosphonate for osteoporosis [17, 76], GH injection for short stature patients with GH secretory insufficiency [16, 77], and statin (hydroxymethylglutaryl CoA reductase inhibitor) administration for hyperlipidemia are proven effective supplementary treatments [78].
For PAP, the usefulness of whole lung lavage for patients has been reported [48]. Recombinant human GM-CSF has also been reported to be effective as a treatment for PAP [66, 79]. On the other hand, there is an opposing opinion that GM-CSF should not be recommended on the basis that an increase in GM-CSF is associated with promotion of granuloma formation coupled with a decrease in the biological activity of surfactant protein D [80], making this treatment controversial.
For nephritis, angiotensin-converting enzyme blocker corticosteroids and mycophenolates are used. Some cases require treatment, such as lupus nephritis.
Vaccinations are recommended. Sometimes re-inoculation may be necessary [23].
In transplantation intervention, there are few reports of renal transplantation for end-stage renal failure [37]. A case of heart–lung transplantation for severe PAP has been reported [81]; although temporarily effective, it was ultimately fatal owing to pulmonary symptoms. Bone marrow transplantation has also been discussed in LPI combined with PAP.
Adulthood
Pregnancy
Pregnant women with LPI tend to have complications including hyperammonemia, anemia progression, toxemia, hemorrhage, placental infarcts at delivery and/or postpartum, and fetal intra-uterine growth retardation [82, 83]. Tanner et al. [82] described the outcome of 18 pregnancies in 9 Finnish LPI women, of whom 4 and 8 pregnancies developed complicated anemia and toxemia, respectively.
During pregnancy and delivery, proper monitoring of vital signs, laboratory findings (especially blood count, renal function, serum calcium, zinc, albumin, amino acid analysis, and urinalysis), and an appropriate diet, including control of protein intake and supplementation of amino acids, such as citrulline, is required. These interventions make it possible to secure the health of mothers and neonates [83].
Other complications
Pulmonary and renal complications are important management issues in adulthood. Progression of these complications cannot be prevented despite amino acid supplementation, resulting in considerable effect on life prognosis [35]. Varicella and common bacterial infections can cause exacerbation of renal and lung disease [21].
Conclusion
We have reviewed the recent pathophysiology of LPI, the practicality of treatment, and problems arising in adulthood. Early diagnosis and intervention in this disease, and countermeasures against hyperammonemia have progressed, enabling patients to live well into adulthood without neurological impairment. Future studies focusing on the pathology of later complications involving the kidneys, lungs, and immune diseases, as well as progress in approaches to treatment would make an important contribution to understanding and treatment of LPI.
References
Mykkänen J, Torrents D, Pineda M, Camps M, Yoldi ME, Horelli-Kuitunen N, et al. Functional analysis of novel mutations in y(+)LAT-1 amino acid transporter gene causing lysinuric protein intolerance (LPI). Hum Mol Genet. 2000;9:431–8.
Sperandeo MP, Bassi MT, Riboni M, Parenti G, Buoninconti A, Manzoni M, et al. Structure of the SLC7A7 gene and mutational analysis of patients affected by lysinuric protein intolerance. Am J Hum Genet. 2000;66:92–9.
Torrents D, Mykkänen J, Pineda M, Feliubadaló L, Estévez R, de Cid R, et al. Identification of SLC7A7, encoding y+ LAT-1, as the lysinuric protein intolerance gene. Nat Genet. 1999;21:293–6.
Borsani G, Bassi MT, Sperandeo MP, De Grandi A, Buoninconti A, Riboni M, et al. SLC7A7, encoding a putative permease-related protein, is mutated in patients with lysinuric protein intolerance. Nat Genet. 1999;21:297–301.
Tringham M, Kurko J, Tanner L, Tuikkala J, Nevalainen OS, Niinikoski H, et al. Exploring the transcriptomic variation caused by the Finnish founder mutation of lysinuric protein intolerance (LPI). Mol Genet Metab. 2102;105:408–15.
