
Abstract
Rare B-cell neoplasms with plasmablastic differentiation may aberrantly express CD3 by immunohistochemical staining, which places a great challenge for diagnosis. We here studied 17 cases of CD3+ plasmablastic B-cell neoplasms, including 12 plasmablastic lymphomas and 5 plasmablastic plasma cell myelomas. All 17 cases occurred in the extranodal sites with a male predominance (13/17). Four cases were initially misinterpreted by outside institutions, among which three were diagnosed as ‘peripheral T-cell lymphoma, not otherwise specified’ and one was classified as ‘poorly differentiated neuroendocrine carcinoma’. The plasmablastic cells were present in all 17 cases diffusely or in a subset of tumor cells. CD3 expression was mostly diffuse (12/17) and moderate to strong (11/16) with a cytoplasmic staining pattern (14/16). Other T-cell markers were nearly absent, including CD2 (0/10), CD4 (1/13), CD5 (0/14), CD7 (0/11), and CD8 (0/13). CD138 was positive in all 17 cases and CD79a was variably positive in 8 of 14 cases. Only one case had immunoreactivity to CD20 (1/17) and PAX5 (1/12). CD56 expression and EBV infection were detected in 8/15 and 6/17, respectively. No HHV8 infection was noted in all 11 cases tested. Most cases (11/13) revealed either kappa or lambda light chain restriction. Of the nine cases studied, six had clonal IGH rearrangements but no clonal TRG rearrangements. Our study further emphasizes that the accurate classification of CD3+ plasmablastic neoplasms requires thorough morphologic examination, incorporation of more B-cell and T-cell markers in addition to CD3 and CD20, frequent addition of CD138 staining, and utilization of necessary molecular and genetic studies.
Similar content being viewed by others
Main
Lineage assignment and classification of non-Hodgkin lymphomas rely heavily on T-cell and B-cell markers, determined by immunohistochemical and/or flow cytometric assays. However, B-cell neoplasms may aberrantly express T-cell markers, particularly expression of CD5 and CD2 in chronic lymphocytic leukemia/small lymphocytic lymphoma and mantle cell lymphoma.1, 2 CD2, CD5, CD7, and CD8 were also occasionally detected in diffuse large B-cell lymphoma.2, 3 Based on our recent study, 53% cases of ALK-positive large B-cell lymphoma expressed CD4, in addition to CD57 positivity in 26% of cases.4 Likewise, rare T-cell lymphomas may have aberrant expression of one or more B-cell markers, including CD19, CD20, and PAX5.5, 6
Aberrant CD3 expression has rarely been described in mature B-cell neoplasms, including diffuse large B-cell lymphoma, classic Hodgkin lymphoma, and follicular lymphoma.3, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18 CD3 expression in CD20+ B-cell lymphomas is usually not problematic since co-expression of these two markers will prompt further workup with more lineage-specific markers and molecular studies. In another study, we noticed that 6–29% cases of primary effusion lymphoma and its solid variant, HHV8+ large B-cell lymphoma, aberrantly expressed CD3;19 however, presence of HHV8 infection in the lymphoma cells provides a strong evidence for primary effusion lymphoma in HIV+ patients.
Rare cases of plasmablastic lymphoma and plasmablastic plasma cell neoplasm have been reported to be CD3+.8, 13, 14, 15, 16, 20 The accurate diagnosis of these cases can be difficult since they are typically negative for common B-cell markers (CD20, CD79a, and PAX5). It is particularly challenging in cases with no or only subtle plasmacytic or plasmablastic morphology, and they can potentially be misclassified as ‘peripheral T-cell lymphoma, not otherwise specified’. Moreover, plasmablastic neoplasms often have CD56 expression and EBV infection, and with co-expression of CD3 they can be mistaken for ‘extranodal NK/T-cell lymphoma, nasal-type’. Therefore, in this study we focus on the CD3+ plasmablastic neoplasms, specifically plasmablastic lymphoma and plasmablastic plasma cell neoplasm, with emphasis on the clinical presentations, morphology, and immunophenotype. We hope to provide further knowledge and draw more attention to this aberrant expression to avoid misinterpretation.
