
Abstract
Neurogenesis during embryonic and adult life is tightly regulated by a network of transcriptional, growth and hormonal factors. Emerging evidence indicates that activation of the stress response, via the associated glucocorticoid increase, reduces neurogenesis and contributes to the development of adult diseases.As corticotrophin-releasing hormone (CRH) or factor is the major mediator of adaptive response to stressors, we sought to investigate its involvement in this process. Accordingly, we found that CRH could reverse the damaging effects of glucocorticoid on neural stem/progenitor cells (NS/PCs), while its genetic deficiency results in compromised proliferation and enhanced apoptosis during neurogenesis. Analyses in fetal and adult mouse brain revealed significant expression of CRH receptors in proliferating neuronal progenitors. Furthermore, by using primary cultures of NS/PCs, we characterized the molecular mechanisms and identified CRH receptor-1 as the receptor mediating the neuroprotective effects of CRH. Finally, we demonstrate the expression of CRH receptors in human fetal brain from early gestational age, in areas of active neuronal proliferation. These observations raise the intriguing possibility for CRH-mediated pharmacological applications in diseases characterized by altered neuronal homeostasis, including depression, dementia, neurodegenerative diseases, brain traumas and obesity.
Similar content being viewed by others


Introduction
Living organisms maintain their physiological homeostasis during development as well as in adult life against constant challenges by internal and environmental stimuli. Although differentiated cells account for the majority of homeostatic functions, stem cells contribute critically to the whole process either by generation of differentiated cells or by changing their own function to adapt to the altered tissue/organ demands.1 Thus, in a number of tissues, including the nervous system, tissue-specific stem cells persist throughout life and give rise to new cells in order to meet the demands of turnover and injury-induced cell loss. Along these lines, emerging evidence has linked changes in adult neurogenesis to the pathogenesis, and often to the success of therapeutic regimens, of major diseases such as depression.2, 3, 4, 5, 6
Neurogenesis occurs constitutively in the embryonic brain and, as has been confirmed lately, to a lesser extent in specific niches of the adult brain.7, 8 In the adult human and rodent brains, proliferation of neural stem/progenitor cells (NS/PCs) persists throughout life in areas such as the subventricular zones (SVZs) and the subgranular hippocampal zones.8 Neurogenesis involves a tightly controlled process of spatiotemporal neuronal proliferation and programmed cell death9 achieved by the orchestrated action of a network of transcription and growth factors. Additional intrinsic factors, such as secreted molecules, and environmental stimuli impact significantly on the potential of NS/PCs for proliferation, differentiation and survival, with mechanisms we start to understand better.10, 11, 12, 13, 14 For example, excess levels of circulating glucocorticoid, such as during prolonged, unopposed stress, are associated with suppressed proliferation12 and decreased survival of NS/PCs in the hippocampus.15, 16
The adaptive response to challenges, otherwise stress or ‘fight-or-flight’response,17 is a well-preserved process intimately associated with survival and development. In mammals, it is driven by the activation of the hypothalamic–pituitary–adrenal axis and the catecholaminergic system.18 The critical step in the development of the stress response is the activation of the neuropeptide corticotropin-releasing hormone (CRH) or factor.19 This response is self-limited as return back to homeostasis is linked to decrease in CRH neurons activation back to their basal state.20 CRH is expressed early in the developing mouse brain, such as on E13.5 in forebrain and on E10 in the cerebellum21, 22 correlating temporally with the birth of the first neurons from the neural progenitor cells of the ventricular layer.22 In addition, in both the developing and the adult brain, CRH is expressed in the neurogenic niche of the hippocampal granular zone, a well-characterized neurogenic area.21, 23 In the central nervous system, CRH has been shown to function as a neurotransmitter/neuromodulator.24 The first indication that CRH may be implicated in processes related to neuronal development and/or differentiation derived from the altered expression of genes involved in myelination and cell proliferation in transgenic mice overexpressing CRH.25 CRH has been also implicated in the differentiation of noradrenergic neurons in the locus coeruleus during brain development.26 CRH acts via binding to CRH receptor-1 (CRH-R1) and -2 (CRH-R2), members of the G-protein-coupled receptors (GPCRs) family.27 Several GPCRs have been implicated in neurogenesis and apoptosis in site- and/or developmental stage-restricted manners.28 In line, neuroprotective effects of CRH have been described during development,23 in primary neuronal cultures exposed to toxic factors,20 or in vivo following oxidative stress.29, 30
Here, we present evidence that CRH regulates neurogenesis, an effect that could be considered as part of the adaptive response of the nervous system to various challenges. This regulatory role of CRH is in line with late reports on the contribution of CRH in the maintenance of peripheral tissue homeostasis following metabolic31 or inflammatory stimuli.32 In particular, we show that CRH through its receptor-1 (CRH-R1) is implicated in the control of proliferation and apoptosis of NS/PCs both in vitro and in vivo. Most importantly, CRH seems sufficient to reverse the glucocorticoid-mediated suppression of proliferation and the associated induction of apoptosis in NS/PCs. Finally, we demonstrate the expression of CRH receptor(s) in the neurogenic areas of mouse and human brain. These data indicate a novel role of CRH and renders CRH as a potential therapeutic target for central nervous system-associated neurological disorders and diseases.