Noguchi A, Nakamura K, Murayama K, Yamamoto S, Komatsu H, Kizu R, et al. Clinical and genetic features of lysinuric protein intolerance in Japan. Pediatr Int. 2016;58:979–83.
Toivonen M, Tringham M, Kurko J, Terho P, Simell O, Heiskanen KM, et al. Interactions of y+ LAT-1 and 4F2hc in the y+l amino acid transporter complex: consequences of lysinuric protein intolerance-causing mutations. Gen Physiol Biophys. 2013;32:479–88.
Kayanoki Y, Kawata S, Yamasaki E, Kiso S, Inoue S, Tamura S, et al. Reduced nitric oxide production by L-arginine deficiency in lysinuric protein intolerance exacerbates intravascular coagulation. Metabolism. 1999;48:1136–40.
Kamada Y, Nagaretani H, Tamura S, Ohama T, Maruyama T, Hiraoka H, et al. Vascular endothelial dysfunction resulting from L-arginine deficiency in a patient with lysinuric protein intolerance. J Clin Invest. 2001;108:717–24.
Takeda T, Watanabe H, Saito T, Saito K, Takeda H, Togashi H, et al. Impaired portal circulation resulting from L-arginine deficiency in patients with lysinuric protein intolerance. Gut. 2006;55:1526–7.
Mannucci L, Emma F, Markert M, Bachmann C, Boulat O, Carrozzo R, et al. Increased NO production in lysinuric protein intolerance. J Inherit Metab Dis. 2005;28:123–9.
Kurko J, Vähä-Mäkilä M, Tringham M, Tanner L, Paavanen-Huhtala S, Saarinen M, et al. Dysfunction in macrophage toll-like receptor signaling caused by an inborn error of cationic amino acid transport. Mol Immunol. 2015;67:416–25.
Sebastio G, Sperandeo MP, Andria G. Lysinuric protein intolerance: reviewing concepts on a multisystem disease. Am J Med Genet C Semin Med Genet. 2011;157C:54–62.
Simell O. Chapter192 Lysinuric protein intolerance and other cationic aminoacidurias. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. New York: McGraw-Hill; 2001. p. 4933–56.
Esposito V, Lettiero T, Fecarotta S, Sebastio G, Parenti G, Salerno M. Growth hormone deficiency in a patient with lysinuric protein intolerance. Eur J Pediatr. 2006;165:763–6.
Niinikoski H, Lapatto R, Nuutinen M, Tanner L, Simell O, Näntö-Salonen K. Growth hormone therapy is safe and effective in patients with lysinuric protein intolerance. JIMD Rep. 2011;1:43–7.
Posey JE, Burrage LC, Miller MJ, Liu P, Hardison MT, Elsea SH, et al. Lysinuric protein intolerance presenting with multiple fractures. Mol Genet Metab Rep. 2014;1:176–83.
Svedström E, Parto K, Marttinen M, Virtama P, Simell O. Skeletal manifestations of lysinuric protein intolerance. A follow-up study of 29 patients. Skelet Radiol. 1993;22:11–6.
Güzel-Ozantürk A1, Ozgül RK, Unal O, Hişmi B, Aydın Hİ, Sivri S. Molecular and clinical evaluation of Turkish patients with lysinuric protein intolerance. Gene. 2013;521:293–5.
Bijarnia-Mahay S, Jain V, Bansal RK, Reddy GM, Haberle J. Lysinuric protein intolerance presenting with recurrent hyperammonemic encephalopathy. Indian Pediatr. 2016;53:732–4.
Lukkarinen M, Näntö-Salonen K, Ruuskanen O, Lauteala T, Säkö S, Nuutinen M, et al. Varicella and varicella immunity in patients with lysinuric protein intolerance. J Inherit Metab Dis. 1998;21:103–11.