Materials and methods
Case Selection, Specimen Processing, and Immunohistochemical Staining
A total of 17 cases of CD3+ plasmablastic neoplasm were retrieved from multiple institutions, including 12 plasmablastic lymphomas and 5 plasma cell myelomas with plasmablastic morphology. CD3 expression in all 17 cases was detected by immunohistochemical staining on the tissue sections. Two previously reported plasmablastic lymphomas were included in this study with further emphasis on the morphologic and CD3 staining features.7 Diagnoses of all cases were confirmed with sufficient clinical data, immunohistochemical stains, and molecular studies. Classification of these plasmablastic neoplasms was based on the ‘2016 Revised World Health Organization Classification of Lymphoid Neoplasms’.21
The tissue for each case was fixed in 10% buffered formalin, embedded in paraffin, and subsequently sectioned at 4.0 μm for hematoxylin & eosin staining and immunohistochemical studies. Immunohistochemical stains were performed on a Dako Autostainer with Envision (+) Detection Kit (Dako, Carpinteria, CA, USA) with satisfactory negative and positive controls. The antibodies, clones, dilutions, and manufacturers are summarized in Table 1. Expression of CD3 in the tumor cells was evaluated based on the staining patterns (membranous and cytoplasmic), intensity (1+, weak; 2+, moderate; and 3+, strong), and percentage of positive cells (0–100%). Ki67 was utilized to assess the proliferation rate of tumor cells (0–100%). The immunostains of other antibodies were graded as: negative (−), weakly and/or focally positive (−/+), and diffusely and strongly positive (+).
In Situ Hybridization
Digoxigenin-labeled probes (DAKO) were used to detect Epstein–Barr virus (EBV)-encoded small RNA-1 and -2 (EBER-1/2) for EBV infection, immunoglobulin kappa light chain (IGK), and immunoglobulin lambda light chain (IGL). The assays were performed on a Ventana Benchmark XT platform with an iViewBlue detection kit according to the manufacturer’s manual (Ventana Medical Systems, Tucson, AZ, USA).
Molecular Assays for Gene Rearrangements
Polymerase chain reaction (PCR) assays were utilized to detect gene rearrangements of immunoglobulin heavy chain (IGH) and T-cell receptor gamma (TRG). Briefly, genomic DNA was extracted from formalin-fixed and paraffin-embedded tissues. Then, the gene rearrangement products for IGH and TRG were amplified by PCR and analyzed on an ABI 310 Genetic Analyzer (Life Technologies, Carlsbad, CA, USA) with appropriate positive and negative controls. For IGH gene rearrangements, 3 sets of primers (frameworks IIA, IIB and IIIA) were used. The primers were specific for the variable and joining regions of the IGH gene, and the sequences were published previously.22 For TRG gene rearrangements, four sets of primers specific for different gamma variable regions were paired with different joining region primers of the TRG gene.23
Results
Clinical Information
The clinical information of the 17 patients is summarized in Table 2. Average age was 58.3 years, ranging from 16 to 94 years. Male patients comprised the majority of the cases (13 of 17). Two of twelve patients tested for HIV were positive (cases 8 and 14), and additional three patients had immunosuppression or dysfunction due to renal transplant (cases 1 and 6) or Wiskott–Aldrich syndrome (case 9). All cases were initially diagnosed in the extranodal sites, including nasal cavity (6 cases), soft tissue (5 cases), gastrointestinal tract (3 cases), and bone (1 case). Two patients initially presented with pleural effusions (cases 4 and 13). Imaging studies and/or bone marrow biopsies were available in 14 patients, and 6 had positive bone scan (cases 5, 6, 14, 15, 16, and 17) including two with current bone marrow involvement (cases 6 and 16). Five of 13 patients (cases 5, 6, 7, 16, and 17) had clonal serum M protein and urine-free light chain demonstrated by serum protein electrophoresis and urine protein electrophoresis. In the 12 patients with clinical information of management, 8 received chemotherapy, local radiation and/or stem cell transplant. The remaining 4 patients had no such treatments due to palliative care for advanced disease or death before initiation of therapy. Totally 5 of 13 patients died of disease with an average follow-up time of 11.9 months.
Pathology Findings
Morphologic features and diagnoses
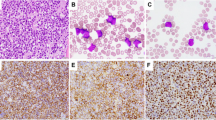
The 17 cases of plasmablastic neoplasm showed a variety of morphologic features with at least a subset of plasmablasts present in all cases. The cases with pure plasmablastic proliferation contained solid sheets of tumor cells, which had a large round nucleus with one prominent nucleolus (Figures 1a and b). The cytoplasm was amphophilic with no perinuclear clearing. The cases with mixed plasmacytic and plasmablastic features revealed a spectrum of morphology; the plasmablasts were variable in number and distributed in a background of atypical plasmacytic cells (Figure 1c). The plasmacytic cells were smaller than plasmablasts and had a round nucleus with clumped chromatin and often perinuclear clearing. On the Wright–Giemsa-stained smears, the plasmablasts possessed increased nucleus to cytoplasm ratio with a round nucleus, moderately condensed chromatin, and a prominent nucleolus (Figure 1d). In addition to plasmablasts, two cases displayed mixed immunoblasts (cases 3 and 4) and three other cases had mixed anaplastic cells (cases 5, 12, and 16).