Materials and methods
Human tissues
Human embryos from 13 weeks old (n=4) were obtained from Alexandra Hospital and from Medical School, University of Athens, Greece. The ethics committees of the participating university and hospital approved tissue collection. Handling of tissue was carried out in accordance with all regulations of the institutional ethics committees.
Animals
Animals housing and care were according to NIH and EU guidelines. The Crh−/− mouse line was generated as described previously.33 Crh–/– mice were raised in C57/Bl6 background and were obtained by crossing of heterozygous, their wildtype littermates, Crh+/+ were used as controls.
Cell culture, fluorescence-activated cell sorting analysis, immunohistochemistry, BrdU, TUNEL assays, reverse transcriptase PCRs and western blotting
Detailed methodology for all these assays is described in Supplementary Materials and Methods.34, 35, 36
Statistical analysis
Results are expressed as mean±s.e.m. Data were analyzed by two-tailed, unpaired, equal variance Student t-test and defined as P<0.05.
For details of animals, cell culture, tissues processing, imaging and quantification, immunohistochemistry, 5-bromo-2-deoxyuridine (BrdU) and terminal transferase dUTP nick-end labeling (TUNEL) assays, PCR analysis and statistical analysis, see Supplementary Materials and Methods.
Results
Evidence for altered neurogenesis in the CRH-deficient mice
To assess the impact of CRH in mouse neurogenesis, we used the Crh-null (Crh−/−) mouse with global CRH deficiency, and the corresponding glucocorticoid insufficiency.33 We used heterozygote pregnancies taking advantage of the fact that in that case all embryos, independent of their genotype, are exposed to similar glucocorticoid levels of maternal origin. Proliferation was assessed by immunohistochemistry of multiple tissue sections following BrdU administration on E14.5 for 2 h. As shown, significantly fewer BrdU-positive NS/PCs were identified in the proliferating layers of the cerebral cortex of Crh−/− mice compared with their littermate wild-type mice (Figures 1a and c). Furthermore, TUNEL analysis revealed significant increase in TUNEL+ cells in the ventricular zones (VZ) and SVZ of the Crh−/− mice (Figures 1b and d). As glucocorticoid insufficiency of the Crh−/− mice was ‘corrected’,37 these findings reflect direct effects of Crh−/− deficiency on neurogenesis in the developing mouse brain.

Altered proliferative and apoptotic properties of neural progenitor cells in the developing brain of Crh−null (Crh−/−) mice. (a) 5-bromo-2-deoxyuridine (BrdU) was administered to the mother at gestational age 14.5 for 2 h. Representative images from brain slices (E14.5) of Crh−/− and wild-type (wt) littermates are shown. Scale bar=20 μm. (c) Graph that shows quantification of the BrdU-positive cell count in 180 × 180 μm2 areas. Data are shown as mean±s.e.m. (n=20 slices of three animals). (b)Representative images from brain slices (E14.5) of Crh−/− and wt littermates after performance of terminal transferase dUTP nick-end labeling (TUNEL) assay. Scale bar=50 μm. (d) Graph that shows quantification of the TUNEL-positive cell count in 180 × 180 μm2 areas. Data are shown as mean±s.e.m. (n=20 slices of four animals) *P<0.05 versus wt mice. vz, svz, LV, lateral ventricle.
Expression of CRH receptors in neural progenitor cells of developing and adult mouse brain
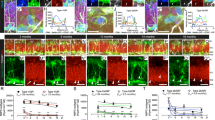
Next, we assessed the expression of CRHRs in the developing mouse brain. Immunohistochemical analysis revealed a broad and strong reactivity for CRH receptors in mouse neuroepithelium on E14.5 (Figures 2a–a′). As shown (Figure 2a), CRH receptors were specifically expressed on NS/PCs, as indicated by the colocalization with the neural stem cell marker nestin. In the adult mouse, we found CRH receptors in the lineage of neuronal progenitors of adult SVZ, a very active neurogenic area, including quiescent radial glia-like cells (nestin+/GFAP+) (Figures 2b–b′ and c–c′), transient amplifying progenitor cells (Mash1+) (Figures 2d–d′) and migrating neuroblasts (DCX+) (Figure 2e–e′).

Expression of CRH receptors (CRHRs) in embryonal and adult neural progenitor cells in vivo and in vitro. (a) Detection of CRH receptors in neural progenitor cells of embryonic (E14.5) forebrain. Co-expression (yellow) of CRHRs and the neural progenitor marker nestin. (b, e) Detection of CRH receptors in neural progenitor cells of adult forebrain. Co-expression of CRHRs (arrows) and the neural progenitor markers nestin (b) and GFAP (c) was observed in the majority of cells lying in the neurogenic areas of mouse brain. Some cells selectively express CRHRs (black arrowheads) or nestin/GFAP (white arrowheads). Co-expression of CRHRs (green) and the neural progenitor markers Mash1 (red, d–d′) and neuroblast marker DCX (red, e–e′) in several areas is shown. (f) PCR analysis revealed mRNA expression of both CRH-R1 and CRH-R2 in neural stem/progenitor cells (NS/PCs). Adult brain mRNA was used as positive control. (g) Detection of CRH receptors in NS/PCs isolated from E13.5 embryonic forebrain. Co-expression of CRHRs and the neural progenitor marker nestin. While the majority of neural progenitor cells express both CRHRs and nestin (white arrows), some cells are selectively positive only to nestin (yellow arrows). 4′6-diamidino-2-phenylindole (DAPI) was used as counterstain. Scale bar=100 μm (a), 50 μm (b–e), 200 μm (g). Figures in the right panel (a′, g′) or lower panels (b′–e′) are magnifications of the figures shown in the left or upper panel, respectively. aSVZ, anterior SVZ; CP, cortical plate; LV, lateral ventricle; pia, pial surface; RMS, rostral migratory stream.