Yoshida Y, Machigashira K, Suehara M, Arimura H, Moritoyo T, Nagamatsu K, et al. Immunological abnormality in patients with lysinuric protein intolerance. J Neurol Sci. 1995;134:178–82.
Lukkarinen M, Parto K, Ruuskanen O, Vainio O, Käyhty H, Olander RM, et al. B and T cell immunity in patients with lysinuric protein intolerance. Clin Exp Immunol. 1999;116:430–4.
Gordon WC, Gibson B, Leach MT, Robinson P. Haemophagocytosis by myeloid precursors in lysinuric protein intolerance. Br J Haematol. 2007;138:1.
Duval M, et al. Intermittent hemophagocytic lymphohistiocytosis is a regular feature of lysinuric protein intolerance. J Pediatr. 1999;134:236–9.
Kamoda T, Nagai Y, Shigeta M, Kobayashi C, Sekijima T, Shibasaki M, et al. Lysinuric protein intolerance and systemic lupus erythematosus. Eur J Pediatr. 1998;157:130–1.
Aoki M, Fukao T, Fujita Y, Watanabe M, Teramoto T, Kato Y, et al. Lysinuric protein intolerance in siblings: complication of systemic lupus erythematosus in the elder sister. Eur J Pediatr. 2001;160:522–3.
Ouederni M, Ben Khaled M, Rekaya S, Ben Fraj I, Mellouli F, Bejaoui M. A nine-month-old-boy with atypical hemophagocytic lymphohistiocytosis. Mediterr J Hematol Infect Dis. 2017;9:e2017057.
Ogier de Baulny H, Schiff M, Dionisi-Vici C. Lysinuric protein intolerance (LPI): a multi organ disease by far more complex than a classic urea cycle disorder. Mol Genet Metab. 2012;106:12–7.
Barilli A, Rotoli BM, Visigalli R, Bussolati O, Gazzola GC, Kadija Z, et al. In lysinuric protein intolerance system y+L activity is defective in monocytes and in GM-CSF-differentiated macrophages. Orphanet J Rare Dis. 2010;26:32. 5
Barilli A, Rotoli BM, Visigalli R, Bussolati O, Gazzola GC, Gatti R, et al. Impaired phagocytosis in macrophages from patients affected by lysinuric protein intolerance. Mol Genet Metab. 2012;105:585–9.
Tringham M, Kurko J, Tanner L, Tuikkala J, Nevalainen OS, Niinikoski H. Exploring the transcriptomic variation caused by the Finnish founder mutation of lysinuric protein intolerance (LPI). Mol Genet Metab. 2012;105:408–15.
Kurko J, Tringham M, Tanner L, Näntö-Salonen K, Vähä-Mäkilä M, Nygren H. Imbalance of plasma amino acids, metabolites and lipids in patients with lysinuric protein intolerance (LPI). Metabolism. 2016;65:1361–75.
Rotoli BM, Barilli A, Visigalli R, Ingoglia F, Milioli M, Di Lascia M, et al. Downregulation of SLC7A7 triggers an inflammatory phenotype in human macrophages and airway epithelial cells. Front Immunol. 2018;19:508.
Mauhin W, Habarou F, Gobin S, Servais A, Brassier A, Grisel C, et al. Update on lysinuric protein intolerance, a multi-faceted disease retrospective cohort analysis from birth to adulthood. Orphanet J Rare Dis. 2017;12:3.
Estève E, Krug P, Hummel A, Arnoux JB, Boyer O, Brassier A. Renal involvement in lysinuric protein intolerance: contribution of pathology to assessment of heterogeneity of renal lesions. Hum Pathol. 2017;62:160–9.
Tanner LM, Näntö-Salonen K, Niinikoski H, Jahnukainen T, Keskinen P, Saha H, et al. Nephropathy advancing to end-stage renal disease: a novel complication of lysinuric protein intolerance. J Pediatr. 2007;150:631–4, 634. e1.