Morphologic features of plasmablastic neoplasms. (a) A CD3+ plasmablastic lymphoma in the distal jejunum with transmural infiltration (Case 1; H&E, original magnification × 20) by sheets of plasmablastic cells (b) (H&E, × 600). (c) A plasma blastic lymphoma with mixed neoplastic plasmacytic cells and plasmablasts (H&E, × 600). (d) A CD3+ plasma cell myeloma with diffuse plasmablasts on the bone marrow aspirate smear (Wright-Giemsa, × 400). (e) The diffuse large B-cell lymphoma in the proximal jejunum and distal ileum of case 1 showed extensive involvement (H&E, × 20) by mostly centroblastic cells (f) (H&E, × 600).
The diagnoses for all 17 cases were confirmed in our institutions, and 12 cases were classified as ‘plasmablastic lymphoma’ (cases 1–4, 8–11, and 13–15) (Table 2). Four cases were initially misinterpreted by outside institutions (Table 2), as ‘peripheral T-cell lymphoma, not otherwise specified’ in three cases (cases 2, 8, and 12) and ‘poorly differentiated neuroendocrine carcinoma’ in one case (case 3). The colectomy specimen in case 2 had significant autolysis due to delayed specimen fixation at the outside institution. Only a thin rim of tumor cells in the periphery of specimen revealed plasmablastic cytology. The tumor cells in case 12 showed mixed anaplastic and plasmablastic features. Therefore, the lack of apparent plasmacytic or plasmablastic morphology may mislead initial workup and then diagnoses in these cases. The patient in case 15 had a history of chronic lymphocytic leukemia for 5.5 years. However, it was not certain whether the chronic lymphocytic leukemia was clonally related to the plasmablastic lymphoma in this case, although direct transformation from chronic lymphocytic leukemia into plasmablastic lymphoma has rarely been reported.24
One case was of particular interest as it showed two distinct but clonally related lymphomas. The patient in case 1 had a history of renal transplant and developed post-transplant lymphoproliferative disorder 17 years later. The single lesion in the distal jejunum revealed plasmablastic lymphoma containing purely plasmablasts (Figures 1a and b); the tumor cells expressed CD3 and CD138 but not CD20 or CD45 (Figures 2a–d) (Table 3). However, the two masses in the proximal jejunum and distal ileum showed diffuse large B-cell lymphoma with mostly centroblastic cells (Figures 1e and f) but no plasmacytic or plasmablastic cells. Interestingly, the lymphoma cells had a different immunophenotype, with strong positivity of CD3, CD20, CD45, and other B-cell markers but not CD138 (Figures 3a–d) (Table 3).

The plasmablastic lymphoma in case 1 had cytoplasmic positivity of CD3 (a) but no expression of CD20 (b) or CD45 (c). CD138 was diffusely positive in the tumor cells (d). (a–d, × 400).

The diffuse large B-cell lymphoma in case 1 co-expressed CD3 with a membranous staining pattern (a), CD20 (b), and CD45 (c) but not CD138 (d). (a–c, × 400; d, × 200).
Five cases were classified as plasma cell myeloma with plasmablastic morphology (cases 5, 6, 7, 16, and 17). Case 5 had a refractory plasma cell myeloma for 9 years and case 6 had a history of monoclonal gammopathy of undetermined significance before renal transplant. Therefore, in both cases the current aggressive plasma cell myelomas likely represented high-grade transformation of the prior tumors. High-grade transformation of plasma cell neoplasm has been report previously.8, 20 For this reason, case 6 was not considered as ‘post-transplant lymphoproliferative disorder’ since the plasma cell myeloma occurred before the renal transplant. The patient in case 7 initially presented with a colonic polyp with no overt plasmacytic or plasmablastic morphology. Notably, the tumor cells had strong expression of CD20 and cyclin D1, closely resembling ‘blastoid mantle cell lymphoma’. Surprisingly, further immunostains showed positive expression of CD138 but negative for CD19, CD45, and SOX11, thus supporting a diagnosis of plasma cell neoplasm. The patient was then found to have bone marrow involvement and monoclonal serum M protein. The patient in case 16 had multiple lytic bone lesions and serum IgA lambda M-protein, and the tumor cells displayed mixed anaplastic and plasmablastic features. The patient in case 17 initially presented with a pathologic fracture of the L5 vertebral body. Further clinical images studies showed multiple lytic bone lesions in the L5 vertebra, S1 vertebra, right rib, mid sternum, and left proximal femur. Biopsy of the L5 lesion demonstrated a diffuse infiltration by plasmablastic cells.