The above findings suggested the possibility that the CRH/CRHRs system may be involved in neurogenesis. We tested this hypothesis in primary cultures of NS/PCs isolated from the cortex of embryonic day 13.5 mouse brain. We first identified expression of CRH receptors in NS/PCs, by reverse transcriptase PCR (Figure 2f) and by immunocytochemistry (Figure 2g–g′). As shown by double staining for nestin and CRHRs, the great majority of nestin-positive NS/PCs (80%) co-express CRHRs (Figure 2g–g′).
CRH blocks glucocorticoid toxicity in NS/PCs and exerts glucocorticoid-independent neuroprotective effects
Glucocorticoid is one of the hormonal factors mostly studied as a negative regulator of neurogenesis via direct pro-apoptotic and, to a lesser extent, anti-proliferative effects on NS/PCs.38 Glucocorticoid release in dependent on CRH activation, an effect severely compromised in Crh− and Crhr1− null mice33, 39 To evaluate the possibility that CRH is involved in the effects of glucocorticoid on NS/PCs, we studied first the proliferation of glucocorticoid-exposed NS/PCs, as revealed by BrdU incorporation. Treatment of NS/PCs with the synthetic glucocorticoid dexamethasone as expected reduced their proliferation by 50%, while co-treatment with CRH (10−7 M) for 24 h abolished the suppressive effect of glucocorticoid (Figures 3a and b). Interestingly, treatment with CRH alone increased the abundance of BrdU-positive NS/PCs, by ∼30% as compared with vehicle treatment (Figures 3a and b). It should be mentioned that according to a recent study, the concentration of CRH in hippocampus can reach 200 nM during stress.23, 40 Next, we evaluated the impact of co-treatment with CRH on the glucocorticoid-induced NS/PCs’ apoptosis by TUNEL assay. As shown, CRH was sufficient to protect NS/PC from dexamethasone-induced apoptosis (Figures 3c and d). Taken together, these results suggest that CRH exerts direct, glucocorticoid-independent effects on mouse NS/PCs. Furthermore, CRH may counteract the negative effect of glucocorticoid on proliferation and survival of NS/PCs by its direct neuroprotective actions.

Effect of CRH in dexamethasone-treated NS/PCs proliferation and apoptosis. (a) Representative figures of 5-bromo-2-deoxyuridine (BrdU)-labeled cells (red) counted 24 h after treatment with or without CRH after pretreatment with dexamethasone. Dexamethasone was added in cell culture 1 h before CRH. 4′6-diamidino-2-phenylindole (DAPI) staining was applied for visualization of total cell abundance. Scale bar=200 μm. (b) Graph that depicts total number of the BrdU-positive cells count in 180 × 180 μm2 areas. Data are shown as mean±s.e.m. (n=4). *P<0.01, **P<0.01 versus non-CRH-treated cells. (c) Effect of CRH on dexamethasone-treated NS/PCs apoptosis induced by serum deprivation for 24 h.Representative images of terminal transferase dUTP nick-end labeling (TUNEL)-stained NS/PCs (red) combined with DAPI nuclear staining (blue) after treatment with or without CRH and/or dexamethasone. Scale bar=200 μm. (d) Graph depicts quantification of the TUNEL-positive cells count in 180 × 180 μm2 areas. Data represent the mean±s.e.m. (n=4). *P<0.05, **P<0.01.
CRH stimulates NS/PCs proliferation and blocks apoptosis via CRH-R1 receptor
To determine which CRH receptor(s) mediate the effects of CRH on NS/PCs, we employed specific antagonists, a very good tool available, especially given the limited specificity of the commercially available antibodies for each specific CRH receptor subtype. We applied the non-peptide CRH-R1 antagonist, antalarmin, or the CRH-R2 antagonist, astressin 2b, 30 min before CRH treatment. When CRH was administrated together with astressin 2b, the total number of BrdU-positive cells was not affected compared with CRH-treated cells alone (Figures 4a–c). In contrast, when CRH was administered in the presence of the antalarmin, the proliferative effects of CRH were abolished (Figures 4a–c), suggesting a CRH-R1-specific effect. Next, we evaluated the effects of the two antagonists on the CRH-induced protection of NS/PCs from apoptosis. As shown by TUNEL assay, it was antalarmin only that reversed the effect of CRH. These findings demonstrate the specificity of the above effects of CRH on NS/PCs mediated by CRH-R1 alone (Figures 4b–d).