Nicolas C, Bednarek N, Vuiblet V, Boyer O, Brassier A, De Lonlay P, et al. Renal involvement in a French paediatric cohort of patients with lysinuric protein intolerance. JIMD Rep. 2016;29:11–17.
Kärki M, Näntö-Salonen K, Niinikoski H, Tanner LM. Urine beta2-microglobulin is an early marker of renal involvement in LPI. JIMD Rep. 2016;25:47–55.
Parto K, Kallajoki M, Aho H, Simell O. Pulmonary alveolar proteinosis and glomerulonephritis in lysinuric protein intolerance: case reports and autopsy findings of four pediatric patients. Hum Pathol. 1994;25:400–7.
DiRocco M, et al. Role of haematological, pulmonary and renal complications in the long-term prognosis of patients with lysinuric protein intolerance. Eur J Pediatr. 1993;152:437–40.
McManus DT, Moore R, Hill CM, Rodgers C, Carson DJ, Love AH. Necropsy findings in lysinuric protein intolerance. J Clin Pathol. 2016;49:345–7.
Riccio E, Pisani A. Fanconi syndrome with lysinuric protein intolerance. Clin Kidney J. 2014;7:599–601.
Benninga MA, Lilien M, de Koning TJ, Duran M, Versteegh FG, Goldschmeding R, et al. Renal Fanconi syndrome with ultrastructural defects in lysinuric protein intolerance. J Inherit Metab Dis. 2007;30:402–3.
Valimahamed-Mitha S, Berteloot L, Ducoin H, Ottolenghi C, de Lonlay P, de Blic J. Lung involvement in children with lysinuric protein intolerance. J Inherit Metab Dis. 2015;38:257–63.
Santamaria F, et al. Early detection of lung involvement in lysinuric protein intolerance: role of high-resolution computed tomography and radioisotopic methods. Am J Respir Crit Care Med. 1996;153:731–5.
Ceruti M, Rodi G, Stella GM, Adami A, Bolongaro A, Baritussio A, et al. Successful whole lung lavage in pulmonary alveolar proteinosis secondary to lysinuric protein intolerance: a case report. Orphanet J Rare Dis. 2007;2:14.
Parenti G, et al. Lysinuric protein intolerance characterized by bone marrow abnormalities and severe clinical course. J Pediatr. 1995;126:246–51.
Parto K, Svedström E, Majurin ML, Härkönen R, Simell O. Pulmonary manifestations in lysinuric protein intolerance. Chest. 1993;104:1176–82.
Shinawi M, Dietzen DJ, White FV, Sprietsma L, Weymann A. Early-onset hepatic fibrosis in lysinuric protein intolerance. J Pedia Gastroenterol Nutr. 2011;53:695–8.
Badizadegan K, Perez-Atayde AR. Focal glycogenosis of the liver in disorders of ureagenesis: its occurrence and diagnostic significance. Hepatology. 1997;26:365–73.
Carpentieri D, Barnhart MF, Aleck K, Miloh T, deMello D. Lysinuric protein intolerance in a family of Mexican ancestry with a novel SLC7A7 gene deletion. Case report and review of the literature. Mol Genet Metab Rep. 1995;2:47–50.
Ghilain V, Wiame E, Fomekong E, Vincent MF, Dumitriu D, Nassogne MC. Unusual association between lysinuric protein intolerance and moyamoya vasculopathy. Eur J Paediatr Neurol. 2016;20:777–81.
Pitkänen HH, Kärki M, Niinikoski H, Tanner L, Näntö-Salonen K, Pikta M, et al. Abnormal coagulation and enhanced fibrinolysis due to lysinuric protein intolerance associates with bleeds and renal impairment. Haemophilia. 2018;24:e312–21.
Sperandeo MP, Annunziata P, Ammendola V, Fiorito V, Pepe A, Soldovieri MV, et al. Lysinuric protein intolerance: identification and functional analysis of mutations of the SLC7A7 gene. Hum Mutat. 2005;25:410.