Immunohistochemistry and in situ hybridization
The data of the immunohistochemical stains and in situ hybridization are summarized in Table 3. Of the 16 cases with detailed evaluation of CD3 staining, 5 cases had a weak (1+) or weak to moderate (1+/2+) positivity, and 11 cases had at least a moderate (≥2+) staining (Figures 4a and b). A diffuse expression (>75% of tumor cells) was noted in 12 of 17 cases (Figure 4b). Most cases (14/16) had a cytoplasmic CD3 staining pattern (Figures 4a and b) including 3 with concurrent membranous staining. While CD3 was positive in all 17 cases, other T-cell markers were essentially absent, including CD2 (0/10), CD5 (0/14), CD7 (0/11), and CD8 (0/13), except focal CD4 expression in 1 of 13 cases. Only one case (case 7) had expression of CD20 (1/17) and PAX5 (1/12), and all other cases stained were negative for these two markers (Figure 4c). Although CD79a was positive in 8 of 14 cases, the staining was generally focal and weak as noted in 4 of 8 positive cases. CD138 and MUM1 were positive in all 17 and 13 cases stained, respectively (Figure 4d). CD56 was detected in 8 of 15 cases with strong expression in four cases (Figure 4e). CD45 was positive in 8 of 13 cases. Three of fifteen cases also showed a variable expression of CD30. Most cases (11/13) demonstrated either kappa or lambda light chain expression by in situ hybridization and/or immunohistochemical staining (Figures 4f and g). Of the 13 cases stained for Ki67, 12 had a high proliferation rate ≥70% and the remaining one had 50%. None of the 11 cases tested for latency-associated nuclear antigen showed any evidence of HHV8 infection. EBV was detected in 6 of 17 cases by in situ hybridization for EBER.

Immunophenotype of plasmablastic neoplasms. The plasmablastic neoplasms mostly had a moderate (a) to diffuse and strong (b) cytoplasmic staining of CD3. CD20 was typically negative (c), whereas CD138 was positive in all 16 cases (d). CD56 was detected in a subset of plasmablastic neoplasms (e). (f, g) A plasmablastic neoplasm with kappa light chain restriction (f) with no expression of lambda light chain (g) by in-situ hybridization. (a, b, and d, × 400; c, e, and f, × 200).
Molecular studies for IGH and TRG gene rearrangements
PCR assays for IGH and TRG gene rearrangements were performed on nine cases. Six cases had clonal IGH rearrangements (cases 1, 3, 4, 7, 8, and 12). None of the nine cases showed clonal TRG rearrangements. The diffuse large B-cell lymphoma and plasmablastic lymphoma in case 1 had an identical peak at 140 bp on the IGH framework 3, indicating the clonal relationship of these two lymphomas (Figures 5a–d). Of the three cases with no clonal IGH rearrangements, case 13 showed indeterminate IGH rearrangement results. The negative finding in the remaining two cases was likely related to poor specimen quality. The colon in case 2 had prominent autolysis due to delayed fixation, and the specimen in case 14 had limited cells from the left-over after flow cytometric assays. Additional immunostains with BOB1 and OCT2 were performed on cases 2 and 14 to confirm the B-cell linage, which demonstrated strong positivity in both cases.

In case 1, the diffuse large B-cell lymphoma and plasmablastic lymphoma had clonal IGH rearrangements (Figures a and b, respectively) but no clonal TRG rearrangements (Figures c and d, respectively). The two lymphomas also revealed an identical peak at 140 bp on the IGH framework 3.
Discussion
Certain terminally differentiated B-cell neoplasms commonly show plasmablastic morphology, including plasmablastic lymphoma, plasmablastic plasma cell myeloma, primary effusion lymphoma, HHV8+ large B-cell lymphoma, and ALK+ large B-cell lymphoma. They have distinct but overlapping clinicopathologic features, which have been discussed in our previous publications.4, 19 Up to 29% cases of primary effusion lymphoma and HHV8+ large B-cell lymphoma have aberrant CD3 expression.19 We here specifically focus on the CD3+ plasmablastic lymphoma and plasma cell myeloma with plasmablastic morphology.