Effect of CRH in NS/PCs proliferation and apoptosis. (a) Representative figures of 5-bromo-2-deoxyuridine (BrdU)-labeled cells (red) counted 24 h after treatment with or without CRH. CRH receptor antagonists or signaling pathway blockers were added in cell culture 1 h before CRH. Antalarmin used as a specific antagonist for CRH-R1, astressin 2B (A2B) as a specific CRH-R2 antagonist, whereas PD98059 and wortmannin were used as blockers of the MAPK and PI3 K pathway, respectively. 4′6-diamidino-2-phenylindole (DAPI) staining was applied for visualization of total cell abundance. Scale bar=200 μm. (b) Effect of CRH on NS/PCs apoptosis induced by serum deprivation for 24 h. Representative images of terminal transferase dUTP nick-end labeling (TUNEL)-stained NS/PCs (red) combined with DAPI nuclear staining (blue) after treatment with or without CRH and/or specific CRH antagonists and signaling blockers as described for panel. Scale bar=200 μm. (c) Graph that depicts total number of the BrdU-positive cells count in 180 × 180 μm2 areas. Data are shown as mean±s.e.m. (n=4). *P<0.01, **P<0.01 versus non-CRH-treated cells. (d) Graph depicts quantification of the TUNEL-positive cells count in 180 × 180 μm2 areas. Data represent the mean±s.e.m. (n=4). *P<0.05, **P<0.01.
To further elucidate the molecular mechanisms mediating the aforementioned effects of CRH and their relevance in human cells, we used the human neuroblastoma cell line, SH-SY5Y.41 Similar to the effects on NS/PCs, CRH induced proliferation of SH-SY5Y cells, whereas analysis of the cell cycle profiles by fluorescence-activated cell sorting showed a higher percentage of cells in the S phase (Supplementary Figure S1A) and significant induction of the expression of cyclin D1 (Supplementary Figure S1B). Serum deprivation followed by TUNEL assay showed reduction of apoptotic bodies by ∼50%, and increased expression of caspase-3 (Supplementary Figure S1C) following CRH treatment. The above data support our findings in NS/PCs and indicate cyclin D1 and caspase-3 as significant factors in the CRH-mediated neuroprotection.
Distinct signaling pathways mediate the effects of CRH/CRH-R1 on NS/PCs
Binding of CRH to CRH-R1 induces the activation of cAMP and, in several cells and tissues, it is shown to engage additional signaling pathways, including mitogen-activated protein kinase (MAPK) and PI3K/Akt.20, 38 To elucidate the contribution of these pathways in the neuroprotective effects of CRH, we applied specific inhibitors. As shown, PD98059, a MAPK inhibitor, blocked the CRH-induced increase of BrdU-positive NS/PCs, while co-treatment with wortmanin, that blocks activation of the PI3 kinase, had no effect (Figures 4a and c). In contrast, wortmanin blocked the anti-apoptotic effect of CRH, whereas no effect of PD98059 was detected (Figures 4b and d). These findings demonstrate that two distinct intracellular signaling pathways, MAPK and PI3K/Akt, are specifically involved in the proliferative and anti-apoptotic effects of CRH/CRH-R1 on NS/PCs.
Expression of CRH receptors in proliferating zones of the developing human brain
To assess the potential implications of our findings in humans, we assessed the expression of CRH receptors in the developing human brain. We performed immunostainings with specific antibody that recognizes both CRH receptors (CRHRs). As shown, we detected wide expression of CRHRs in the fetal human brain from 13 weeks embryo (Figure 5a). Most importantly, there were high levels of expression staining in the ventricular and SVZ (Figures 5b–b′), both rich in proliferating cells as revealed by staining for Ki67 (Figure 5c). Our findings demonstrate for the first time CRH receptors in the human brain in the proliferating neuronal cells from early fetal age and provide evidence for the possible relevance of our findings in human neurogenesis.

Expression of CRH receptors (CRHRs) in fetal human brain. (a) Detection of CRH receptors in differentiated neurons of the primordial plexiform layer (PPL) of the thalamus at 13 gestational week (arrows). (b) In the ventricular (VZ) and subventricular (SVZ) neurogenic areas there are many CRH receptor-expressing cells belonging to the neural stem/progenitor cells population as revealed by the Ki67 immunoreactivity in consecutive sections (c). Scale bar=100 μm (a), 100 μm (b, c). Figure b′ is a magnification of figure b. VZ, ventricular zone.
Discussion
In this study, we demonstrate that CRH, identified as the major mediator of the stress response and glucocorticoid release in mammals, exerts protective effects on mouse neural progenitors/stem cells. We show that CRH receptors are expressed in neurogenic areas of fetal and adult mouse brain and that CRH-R1 mediates the above effects of CRH, via activation of distinct signaling pathways, MAPK and PI3K. We also provide evidence that CRH can oppose the neurotoxic effects of excess glucocorticoid on neuronal progenitors. Finally, we demonstrate that CRHRs are expressed in the dividing human fetal brain cells.
