
Abstract
Macroscopic quantum coherence has been observed in some many-body systems including superconductors, quantum liquids1 and cold atom condensates2, but never for a single quasi-particle state. In an ideal semiconductor, excitons (electron–hole pairs bound by the Coulomb interaction) can, in principle, exist as delocalized plane waves extending over the entire volume. However, any kind of disorder prevents long-range spatial coherence from emerging. There has been evidence for the formation of macroscopic coherent states only in condensate phases such as in the case of microcavity polaritons condensation3,4 or in a dense quasi-two-dimensional exciton gas5. It is unclear however, whether in this latter case the observations are really related to macroscopic coherence6. Here, we show that a single exciton state in an individual ordered conjugated polymer chain7,8, shows macroscopic quantum spatial coherence reaching tens of micrometres, limited by the chain length. The spatial coherence of the k=0 exciton state is demonstrated by selecting two spatially separated emitting regions of the chain and observing their interference.
Similar content being viewed by others

Main
Polydiacetylene (PDA) chains form a model system to investigate the electronic properties of conjugated polymers9. Their optical excitation, an exciton, is usually considered as a neutral excited state of the chain10. However, a one-dimensional excitonic energy band is necessary to account for the optical properties of PDA7,8. Here, we demonstrate that these macromolecules are actually more than model conjugated polymers: the chains are almost perfect semiconductor quantum wires allowing macroscopic delocalization of excitons. In the experiments reported here, a single organic quantum wire, a PDA chain, is studied at low temperatures by means of microscopic imaging spectroscopy using our microphotoluminescence setup with an imaging spectrometer (Fig. 1a). The wire excitonic fluorescence, when imaged, shows large spatial extension over tens of micrometres. When overlapping the emission coming from two spatially well-separated regions of a single chain, interference fringes are observed, demonstrating a macroscopic spatial coherence length of the radiative exciton.

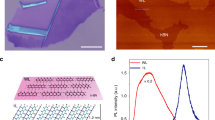
a, Schematic of the microphotoluminescence setup. A highly diluted PDA sample, with a concentration of red chains less than 10−7 in weight, is mounted on the cold finger of a helium cryostat and cooled down to 10 K. Using a f=7.8 mm microscope objective with a 0.6 numerical aperture mounted on three-dimensional piezo-actuators, a single isolated red chain is excited along 1 μm. The overall spectral and spatial resolutions are 50 μeV and 1 μm, respectively. The chain excitonic fluorescence is collected by the same objective and either directly imaged on the nitrogen cooled charge-coupled device (CCD) camera (2000×800 pixels, Jobin-Yvon) of an imaging spectrometer (Triax 550, Jobin-Yvon), or imaged with an 800 mm lens on the slits for the interference experiment. The magnification is around ×100 whereas the slits have a 20 μm width and their separation can be varied, from 200 to 900 μm. Therefore, two 1 μm regions of the chain, length limited by the spatial resolution, are selected and their separation can be tuned from 2 to 9 μm. The inset in the figure shows schematically how the chain fluorescence is imaged on the slits. a is the variable distance between the two slits. b, Spectrally resolved fluorescence image for a single isolated red chain. The vertical axis is the position along the chain in micrometres. The horizontal axis is the emission energy in electronvolts. The excitation laser spot (which is referred to as being at x=0 in the text) is centred at the position 50 μm. c, Normalized fluorescence spectral profile taken from cross-sections at 60 and 55 μm positions (grey circles and squares, respectively) out of the image in b. The curve corresponds to a lorentzian fit with a full-width at half-maximum of 510 μeV. d, Normalized fluorescence spatial profile when moving the laser excitation spot along the chain. The resulting profiles are obtained for an excitation spot position centred at 50, 55 and 60 μm represented by the circles, triangles and squares, respectively.