Summary of CD3+ Plasmablastic Neoplasms from Our Study and the Literature
The 17 cases of plasmablastic neoplasm in our study revealed plasmablastic morphology and a high proliferation rate. The CD3 expression was mostly moderate to strong with a cytoplasmic staining pattern. However, other T-cell-associated markers were nearly absent, including CD2, CD4, CD5, CD7, and CD8. Therefore, based on our study and the published data, CD3 expression in plasmablastic neoplasms was mostly in the cytoplasm and as a sole aberrant T-cell marker.7, 8 In contrast, aberrant CD3 expression in diffuse large B-cell lymphomas was often membranous, with frequent co-expression of one or multiple other T-cell markers.7, 8 In our case 1, CD3 indeed showed a cytoplasmic staining pattern in the plasmablastic lymphoma (Figure 2a) but a membranous pattern in the diffuse large B-cell lymphoma (Figure 3a), despite the fact that these two lymphomas were clonally related. In our study, most plasmablastic neoplasms had kappa or lambda light chain restriction and/or clonal IGH rearrangements with no clonal TRG rearrangements, which essentially confirmed the B-cell lineage and excluded the possibility of a T-cell neoplasm.
Additional 11 cases of CD3+ plasma cell neoplasm (7 cases) and plasmablastic lymphoma (4 cases) were retrieved from the literature,8, 13, 14, 15, 16, 20 which showed similar clinicopathologic features to those noted in our 17 cases. Characteristics of the 11 cases in the literature (also Table 4) were as follows: the patients were mostly male (10/11) with extranodal infiltration in all of them. The morphologic features included plasmablastic (5 cases), plasmacytic (3 cases), anaplastic (1 case), immunoblastic (1 case), and high-grade lymphoid (1 case). The plasmablastic neoplasms revealed a high-grade morphology with frequent mitoses and high proliferation indices. All 11 cases had a cytoplasmic CD3 staining pattern, and CD4 was detected in 4 of 8 cases. Other T-cell markers tested were all negative, including CD2 (0/8), CD5 (0/8), CD7 (0/7), and CD8 (0/6). CD138 and MUM1 were detected in all nine and eight cases reported, respectively. CD79a was positive in five of nine cases with a variable expression, whereas other B-cell markers were mostly negative, including CD20 (1/11) and PAX5 (1/9). Four of five cases were positive for CD56. EBV infection was detected in 4 of 10 cases. Molecular studies were performed in 4 cases, all of which were positive for IGH but not TRG gene rearrangements.
Difficulties in Diagnosis of CD3+ Plasmablastic Neoplasms and Advice to Avoid Misclassification
The accurate diagnosis of CD3+ plasmablastic neoplasms can be very challenging in the absence of common B-cell markers (CD20, CD79a, and PAX5). Particularly, these cases may potentially be misclassified as ‘peripheral T-cell lymphoma, not otherwise specified’, as noted in the three cases from our series and one in the literature.14 The difficulties in diagnosis are likely due to the following facts: plasmablastic neoplasms may not always show apparent plasmacytic and/or plasmablastic morphology. The tumor cells can display prominent immunoblastic and/or anaplastic features, as seen in our cases 3–5, 9, and 16. In addition, the commonly used needle core biopsies often constrain pathologists with limited amounts of tissue and marked biopsy artifacts. Furthermore, poor specimen fixation and suboptimal specimen processing may significantly compromise morphologic evaluation. The colon specimen in our case 2 had significant autolysis due to poor fixation, with only a thin rim of tumor cells in the periphery showing satisfactory morphology. Notably, plasmablastic neoplasms commonly express CD56 (totally 12/20 in our study and the literature), which can be easily misinterpreted as ‘NK/T-cell lymphoma, nasal type’, in conjunction with CD3 positivity and presence of EBV infection. Last and most importantly, CD138 is not commonly used during initial workup for high-grade neoplasms in some institutions, particularly in the cases already positive for CD3 or CD20. Without addition of CD138, our case 7 would likely have been rendered as ‘blastoid mantle cell lymphoma’ due to strong co-expression of CD20 and cyclin D1. If CD138 could have been included in the initial studies, the 3 cases in our study and the literature would have not been misclassified as ‘peripheral T-cell lymphoma, not otherwise specified’.
Therefore, a scrutinized morphologic examination is essential for the pathologists to search for evidences of plasmacytic and/or plasmablastic differentiation particularly on a high-grade lymphoid neoplasm. We also advocate using CD138 antibody during initial workup for a high-grade lymphoma, in addition to other lineage-specific markers. However, CD138 is also a sensitive marker for carcinoma, and co-expression of CD138 and CD56 may be seen in both plasmablastic neoplasms and neuroendocrine carcinomas. Therefore, for a poorly differentiated neoplasm with CD138 expression, other markers are necessary to confirm the cell origin, including more plasma cell markers, in situ hybridization for kappa and lambda light chains, and epithelial markers. Moreover, the staining quality of CD138 is sensitive to tissue autolysis and necrosis, similar to what noted in many nuclear antibodies; therefore, it should be very cautious to interpret CD138 staining during these circumstances. In our case 2 with prominent autolysis, although CD138 was incorporated in the initial workup, the tumor cells had an unusual granular cytoplasmic staining pattern, which was considered as ‘negative with nonspecific staining’ by the outside institution. However, we noticed that the reactive plasma cells in the mucosa also had a similar ‘granular’ staining pattern of CD138, which was clearly due to poor specimen quality. We hereby recommend frequent comparison of immunoreactivity of antigens on tumors with normal cells not just in poorly processed tissues, but in all tissues for different antibodies.