We found compromised neuronal proliferation and increased rates of apoptosis in the Crh−/− fetal mouse brain as compared with wild-type tissue (Figure 1). CRH receptors are broadly expressed in the developing mouse neuroepithelium, particularly in actively proliferating, nestin-positive NS/PCs (Figure 2), around the time window that neurogenesis occurs. Significant expression of CRH receptors in neurogenic niches persisted in adult brain, with the strongest staining in the SVZ/rostral migratory stream. This area is considered critical for recovery from ischemia, generation of GABAergic neurons and olfaction.41, 42 As shown (Figure 2), a good proportion of all distinct cell types in the lineage of adult neurogenesis, including GFAP+, Nestin+, Mash1+ and DCX+ cells, were also positive for CRHRs staining. The high abundance of CRH receptors in the mouse cortical neurogenic areas (Figure 2) and the altered cell proliferation and apoptosis in the same areas of the Crh−/− brain (Figure 1) suggested the possibility that CRH may have direct effects on NS/PCs. Indeed, culture of NS/PCs in the presence of CRH resulted in dramatic enhancement, by ∼60%, of the proportion of proliferating cells (Figures 3 and 4). It seems that CRH exerts a dual effect on NS/PCs, by increasing the proportion of cells that will remain in the proliferating state and enhancing their ability to overcome apoptosis (Figure 4). Although by reverse transcriptase PCR both CRH receptors are expressed in NS/PCs, it appears that the neuroprotective effects of CRH on these cells are mediated by CRH-R1, as they were completely abolished by co-treatment with antalarmin, a CRH-R1-specific antagonist (Figure 4). Recent studies report different effects of CRH acting via CRH-R1 on proliferation and survival of cells from a variety of origins. Thus, both inhibition of the proliferation of epidermal keratinocytes,43, 44 human breast cancer and endometrial adenocarcinoma cells,45 and stimulation of the proliferation of β-cells in the pancreas31 were shown. In our hands, CRH induced proliferation and protected from apoptosis the human neuroblastoma SH-SY5Y cells, that express CRHRs and have been used to study neuroprotection (Supplementary Figure S1). Several reasons may account for the discrepancies between the reported effects of the CRH/CRH-R1 on cell proliferation, such as tissue-specific factors, co-expression of CRH-R2 and/or additional ligands, the cell type, that is, primary versus stable cell line.
Neural mitogenic signaling has been primarily associated with the activation of tyrosine kinase receptors,46, 47, 48 whereas emerging evidence points to similar effects following activation of several GPCRs.49, 50 CRH receptors belong to the class B subfamily of GPCRs that in addition to cAMP activation, in several tissues or cells, act through induction of NFκB, MAPK and PI3K.51, 52, 53 Modulation of neuronal survival analogous to that we found for CRH has been described for PACAP, VIP and MIP-254, 55 ligands of other GPCRs. Our present data suggest that the dual effects, mitogenic and anti-apoptotic, of CRH on neural progenitor populations are specific and achieved via distinct signal transduction pathways, MAPK and PI3K, respectively (Figure 4). These findings were replicated in the SH-SY5Y cells, that provided a tool to show specific effects of CRH in cell cycle events and induction of cyclin D1 (Supplementary Figure S1). A similar mechanism has been postulated for the neuroprotective effects of VEGF.56 Using the same system, we found inhibition of the activation of caspase-3 by CRH (Supplementary Figure S1), similar to previous reports on VIP and PACAP.54
Neuroprotective effects of CRH/CRH-R1 have been described in primary and permanent cell lines as well as in cultured brain slices,20 following exposure to neurotoxic agents such as amyloid,30 glutamate and lipid peroxides or hypoxia.57, 58 On the other side, there is a body of reports on the hazardous effects of CRH/CRHR1 in the brain in association with chronic stress and the corresponding increase in glucocorticoid.59 Glucocorticoid receptors are expressed in the developing neuroepithelium, and the proliferation of NS/PCs has been decreased dramatically following dexamethasone or corticosterone treatment.60, 61 Furthermore, high levels of glucocorticoid has been considered as the major cause of the stress-induced neuronal death,60, 62 evidenced, for example, by reduction of the volume of the dentate gyrus during chronic stress.63 Here, we demonstrate blockade of these effects of glucocorticoid following co-treatment with CRH (Figure 3). Based on the above, it is possible that CRH and glucocorticoid have exerted opposing effects on the proliferation of NS/PCs. Along these lines, it was recently shown glucocorticoid inhibit cyclin D1,60, 61 an effect opposite to our current findings with CRH.
To our knowledge, this is the first study that demonstrates specific effects of CRH on physiological neurogenesis and the mechanisms involved. In agreement with our results, it has been shown that connexin 43, a factor expressed in fetal brain and involved in neurogenesis, mediates the neuroprotective effects of CRH.64 It is in support of the intriguing possibility of the applicability of our findings in human neurogenesis, the identification of CRHR in human fetal dividing neurons (Figure 5). Furthermore, detailed analysis of the expression of the CRHR1 and CRHR2 genes in human tissues showed expression for both in the hippocampus to levels similar or even higher than in the amygdala, one of the main areas for CRH action.65 In a recent study, looking at the effect of CRH specifically in hippocampal pyramidal cells, the authors suggested that physiological release of low levels of CRH seem to be required for normal function of differentiated neurons.40
In summary, our findings demonstrate stimulatory effects of CRH on mouse neurogenesis and indicate a direct homeostatic role for CRH in antagonizing the negative effects of glucocorticoid in neuronal survival. Our working hypothesis is that CRH exerts direct, beneficial effects on neuronal progenitors, via its specific receptor CRH-R1. These effects are unmasked in states of severe stress owing to the prolonged and significant rise in glucocorticoid and the associated inhibition of CRH expression.66 More studies are needed to provide further insights on the role of this peptide in human neuronal stem and progenitor cells. Our study raises the possibility for potential therapeutic application of CRH/CRHR1 in the treatment of brain and neurodegenerative disorders by support of specific neuronal actions.