PDA chains diluted in their monomer crystalline matrix are very long, highly regular polymer chains, isolated from one another. They can take either of two conformations11, conventionally named ‘blue’ and ‘red’, which correspond to two electronic configurations with identical molecular structures but slightly different geometrical conformations. This work exclusively deals with ‘red’ chains. Disorder is very small, the inhomogeneous broadening of the absorption or emission of an ensemble of red chains being of the order of 2 meV (refs 1112). These chains emit a strong resonance fluorescence around 2.28 eV (at a temperature T=10 K), demonstrating the absence of self-trapping effects, and several vibronic replicas that are two or more orders of magnitude weaker. The emission is polarized parallel to the chain axis, demonstrating that indeed it comes from an isolated single chain and not from a pair or a stack of chains, because in that case the emission would be differently polarized. Microphotoluminescence experiments are possible, and thanks to high dilution and to the intense and narrow resonance emission, a single isolated chain can be studied12. In this experiment, the chain is excited along ≈1 μm, which is much smaller than the overall chain length. The average length of the chains has been measured by light scattering and is of the order of 10 μm. This is in agreement with the observed spatial extension in the fluorescence experiment. We have shown that a single red chain has the characteristic properties of a one-dimensional semiconducting quantum wire. Indeed, the optical excitation is a highly bound exciton as discussed in ref. 10 (p. 318), described by a one-dimensional energy band with the predicted  (where E is the exciton kinetic energy) behaviour of the density of states7, leading to an exciton effective lifetime scaling with temperature as
(where E is the exciton kinetic energy) behaviour of the density of states7, leading to an exciton effective lifetime scaling with temperature as  (ref. 8). The single-chain fluorescence comes from the radiatively coupled exciton states near wavevector k=0. It is lorentzian with an homogeneous width Γh governed by the interaction between the exciton and the three-dimensional longitudinal acoustic phonons of the monomer matrix surrounding the chain7,12 (Γh≈500 μeV at 10 K, see Fig. 1c).
(ref. 8). The single-chain fluorescence comes from the radiatively coupled exciton states near wavevector k=0. It is lorentzian with an homogeneous width Γh governed by the interaction between the exciton and the three-dimensional longitudinal acoustic phonons of the monomer matrix surrounding the chain7,12 (Γh≈500 μeV at 10 K, see Fig. 1c).
For resonant excitation, when pumping the radiating k≈0 exciton states directly, the resonant fluorescence is much weaker than the strong scattered laser light, making its detection difficult to achieve. Therefore, we choose to photocreate excitons by pumping within the homogeneous linewidth of the absorption of the strongest vibronic replica at 2.47 eV (the so-called D-absorption line corresponding to the double-bond stretch optical phonon). Owing to the flat dispersion of the optical phonon branch and to the small exciton effective mass7, energy and momentum conservation arguments require that the photocreated vibron relaxes into a k=0 exciton and a k=0 phonon. This relaxation is quasi-instantaneous. Indeed, the rise time of the fluorescence signal of an ensemble of PDA chains is at most 50 fs (ref. 13).
Figure 1b shows a spectrally resolved single-chain fluorescence image obtained with a low-temperature setup of high mechanical stability12. This microscopic imaging setup allows us to remain focused on a single chain during a long enough time so that very low pump power experiments can be performed. Knowing the absorption coefficient under non-resonant excitation at 2.47 eV (ref. 14) the low excitation power (≈1–20 μW) ensures that the probability of photocreating an exciton in the chain during its effective lifetime, is less than unity. Hereafter, we therefore deal with the properties of a single exciton. The chain emission is extended over 20 μm (for Fig. 1b), which is probably the chain length in this case. Its spectral profile does not vary along the fluorescence spatial extension (Fig. 1c) proving that the potential confining the red exciton is highly regular, even on a macroscopic scale. The regularity of the confinement potential is also confirmed by the microphotoluminescence results. Note that one chain is much longer (≈10 μm) than the mean interchain distance (≈1 μm). If localization was present then we would not observe a single homogeneously broadened emission line all along the chain. Moreover, the excitation position does not affect the spectral intensity profile or the spatial intensity profile. Indeed, when moving the latter up to 10 μm along the chain, the fluorescence image remains identical (Fig. 1d). These experimental facts show that the emission spatial profile is characteristic of the chain and is established very rapidly, on a short timescale compared with the 90 ps effective lifetime of the exciton8.
To account for this, one could invoke conventional transport mechanisms for the exciton, such as diffusive or ballistic transport. A diffusive transport of the exciton would require a diffusion coefficient D=L2/τ≈104 cm2 s−1, where L is the fluorescence spatial extension (≈20 μm) and τ is the effective lifetime (≈100 ps at 10 K). This diffusion coefficient, deduced to match the experimental data, is several orders of magnitude larger than values measured in organic or inorganic semiconductors15. A ballistic transport would require an exciton kinetic energy E=(1/2)mX*(L/τ)2≈10 meV (the exciton effective mass mX*≈0.1m0 (ref. 8), with m0 the free electron mass). However, at 10 K, the exciton average kinetic energy is of the order of 1 meV, as it has been shown to be in thermodynamic equilibrium with the lattice7. Therefore, neither of these two conventional transport mechanisms can explain the ultrafast macroscopic spatial extension of the fluorescence.