Furthermore, for a high-grade lymphoma with expression of CD3 or CD20, it may be necessary to add more T-cell and/or B-cell markers to confirm the lineage and to exclude the possibility of aberrant expression. We recently demonstrated that OCT2 and BOB1 are very sensitive and specific to confirm the B-cell lineage of poorly differentiated B-cell neoplasms without expression of common B-cell markers (CD20, CD79a, and Pax5).25
Possible Explanation of Aberrant CD3 Expression
The etiology of aberrant CD3 expression in B-cell neoplasms is still uncertain. Anti-CD3 antibody reacts to the cytoplasmic domain of the non-glycosylated epsilon chain of CD3. The immunostain typically shows membranous and/or cytoplasmic staining and is routinely used to identify normal and neoplastic T cells. Several possible mechanisms have been proposed to explain the aberrant expression of CD3 in B-cell neoplasms. EBV infection has frequently been observed in B-cell neoplasms with CD3 expression, including primary effusion lymphoma, HHV8+ large B-cell lymphoma, and diffuse large B-cell lymphoma associated with chronic inflammation.11, 17, 19 However, in our study and review, only 10 of 25 plasmablastic neoplasms had EBV infection (Table 4). In addition, EBV-negative diffuse large B-cell lymphomas with aberrant CD3 expression have also been reported.12 Therefore, EBV infection may only partially account for the aberrant CD3 expression.
Lineage ambiguity, infidelity, and promiscuity have been recognized in hematopoietic neoplasms, including acute leukemia with ambiguous lineage and mature B-cell or T-cell lymphoma with aberrant expression. Moreover, mature B-cell or T-cell lymphomas can transdifferentiate into other types of neoplasms, particularly dendritic cell and histiocytic sarcomas.26, 27, 28, 29 In the reported cases, the transdifferentiated dendritic cell and histiocytic sarcomas had the typical morphology and immunophenotype of dendritic cell and histiocytic neoplasms, although they still retained the same chromosomal translocations and IGH or TRG gene rearrangements present in the original B-cell or T-cell lymphomas. However, in our opinion, the aberrant CD3 expression in our plasmablastic neoplasms was not related to transdifferentiation. The CD3+ plasmablastic neoplasms still kept the plasmablastic and plasmacytic morphology with expression of plasmacytic markers, and CD3 was mostly expressed as the sole aberrant T-cell marker.
Another proposed mechanism was downregulation of B-cell transcriptional factors, particularly PAX5. PAX5 is an important regulator of B-cell differentiation, and loss of this protein in plasmablastic neoplasms may allow aberrant T-cell antigen expression.30
The cytoplasmic CD3 staining in the plasmablastic neoplasms is probably not attributed to the background staining or cross reactivity with the cytoplasmic immunoglobulin. In our series, we realized that only the neoplastic cells but not the background plasma cells stained positively for CD3. Furthermore, aberrant CD3 expression was preferentially detected in the cases with immunosuppression and/or EBV infection. Particularly, we noticed that CD3 can be detected in HHV8+ large B-cell lymphomas without detectable cytoplasmic kappa or light chain (Table 4 in reference #19: cases 2, 5, 45, and 46).
Another consideration of aberrant CD3 expression in plasmablastic neoplasms is the sensitivity and specificity of CD3 antibody. Multiple CD3 antibody clones have been used in the studies reporting CD3 expression in B-cell neoplasms, including Ventana Rabbit monoclonal 26V6, Novocastra mouse monoclonal PS1, and Dako F7.2.38.7, 8, 10 Oliveira et al.8 stained their CD3+ B-cell neoplasms with two different CD3 antibody clones (Novocastra PS1 clone and Dako F7.2.38), which showed similar staining patterns. Hence, this finding further indicated that CD3 expression in B-cell neoplasms was not due to background staining or cross reactivity to immunoglobulin.