References
Nakada D, Levi BP, Morrison SJ . Integrating physiological regulation with stem cell and tissue homeostasis. Neuron 2011; 70: 703–718.
Sharp FR, Liu J, Bernabeu R . Neurogenesis following brain ischemia. Brain Res Dev Brain Res 2002; 134: 23–30.
Geraerts M, Krylyshkina O, Debyser Z, Baekelandt V . Concise review: therapeutic strategies for Parkinson disease based on the modulation of adult neurogenesis. Stem Cells 2007; 25: 263–270.
Hitoshi S, Maruta N, Higashi M, Kumar A, Kato N, Ikenaka K . Antidepressant drugs reverse the loss of adult neural stem cells following chronic stress. J Neurosci Res 2007; 85: 3574–3585.
Beauquis J, Saravia F, Coulaud J, Roig P, Dardenne M, Homo-Delarche F et al. Prominently decreased hippocampal neurogenesis in a spontaneous model of type 1 diabetes, the nonobese diabetic mouse. Exp Neurol 2008; 210: 359–367.
Stranahan AM, Arumugam TV, Cutler RG, Lee K, Egan JM, Mattson MP . Diabetes impairs hippocampal function through glucocorticoid-mediated effects on new and mature neurons. Nat Neurosci 2008; 11: 309–317.
Li G, Pleasure SJ . Ongoing interplay between the neural network and neurogenesis in the adult hippocampus. Curr Opin Neurobiol 2010; 20: 126–133.
Zhao C, Deng W, Gage FH . Mechanisms and functional implications of adult neurogenesis. Cell 2008; 132: 645–660.
Blaschke AJ, Staley K, Chun J . Widespread programmed cell death in proliferative and postmitotic regions of the fetal cerebral cortex. Development 1996; 122: 1165–1174.
Tanapat P, Hastings NB, Gould E . Ovarian steroids influence cell proliferation in the dentate gyrus of the adult female rat in a dose- and time-dependent manner. J Comp Neurol 2005; 481: 252–265.
Tanapat P, Hastings NB, Reeves AJ, Gould E . Estrogen stimulates a transient increase in the number of new neurons in the dentate gyrus of the adult female rat. J Neurosci 1999; 19: 5792–5801.
Cameron HA, Gould E . Adult neurogenesis is regulated by adrenal steroids in the dentate gyrus. Neuroscience 1994; 61: 203–209.
Karishma KK, Herbert J . Dehydroepiandrosterone (DHEA) stimulates neurogenesis in the hippocampus of the rat, promotes survival of newly formed neurons and prevents corticosterone-induced suppression. Eur J Neurosci 2002; 16: 445–453.
Baud O, Verney C, Evrard P, Gressens P . Injectable dexamethasone administration enhances cortical GABAergic neuronal differentiation in a novel model of postnatal steroid therapy in mice. Pediatr Res 2005; 57: 149–156.
Wong EY, Herbert J . The corticoid environment: a determining factor for neural progenitors’ survival in the adult hippocampus. Eur J Neurosci 2004; 20: 2491–2498.
Wong EY, Herbert J . Raised circulating corticosterone inhibits neuronal differentiation of progenitor cells in the adult hippocampus. Neuroscience 2006; 137: 83–92.
Cannon W . Bodily Changes in Pain, Hunger, Fear and Rage (2nd edn). Harper & Row: New York, 1929.
Selye H . Stress in Health and Disease. Butterworths: Reading, MA, 1976.
Vale W, Spiess J, Rivier C, Rivier J . Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science 1981; 213: 1394–1397.
Bayatti N, Zschocke J, Behl C . Brain region-specific neuroprotective action and signaling of corticotropin-releasing hormone in primary neurons. Endocrinology 2003; 144: 4051–4060.
Keegan CE, Herman JP, Karolyi IJ, O'Shea KS, Camper SA, Seasholtz AF . Differential expression of corticotropin-releasing hormone in developing mouse embryos and adult brain. Endocrinology 1994; 134: 2547–2555.
Bishop GA, King JS . Corticotropin releasing factor in the embryonic mouse cerebellum. Exp Neurol 1999; 160: 489–499.
Chen Y, Bender RA, Brunson KL, Pomper JK, Grigoriadis DE, Wurst W et al. Modulation of dendritic differentiation by corticotropin-releasing factor in the developing hippocampus. Proc Natl Acad Sci USA 2004; 101: 15782–15787.
Heinrichs SC, Koob GF . Corticotropin-releasing factor in brain: a role in activation, arousal, and affect regulation. J Pharmacol Exp Ther 2004; 311: 427–440.
Peeters PJ, Fierens FL, van den Wyngaert I, Goehlmann HW, Swagemakers SM, Kass SU et al. Gene expression profiles highlight adaptive brain mechanisms in corticotropin releasing factor overexpressing mice. Brain Res Mol Brain Res 2004; 129: 135–150.
Traver S, Marien M, Martin E, Hirsch EC, Michel PP . The phenotypic differentiation of locus coeruleus noradrenergic neurons mediated by brain-derived neurotrophic factor is enhanced by corticotropin releasing factor through the activation of a cAMP-dependent signaling pathway. Mol Pharmacol 2006; 70: 30–40.