Shoji Y, Noguchi A, Shoji Y, Matsumori M, Takasago Y, Takayanagi M, et al. Five novel SLC7A7 variants and y+L gene-expression pattern in cultured lymphoblasts from Japanese patients with lysinuric protein intolerance. Hum Mutat. 2002;20:375–81.
Sperandeo MP, Andria G, Sebastio G. Lysinuric protein intolerance: update and extended mutation analysis of the SLC7A7 gene. Hum Mutat. 2008;29:14–21.
Nunes V & Niinikoski H. Lysinuric protein intolerance. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle, 2018; p. 1993–2018.
Noguchi A, Shoji Y, Koizumi A, Takahashi T, Matsumori M, Kayo T, et al. SLC7A7 genomic structure and novel variants in three Japanese lysinuric protein intolerance families. Hum Mutat. 2000;15:367–72.
Koizumi A, Shoji Y, Nozaki J, Noguchi A, Dakeishi M, Ohura T, et al. A cluster of lysinuric protein intolerance (LPI) patients in a northern part of Iwate, Japan due to a founder effect. The Mass Screening Group. Hum Mutat. 2000;16:270–1.
Koizumi A, Matsuura N, Inoue S, Utsunomiya M, Nozaki J, Inoue K, et al. Evaluation of a mass screening program for lysinuric protein intolerance in the northern part of Japan. Genet Test. 2003;7:29–35.
Ko JM, Shin CH, Yang SW, Seong MW, Park SS, Song J. The first Korean case of lysinuric protein intolerance: presented with short stature and increased somnolence. J Korean Med Sci. 2012;27:961–4.
Güzel-Ozantürk A, Ozgül RK, Unal O, Hişmi B, Aydın Hİ, Sivri S, et al. Molecular and clinical evaluation of Turkish patients with lysinuric protein intolerance. Gene. 2013;521:293–5.
Esseghir N, Bouchlaka CS, Fredj SH, Ben Chehida A, Azzouz H, Fontaine M, et al. 1471 delTTCT a common mutation of Tunisian patients with lysinuric protein intolerance. Clin Lab. 2015;61:1973–7.
Habib A, Azize NA, Yakob Y, Md Yunus Z, Wee TK. Biochemical and molecular characteristics of Malaysian patients with lysinuric protein intolerance. Malays J Pathol. 2016;38:305–10.
Zhang G, Cao L. New mutations in the SLC7A7 gene of two chinese sisters with lysinuric protein intolerance. Pediatr Pulmonol. 2017;52:E94–E96.
Font-Llitjós M, Rodríguez-Santiago B, Espino M, Sillué R, Mañas S, Gómez L, et al. Novel SLC7A7 large rearrangements in lysinuric protein intolerance patients involving the same AluY repeat. Eur J Hum Genet. 1999;17:71–9.
Esseghir N, Bouchlaka CS, Fredj SH, Chehida AB, Azzouz H, Fontaine M, et al. First report of a molecular prenatal diagnosis in a tunisian family with lysinuric protein intolerance. JIMD Rep. 2001;1:37–8.
Maines E, Comberiati P, Piacentini GL, Boner AL, Peroni DG. Lysinuric protein intolerance can be misdiagnosed as food protein-induced enterocolitis syndrome. Pedia Allergy Immunol. 2013;24:509–10.
Tanner LM, Näntö-Salonen K, Venetoklis J, Kotilainen S, Niinikoski H, Huoponen K, et al. Nutrient intake in lysinuric protein intolerance. J Inherit Metab Dis. 2007;30:716–21.
Rajantie J, Simell O, Rapola J, Perheentupa J. Lysinuric protein intolerance: a two-year trial of dietary supplementation therapy with citrulline and lysine. J Pediatr. 1980;97:927–32.
Takada G, Goto A, Komatsu K, Goto R. Carnitine deficiency in lysinuric protein intolerance: lysine-sparing effect of carnitine. Tohoku J Exp Med. 1987;153:331–4.