The delocalization of the red exciton centre of mass over the entire chain is actually an attractive picture. In such a case, the exciton would be a quantum state, with a wavefunction having a macroscopic coherence length. To investigate this spatial coherence, an interference experiment was set up (see Fig. 1a and inset). Two slits are used for selecting two spatially well-separated emitting regions of the chain fluorescence and observing the interferences between them. This is the main difference to the classical Young’s interference experiment where a single light source is split into two. High-contrast interference fringes are observed, demonstrating extended spatial coherence of the radiating states over a length comparable to the total chain length. For a single 10-μm-long chain, the resulting interference pattern is presented in Fig. 2a,b. The fringe period scales as λ f/a, where λ is the fluorescence wavelength, f the focal length of the spectrometer entrance lens and a the distance between the slits (see Fig. 2c). Such an interference pattern is always observed when a single chain is excited. In some cases when two chains are present within the laser spot, two interference patterns appear at slightly different energies. However, when several chains are present, the patterns overlap and the interferences are therefore blurred.

a, Interference pattern obtained from two 1-μm-wide emitting regions of a 10-μm-long chain. This interference pattern is observed in the Fourier plane formed in the focal plane of the spectrometer entrance lens. The pattern length is limited by the collecting optics. The separation between the two sources taken out of the chain is about 2 μm. The corresponding distance in the plane of the slits is around 220 μm leading to the fringe period of about 225 μm. b, Cross-section of the previous interference pattern at 2.2875 eV (see arrows in Fig. 2a) showing the intensity profile which gives a value of the contrast C=(Imax−Imin)/(Imax+Imin) of 75%. Imin and Imax and the minimum and maximum intensity respectively. c, Plot of the fringes period as a function of the distance between the two interfering regions. The plot shows the results obtained for three experiments each performed on an individual chain (squares, circles and crosses). The fringe period scales as λ f/a as shown by the straight line. The scaling factor λ f only depends on the wavelength λ of the emission and on the focal length f of the spectrometer entrance lens. The error bar on the value of the fringes period is related to the Fourier transform analysis, which gives the value of the spatial frequency. The uncertainty is given by the separation between two points in the Fourier transform. It leads to a varying uncertainty on the fringe period. The error bar on 1/a corresponds to a constant 5-μm reading uncertainty on a. d, Fourier transform of the cross-section presented in b. A single spatial frequency appears corresponding to the fringe period.
The formation of a macroscopically coherent state needs further understanding. A probable microscopic scheme that we may invoke is the following. First of all, let us stress that in this system a strong exciton–photon coupling regime can be reached, that is, the eigenstate is a polariton16 (see also ref. 14; and http://tel.ccsd.cnrs.fr). According to energy and momentum conservation requirements, only exciton states with a wavevector near k=0 are coupled to light. Taking into account that incident light is focused within an angle θ of 36∘, the incident photons have a wavevector dispersion around kν=0 equal to kνsinθ/2≈5×10−4 Å−1 (kν=n Eν/ħc is the photon wavevector in the material with refractive index n=1.5 (ref. 13), Eν=2.28 eV is the exciton transition energy, ħ is the reduced Planck’s constant and c is the speed of light). Thus, the excitons coupled to light have a wavevector along the chain axis such that k≤kνsinθ/2. If we compare this with the edge π/d of the first Brillouin zone, where d is the chain unit cell (≈10 Å), the wavevector of the photocreated exciton is very close to k=0. Only excitons within an energy range Δrad=ħ2kν2/2mX≈10 μeV can then recombine radiatively assuming that all the lowest-lying states in the band are thermally populated17. For a given chain length L (10 μm in the case of the chain studied in Fig. 2) the accessible momentum states are quantized and separated by Δk=π/L so that about 20 states are photocreated. Considering the initial excitation (at x=0 and time t=0), a coherent superposition of those different k-states, being in phase at x=0, is created, leading to the formation of a state with a pronounced maximum at x=0 and extending over the area of the exciting incident light (≈1 μm). This picture is similar to the formation of a quantum ‘wavepacket’. Then, in a picosecond time range7, the interaction with acoustical phonons dephase the different k-modes. Thus, the final state spreads over the total length of the chain and has a homogeneous broadening Γh inversely proportional to this interaction time. This microscopic picture of the exciton state formation corresponds to a transient regime that cannot be observed in the continuous-wave experiments presented above. Therefore, what we do observe when averaging in time is a stationary macroscopic quantum state of given energy, with a wavefunction of given phase and coherence length limited by the chain length. When probing the spatial coherence between two well-separated positions on the chain, the dephasing is stationary in time and depends only on the difference in the optical path, leading to an interference pattern like that shown in Fig. 2a.