De Souza EB, Battaglia G . Corticotropin-releasing hormone (CRH) receptors in brain. Adv Exp Med Biol 1988; 245: 123–136.
Martin B, Lopez de Maturana R, Brenneman R, Walent T, Mattson MP, Maudsley S . Class II G protein-coupled receptors and their ligands in neuronal function and protection. Neuromolecular Med 2005; 7: 3–36.
Fox MW, Anderson RE, Meyer FB . Neuroprotection by corticotropin releasing factor during hypoxia in rat brain. Stroke 1993; 24: 1072–1075.
Lezoualc’h F, Engert S, Berning B, Behl C . Corticotropin-releasing hormone-mediated neuroprotection against oxidative stress is associated with the increased release of non-amyloidogenic amyloid beta precursor protein and with the suppression of nuclear factor-kappaB. Mol Endocrinol 2000; 14: 147–159.
Huising MO, van der Meulen T, Vaughan JM, Matsumoto M, Donaldson CJ, Park H et al. CRFR1 is expressed on pancreatic beta cells, promotes beta cell proliferation, and potentiates insulin secretion in a glucose-dependent manner. Proc Natl Acad Sci USA 2010; 107: 912–917.
Chaniotou Z, Giannogonas P, Theoharis S, Teli T, Gay J, Savidge T et al. Corticotropin-releasing factor regulates TLR4 expression in the colon and protects mice from colitis. Gastroenterology 2010; 139: 2083–2092.
Muglia L, Jacobson L, Dikkes P, Majzoub JA . Corticotropin-releasing hormone deficiency reveals major fetal but not adult glucocorticoid need. Nature 1995; 373: 427–432.
Elkouris M, Balaskas N, Poulou M, Politis PK, Panayiotou E, Malas S et al. Sox1 maintains the undifferentiated state of cortical neural progenitor cells via the suppression of Prox1-mediated cell cycle exit and neurogenesis. Stem Cells 2011; 29: 89–98.
Remboutsika E, Elkouris M, Iulianella A, Andoniadou CL, Poulou M, Mitsiadis TA et al. Flexibility of neural stem cells. Front Physiol 2011; 2: 16.
Kaltezioti V, Kouroupi G, Oikonomaki M, Mantouvalou E, Stergiopoulos A, Charonis A et al. Prox1 regulates the notch1-mediated inhibition of neurogenesis. PLoS Biol 2010; 8: e1000565.
Venihaki M, Carrigan A, Dikkes P, Majzoub JA . Circadian rise in maternal glucocorticoid prevents pulmonary dysplasia in fetal mice with adrenal insufficiency. Proc Natl Acad Sci USA 2000; 97: 7336–7341.
Punn A, Levine MA, Grammatopoulos DK . Identification of signaling molecules mediating corticotropin-releasing hormone-R1alpha-mitogen-activated protein kinase (MAPK) interactions: the critical role of phosphatidylinositol 3-kinase in regulating ERK1/2 but not p38 MAPK activation. Mol Endocrinol 2006; 20: 3179–3195.
Timpl P, Spanagel R, Sillaber I, Kresse A, Reul JM, Stalla GK et al. Impaired stress response and reduced anxiety in mice lacking a functional corticotropin-releasing hormone receptor 1. Nat Genet 1998; 19: 162–166.
Maras PM, Baram TZ . Sculpting the hippocampus from within: stress, spines, and CRH. Trends Neurosci 2012; 35: 315–324.
Whitman MC, Greer CA . Adult neurogenesis and the olfactory system. Prog Neurobiol 2009; 89: 162–175.
Ming GL, Song H . Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron 2011; 70: 687–702.
Mitsuma T, Matsumoto Y, Tomita Y . Corticotropin releasing hormone stimulates proliferation of keratinocytes. Life Sci 2001; 69: 1991–1998.
Zbytek B, Slominski AT . Corticotropin-releasing hormone induces keratinocyte differentiation in the adult human epidermis. J Cell Physiol 2005; 203: 118–126.
Graziani G, Tentori L, Muzi A, Vergati M, Tringali G, Pozzoli G et al. Evidence that corticotropin-releasing hormone inhibits cell growth of human breast cancer cells via the activation of CRH-R1 receptor subtype. Mol Cell Endocrinol 2007; 264: 44–49.
Aberg MA, Aberg ND, Hedbacker H, Oscarsson J, Eriksson PS . Peripheral infusion of IGF-I selectively induces neurogenesis in the adult rat hippocampus. J Neurosci 2000; 20: 2896–2903.
Richardson RL, Hausman GJ, Gaskins HR . Effect of transforming growth factor-beta on insulin-like growth factor 1- and dexamethasone-induced proliferation and differentiation in primary cultures of pig preadipocytes. Acta Anat 1992; 145: 321–326.
Reynolds BA, Tetzlaff W, Weiss S . A multipotent EGF-responsive striatal embryonic progenitor cell produces neurons and astrocytes. J Neurosci 1992; 12: 4565–4574.
Cazillis M, Gonzalez BJ, Billardon C, Lombet A, Fraichard A, Samarut J et al. VIP and PACAP induce selective neuronal differentiation of mouse embryonic stem cells. Eur J Neurosci 2004; 19: 798–808.