Tanner LM, Näntö-Salonen K, Rashed MS, Kotilainen S, Aalto M, Venetoklis J, et al. Carnitine deficiency and L-carnitine supplementation in lysinuric protein intolerance. Metabolism. 2008;57:549–54.
Lukkarinen M, Näntö-Salonen K, Pulkki K, Aalto M, Simell O. Oral supplementation corrects plasma lysine concentrations in lysinuric protein intolerance. Metabolism. 2003;52:935–8.
Tanner LM, Näntö-Salonen K, Niinikoski H, Huoponen K, Simell O. Long-term oral lysine supplementation in lysinuric protein intolerance. Metabolism. 2007;56:185–9.
Gömez L, García-Cazorla A, Gutiérrez A, Artuch R, Varea V, Martín J, et al. Treatment of severe osteoporosis with alendronate in a patient with lysinuric protein intolerance. J Inherit Metab Dis. 2006;29:687.
Evelina M, Grazia M, Francesca O, Marta C, Paolo C, Rossella G. Growth hormone deficiency and lysinuric protein intolerance: case report and review of the lliterature. JIMD Rep. 2015;19:35–41.
Tanner LM, Niinikoski H, Näntö-Salonen K, Simell O. Combined hyperlipidemia in patients with lysinuric protein intolerance. J Inherit Metab Dis. 2010;Suppl 3:S145–50.
Tanner LM, Kurko J, Tringham M, Aho H, Mykkänen J, Näntö-Salonen K, et al. Inhaled sargramostim induces resolution of pulmonary alveolar proteinosis in lysinuric protein intolerance. JIMD Rep. 2017;34:97–104.
Douda DN, Farmakovski N, Dell S, Grasemann H, Palaniyar N. SP-D counteracts GM-CSF-mediated increase of granuloma formation by alveolar macrophages in lysinuric protein intolerance. Orphanet J Rare Dis. 2009;23:29. 4
Santamaria F, Brancaccio G, Parenti G, Francalanci P, Squitieri C, Sebastio G, et al. Recurrent fatal pulmonary alveolar proteinosis after heart-lung transplantation in a child with lysinuric protein intolerance. J Pediatr. 2004;145:268–72.
Tanner L, Näntö-Salonen K, Niinikoski H, Erkkola R, Huoponen K, Simell O. Hazards associated with pregnancies and deliveries in lysinuric protein intolerance. Metabolism. 2006;55:224–31.
Unal O, Coşkun T, Orhan D, Tokatl A, Dursun A, Hişmi B, et al. Pregnancy and lactation outcomes in a Turkish patient with lysinuric protein intolerance. JIMD Rep. 2014;13:3.
Acknowledgements
We are grateful to the patients for their cooperation, and to our staff of Akita University School of Medicine for their support during the study. We thank Editage (www.editage.jp) for English language editing.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Noguchi, A., Takahashi, T. Overview of symptoms and treatment for lysinuric protein intolerance. J Hum Genet 64, 849–858 (2019). https://doi.org/10.1038/s10038-019-0620-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-019-0620-6
This article is cited by
-
Whole lung lavage and GM-CSF use for pulmonary alveolar proteinosis in an infant with lysinuric protein intolerance: a case report
Italian Journal of Pediatrics (2024)
-
The Great Masquerade: Varying Manifestations of Lysinuric Protein Intolerance
Indian Journal of Pediatrics (2024)
-
Lysinuric protein intolerance presenting as pancytopenia and splenomegaly mimicking acute leukaemia: a case report
BMC Pediatrics (2023)
-
Characteristics and genetic analysis of patients suspected with early-onset systemic lupus erythematosus
Pediatric Rheumatology (2022)
-
Familial hemophagocytic lymphohistiocytosis syndrome due to lysinuric protein intolerance: a patient with a novel compound heterozygous pathogenic variant in SLC7A7
International Journal of Hematology (2022)