Nevertheless, although we do not know the exact mechanisms of CD3 expression in B-cell neoplasms, detection of this aberrant expression indicates that immunostains with only CD20 and CD3 to establish the cell lineage may lead to misclassification of hematopoietic neoplasms. Therefore, in the initial workup of high-grade lymphomas with unusual clinical presentations and morphologic features, we recommend combination of one or more T-cell markers (CD2, CD4, CD5, CD7, and CD8) and B-cell markers (CD79a, PAX5, BOB1, and OCT2), in addition to CD3 and CD20. Particularly, CD138 should be frequently stained, even in cases without obvious plasmacytic or plasmablastic cytology. Molecular analyses for IGH and TRG rearrangements are also very helpful in determining the cell origin and assisting in accurate classification of these unusual neoplasms.
Conclusions
Plasmablastic lymphoma and plasmablastic plasma cell myeloma with aberrant CD3 expression almost exclusively occurred in the extranodal sites with a male predominance. The tumor cells typically showed a high-grade morphology with a high proliferation rate. Aberrant CD3 expression in these plasmablastic neoplasms was mostly in a cytoplasmic staining pattern and as the sole aberrant T-cell marker. Common B-cell markers (CD20, CD79a, and PAX5) were mostly negative or only partially expressed. Therefore, it is essential to avoid misclassification of CD3+ plasmablastic neoplasms as ‘peripheral T-cell lymphoma, not otherwise specified’ or ‘“extranodal NK/T-cell lymphoma, nasal type’ by scrutinized morphologic examination, frequent incorporation of CD138 immunostaining, and utilization of more B-cell and T-cell markers in addition to CD3 and CD20, in conjunction with necessary molecular studies.
References
Thakral B, Medeiros LJ, Desai P et al. Prognostic impact of CD5 expression in diffuse large B-cell lymphoma in patients treated with rituximab-EPOCH. Eur J Haematol 2016;98:415–421.
Kaleem Z, White G, Zutter MM . Aberrant expression of T-cell-associated antigens on B-cell non-Hodgkin lymphomas. Am J Clin Pathol 2001;115:396–403.
Suzuki Y, Yoshida T, Wang G et al. Incidence and clinical significance of aberrant T-cell marker expression on diffuse large B-cell lymphoma cells. Acta Haematol 2013;130:230–237.
Pan Z, Hu S, Li M et al. ALK-positive large B-cell lymphoma: a clinicopathologic study of 26 cases with review of additional 108 cases in the literature. Am J Surg Pathol 2017;41:25–38.
Matnani RG, Stewart RL, Pulliam J et al. Peripheral T-cell lymphoma with aberrant expression of CD19, CD20, and CD79a: case report and literature review. Case Rep Hematol 2013;2013:183134.
Tachibana T, Tomita N, Furuya M et al. Aberrant CD20 expression in angioimmunoblastic T-cell lymphoma. Intern Med 2011;50:495–499.
Wang J, Chen C, Lau S et al. CD3-positive large B-cell lymphoma. Am J Surg Pathol 2009;33:505–512.
Oliveira JL, Grogg KL, Macon WR et al. Clinicopathologic features of B-cell lineage neoplasms with aberrant expression of CD3: a study of 21 cases. Am J Surg Pathol 2012;36:1364–1370.
Wang E, Stoecker M . Primary mediastinal (thymic) large B cell lymphoma with aberrant expression of CD3: a case report with review of the literature. Int J Hematol 2010;91:509–515.
Wallentine JC, Perkins SL, Tripp SR et al. Diffuse large B-cell lymphoma with coexpression of CD3 in a pediatric patient: a case report, review of the literature, and tissue microarray study. J Pediatr Hematol Oncol 2009;31:124–127.
Lee M, Cha HJ, Yoon DH et al. EBV-positive diffuse large B-cell lymphoma of the elderly with aberrant expression of CD3 and TIA-1. Blood Res 2013;48:156–160.
Wu B, Vallangeon B, Galeotti J et al. Epstein-Barr virus-negative diffuse large B cell lymphoma with aberrant expression of CD3 and other T cell-associated antigens: report of three cases with a review of the literature. Ann Hematol 2016;95:1671–1683.
Luo X, Kuklani R, Bains A . Dual CD3 and CD4 positive plasma cell neoplasm with indistinct morphology: a diagnostic pitfall. Pathology 2016;48:378–380.
Tang YL, Chau CY, Yap WM et al. CD3 expression in plasma cell neoplasm (multiple myeloma): a diagnostic pitfall. Pathology 2012;44:668–670.
Sun J, Medeiros LJ, Lin P et al. Plasmablastic lymphoma involving the penis: a previously unreported location of a case with aberrant CD3 expression. Pathology 2011;43:54–57.
Suzuki Y, Yoshida T, Nakamura N et al. CD3- and CD4-positive plasmablastic lymphoma: a literature review of Japanese plasmablastic lymphoma cases. Intern Med 2010;49:1801–1805.