Barzi M, Kostrz D, Menendez A, Pons S . Sonic Hedgehog-induced proliferation requires specific Galpha inhibitory proteins. J Biol Chem 2011; 286: 8067–8074.
Naor Z, Benard O, Seger R . Activation of MAPK cascades by G-protein-coupled receptors: the case of gonadotropin-releasing hormone receptor. Trends Endocrinol Metab 2000; 11: 91–99.
Patial S, Luo J, Porter KJ, Benovic JL, Parameswaran N . G-protein-coupled-receptor kinases mediate TNFalpha-induced NFkappaB signaling via direct interaction with and phosphorylation of IkappaB alpha. Biochem J 2009; 425: 169–178.
New DC, Wu K, Kwok AW, Wong YH . G protein-coupled receptor-induced Akt activity in cellular proliferation and apoptosis. FEBS J 2007; 274: 6025–6036.
Dejda A, Sokolowska P, Nowak JZ . Neuroprotective potential of three neuropeptides PACAP, VIP and PHI. Pharmacol Rep 2005; 57: 307–320.
Scharf E, May V, Braas KM, Shutz KC, Mao-Draayer Y . Pituitary adenylate cyclase-activating polypeptide (PACAP) and vasoactive intestinal peptide (VIP) regulate murine neural progenitor cell survival, proliferation, and differentiation. J Mol Neurosci 2008; 36: 79–88.
Zhu Y, Jin K, Mao XO, Greenberg DA . Vascular endothelial growth factor promotes proliferation of cortical neuron precursors by regulating E2F expression. FASEB J 2003; 17: 186–193.
Facci L, Stevens DA, Pangallo M, Franceschini D, Skaper SD, Strijbos PJ . Corticotropin-releasing factor (CRF) and related peptides confer neuroprotection via type 1 CRF receptors. Neuropharmacology 2003; 45: 623–636.
Madtes P, Lee KH, King JS, Burry RW . Corticotropin releasing factor enhances survival of cultured GABAergic cerebellar neurons after exposure to a neurotoxin. Brain Res Dev Brain Res 2004; 151: 119–128.
Ivy AS, Rex CS, Chen Y, Dube C, Maras PM, Grigoriadis DE et al. Hippocampal dysfunction and cognitive impairments provoked by chronic early-life stress involve excessive activation of CRH receptors. J Neurosci 2010; 30: 13005–13015.
Sundberg M, Savola S, Hienola A, Korhonen L, Lindholm D . Glucocorticoid hormones decrease proliferation of embryonic neural stem cells through ubiquitin-mediated degradation of cyclin D1. J Neurosci 2006; 26: 5402–5410.
Bose R, Moors M, Tofighi R, Cascante A, Hermanson O, Ceccatelli S . Glucocorticoids induce long-lasting effects in neural stem cells resulting in senescence-related alterations. Cell Death Dis 2010; 1: e92.
Li WZ, Li WP, Yao YY, Zhang W, Yin YY, Wu GC et al. Glucocorticoids increase impairments in learning and memory due to elevated amyloid precursor protein expression and neuronal apoptosis in 12-month old mice. Eur J Pharmacol 2010; 628: 108–115.
Yu S, Patchev AV, Wu Y, Lu J, Holsboer F, Zhang JZ et al. Depletion of the neural precursor cell pool by glucocorticoids. Ann Neurol 2010; 67: 21–30.
Hanstein R, Trotter J, Behl C, Clement AB . Increased connexin 43 expression as a potential mediator of the neuroprotective activity of the corticotropin-releasing hormone. Mol Endocrinol 2009; 23: 1479–1493.
Hiroi N, Wong ML, Licinio J, Park C, Young M, Gold PW et al. Expression of corticotropin releasing hormone receptors type I and type II mRNA in suicide victims and controls. Mol Psychiatry 2001; 6: 540–546.
Opendak M, Gould E . New neurons maintain efficient stress recovery. Cell Stem Cell 2011; 9: 287–288.
Acknowledgements
This work was supported by intramural funding of BRFAA to KPK and PKP and a Regpot Grant (TranSMed) from the EU (KPK).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest
Additional information
Supplementary Information accompanies the paper on the Molecular Psychiatry website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Koutmani, Y., Politis, P., Elkouris, M. et al. Corticotropin-releasing hormone exerts direct effects on neuronal progenitor cells: implications for neuroprotection. Mol Psychiatry 18, 300–307 (2013). https://doi.org/10.1038/mp.2012.198
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mp.2012.198
Keywords
This article is cited by
-
Shared molecular genetic factors influence subcortical brain morphometry and Parkinson’s disease risk
npj Parkinson's Disease (2023)
-
Stress-inducible-stem cells: a new view on endocrine, metabolic and mental disease?
Molecular Psychiatry (2019)
-
Corticotropin-releasing factor 1 receptor haplotype and cognitive features of major depression
Translational Psychiatry (2018)
-
cAMP-dependent cell differentiation triggered by activated CRHR1 in hippocampal neuronal cells
Scientific Reports (2017)
-
The Placenta as a Mediator of Stress Effects on Neurodevelopmental Reprogramming
Neuropsychopharmacology (2016)

{kind=link}