The fringe contrast can be very high, up to 75% (Fig. 2b). However, variations of the fringe contrast have been observed depending on the chosen positions on the chain. A possible explanation for contrast variations could lie in the non-uniform spatial emission profile that is usually observed (Fig. 1d). Indeed, if the relative intensity of the light emitted from the two regions were different, the interference contrast would be reduced. Actually, the spatial profile of the fluorescence can be influenced by local disorder present along the chain (mostly associated with elastic stress in the crystal). This would induce small-amplitude potential fluctuations creating a non-uniform emission profile along the chain without destroying the spatial coherence of the state.
References
Yarmuck, E. J., Gordon, M. J. V. & Packard, R. E. Observation of stationary vortex arrays in rotating superfluid helium. Phys. Rev. Lett. 43, 214–217 (1979).
Simsarian, J. E. et al. Imaging the phase of an evolving Bose-Einstein condensate wave function. Phys. Rev. Lett. 85, 2040–2043 (2000).
Savvidis, P. G. & Lagoudakis, P. G. Teaching polaritons new tricks. Semicond. Sci. Technol. 18, S311–S318 (2003).
Bloch, J. et al. Monitoring the dynamics of a coherent cavity polariton population. Phys. Rev. B 71, 155311 (2005).
Butov, L. V., Gossard, A. C. & Chemla, D. S. Macroscopically ordered state in an exciton system. Nature 418, 751–754 (2002).
Snoke, D., Denev, S., Liu, Y., Pfeiffer, L. & West, K. Long-range transport in excitonic dark states in coupled quantum wells. Nature 418, 754–757 (2002).
Dubin, F. et al. Optical evidence of a purely one-dimensional exciton density of states in a single conjugated polymer chain. Phys. Rev. B 66, 113202 (2002).
Lécuiller, R. et al. Fluorescence yield and lifetime of isolated polydiacetylene chains: evidence of a one-dimensional exciton band in a conjugated polymer. Phys. Rev. B 66, 125205 (2002).
Schott, M. in Photophysics of Molecular Materials. From Single Molecules to Single Crystals (ed. Lanzani, G.) (Wiley–VCH, Berlin, in the press).
Sariciftci, N. S. (ed.) Primary Photoexcitations in Conjugated Polymers: Molecular Excitons Versus Semiconductor Band Model (World Scientific, Singapore, 1997).
Lécuiller, R., Berréhar, J., Lapersonne-Meyer, C. & Schott, M. Dual resonance fluorescence of polydiacetylene chains isolated in their crystalline monomer matrix. Phys. Rev. Lett. 80, 4068–4071 (1998).
Guillet, T. et al. Emission of a single conjugated polymer chain isolated in its single crystal monomer matrix. Phys. Rev. Lett. 87, 087401 (2001).
Haacke, S., Berréhar, J., Lapersonne-Meyer, C. & Schott, M. Dynamics of singlet excitons in one-dimensional conjugated polydiacetylene chains: a femtosecond fluorescence study. Chem. Phys. Lett. 308, 363–368 (1999).
Dubin, F., Berréhar, J., Grousson, R., Schott, M. & Voliotis, V. Evidence of polariton induced transparency in a single organic quantum wire. Preprint at < http://arxiv.org/abs/cond-mat/0508602> 2005.
Pulizzi, F., Thijssen, W. H. A., Christianen, P. C. M. & Maan, J. C. Diffusion of two-dimensional magnetoexcitons. Physica B 298, 441–445 (2001).
Hopfield, J. J. Theory of the contribution of excitons to the complex dielectric constant of crystals. Phys. Rev. 112, 1555–1567 (1958).
Citrin, D. S. Long intrinsic radiative lifetimes of excitons in quantum wires. Phys. Rev. Lett. 69, 3393–3396 (1992) ibid Phys. Rev. Lett. 70, 1186 (1993).
Acknowledgements
This work has been supported by the Region Ile de France (SESAME N∘E. 1751).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Dubin, F., Melet, R., Barisien, T. et al. Macroscopic coherence of a single exciton state in an organic quantum wire. Nature Phys 2, 32–35 (2006). https://doi.org/10.1038/nphys196
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nphys196
This article is cited by
-
Energy transfer in N-component nanosystems enhanced by pulse-driven vibronic many-body entanglement
Scientific Reports (2023)
-
Detecting photoelectrons from spontaneously formed excitons
Nature Physics (2021)
-
Nanoscale π-conjugated ladders
Nature Communications (2021)
-
Microcavity-like exciton-polaritons can be the primary photoexcitation in bare organic semiconductors
Nature Communications (2021)
-
Quantum stochastic transport along chains
Scientific Reports (2020)