Petitjean B, Jardin F, Joly B et al. Pyothorax-associated lymphoma: a peculiar clinicopathologic entity derived from B cells at late stage of differentiation and with occasional aberrant dual B- and T-cell phenotype. Am J Surg Pathol 2002;26:724–732.
Tzankov A, Bourgau C, Kaiser A et al. Rare expression of T-cell markers in classical Hodgkin's lymphoma. Mod Pathol 2005;18:1542–1549.
Pan ZG, Zhang QY, Lu ZB et al. Extracavitary KSHV-associated large B-Cell lymphoma: a distinct entity or a subtype of primary effusion lymphoma? Study of 9 cases and review of an additional 43 cases. Am J Surg Pathol 2012;36:1129–1140.
Mishra P, Kakri S, Gujral S . Plasmablastic transformation of plasma cell myeloma with heterotropic expression of CD3 and CD4: a case report. Acta Clin Belg 2017;72:250–253.
Swerdlow SH, Campo E, Pileri SA et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016;127:2375–2390.
Segal GH, Jorgensen T, Masih AS et al. Optimal primer selection for clonality assessment by polymerase chain reaction analysis: I. Low grade B-cell lymphoproliferative disorders of nonfollicular center cell type. Hum Pathol 1994;25:1269–1275.
Greiner TC, Raffeld M, Lutz C et al. Analysis of T cell receptor-gamma gene rearrangements by denaturing gradient gel electrophoresis of GC-clamped polymerase chain reaction products. Correlation with tumor-specific sequences. Am J Pathol 1995;146:46–55.
Pan Z, Xie Q, Repertinger S et al. Plasmablastic transformation of low-grade CD5+ B-cell lymphoproliferative disorder with MYC gene rearrangements. Hum Pathol 2013;44:2139–2148.
Yin L, Xu J, Li M et al. Oct2 and Bob1 are sensitive and specific markers in lineage determination of B-cell lymphomas with no expression of conventional B-cell markers. Histopathology 2016;69:775–783.
Shao H, Xi L, Raffeld M et al. Clonally related histiocytic/dendritic cell sarcoma and chronic lymphocytic leukemia/small lymphocytic lymphoma: a study of seven cases. Mod Pathol 2011;24:1421–1432.
Wang E, Papalas J, Hutchinson CB et al. Sequential development of histiocytic sarcoma and diffuse large b-cell lymphoma in a patient with a remote history of follicular lymphoma with genotypic evidence of a clonal relationship: a divergent (bilineal) neoplastic transformation of an indolent B-cell lymphoma in a single individual. Am J Surg Pathol 2011;35:457–463.
West DS, Dogan A, Quint PS et al. Clonally related follicular lymphomas and Langerhans cell neoplasms: expanding the spectrum of transdifferentiation. Am J Surg Pathol 2013;37:978–986.
Mehrotra S, Pan Z . Fine needle aspiration cytology of histiocytic sarcoma with dendritic cell differentiation: a case of transdifferentiation from low-grade follicular lymphoma. Diagn Cytopathol 2015;43:659–663.
Lazzi S, Bellan C, Onnis A et al. Rare lymphoid neoplasms coexpressing B- and T-cell antigens. The role of PAX-5 gene methylation in their pathogenesis. Hum Pathol 2009;40:1252–1261.
Acknowledgements
We thank Ms. Lisa Litzenberger (Department of Pathology, University of Colorado Denver, Aurora, CO, USA) for her assistance in assembling the digital figures for this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Pan, Z., Chen, M., Zhang, Q. et al. CD3-positive plasmablastic B-cell neoplasms: a diagnostic pitfall. Mod Pathol 31, 718–731 (2018). https://doi.org/10.1038/modpathol.2017.177
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2017.177
This article is cited by
-
CD3+CD4+ lymphoid neoplasm: diagnostic clues for plasmablastic lymphoma instead of peripheral T-cell lymphoma
Annals of Hematology (2022)
-
CD3+/CD56+ EBV+ neoplasms in the nose and upper aerodigestive tract: potential misdiagnosis of plasma cell malignancies as NK/T cell lymphoma
Annals of Hematology (2021)
-
Heterogeneity in the diagnosis of plasmablastic lymphoma, plasmablastic myeloma, and plasmablastic neoplasm: a scoping review
International Journal of Hematology (2021)
-
The broad and challenging landscape of extranodal lymphoproliferations
Virchows Archiv (2020)
-
Lymphomas arising in immune-privileged sites: insights into biology, diagnosis, and pathogenesis
Virchows Archiv (2020)
