
Abstract
Although largely deprived from exogenous stimuli in utero, the mucosal barriers of the neonate after birth are bombarded by environmental, nutritional, and microbial exposures. The microbiome is established concurrently with the developing immune system. The nature and timing of discrete interactions between these two factors underpins the long-term immune characteristics of these organs, and can set an individual on a trajectory towards or away from disease. Microbial exposures in the gastrointestinal and respiratory tracts are some of the key determinants of the overall immune tone at these mucosal barriers and represent a leading target for future intervention strategies. In this review, we discuss immune maturation in the gut and lung and how microbes have a central role in this process.
Similar content being viewed by others
Introduction
Mucosal body surfaces such as the respiratory and gastrointestinal tract have to perform a complex task—they must remain tolerant against innocuous environmental, nutritional, and microbial antigens to ensure organ function, but must also mount efficacious immune responses against invading pathogens. In the adult intestine, this dual role is accomplished by a diverse and competitive microbiota, a structured and effector protein-enriched mucus layer, a rapidly renewing, reactive epithelium and a tightly regulated adaptive immune system. This allows compartmentalization of the majority of commensal bacteria within the gut lumen, rapid responsiveness to disrupted barrier integrity and tissue regeneration, and the generation of long-lasting highly effective adaptive immune effector functions. Similarly, the healthy adult lung effectively manages the incessant exposure to environmental antigens, is in constant cross-talk with its steady-state microbiota, and has effective immune compartments for rapid responsiveness against pathogens. In both organs, however, the situation fundamentally differs in neonates. In the neonatal intestine, rich nutrients and low competitiveness of the microbiota provide an open habitat and niches for incoming pathogens. Both the innate and adaptive immune system still undergo differentiation and adaptation and are impaired in their response to microbial challenge leaving the host susceptible to a variety of inflammatory and infectious diseases. Similarly, immune cells in the neonatal lung differ in quantity and quality and, hence, react differently to environmental and microbial exposures. In a constant process of immune education during postnatal development, lung-resident immune cells are faced with the massive challenge of reacting in a manner that protects the organ’s critical gas exchange machinery. In the following chapters, we will describe what is known about intestinal and pulmonary homeostasis during pre- and postnatal development in mice and man. We will highlight functional differences between the infant and adult scenarios, and survey the mechanisms that lead to an age-dependent susceptibility to infectious and immune-mediated diseases.
Prenatal development and birth
Tissue morphogenesis and innate immunity in the prenatal intestine
Gut tissue embryogenesis follows a similar order of events in most vertebrates.1 However, the length of the gestational period and the tissue maturity at birth differ significantly. Following a long gestational period, the human gut mucosa exhibits a fully differentiated intestinal architecture with crypts and villi and a complex cellular composition at birth.2 In contrast, the short gestational period of only 20 days in mice results in an incomplete maturation of the intestinal mucosa at birth and pronounced functional and structural changes during the postnatal period.1 Although these differences might limit the value of the murine model for a better understanding of the ontogeny of host–microbial interactions, both humans and mice have to facilitate the transition between the protected situation in utero and the intimate interaction with both commensal and pathogenic microorganisms after birth. Moreover, murine samples can be acquired in a well-controlled manner, whereas medical and ethical concerns limit access to human mucosal tissue samples from both healthy and diseased neonates. The occurrence of premature delivery in a significant portion of human neonates additionally stresses the potential value of considering the functional influence of tissue ontogeny on neonatal immunology. We will therefore consider results from both murine and human studies in the present review but highlight developmental differences and their potential influence.
In mice, small intestinal villi emerge at gestational day 15 (E15) and proliferating epithelial cells become confined to the intervillus region at E17.1 Three epithelial cell types namely absorptive enterocytes, goblet, and enteroendocrine cells appear with the emergence of villi in the fetus. Although the overall tissue architecture has formed in the fetus, the epithelium at birth is devoid of the regenerative unit of the small intestinal crypt as well as crypt-based Paneth cells (Figure 1).3, 4 This may have major implications since the crypt epithelium in adult mice harbors the constantly proliferating transit amplifying cells. Together with epithelial migration along the crypt–villus axis and exfoliation at the villus tip, this facilitates constant epithelial cell turnover. This ongoing surface renewal might significantly contribute to protection from invasion or attachment of enteric pathogens.5 Furthermore, the lack of mature Paneth cells and Paneth cell-derived antimicrobial peptides could render the neonatal intestine susceptible to infection and an altered composition of the enteric microbiota.6 Instead, expression of the cathelicidin Cramp was reported in the neonatal intestinal epithelium.7 This is interesting as Cramp expression has also been found in skin, breast milk, and vernix caseosa in mice and man, and might thus represent a universal neonatal antimicrobial peptide.8, 9

Development of the intestinal mucosal immune system. Before birth, Peyer’s patch anlagen develop via a crosstalk between lymphoid tissue inducer (LTi) and stroma cells within the sterile environment of the womb. The small intestinal mucosa is populated by the fetal wave of γδ T lymphocytes. At birth, the murine intestinal tissue architecture is immature and undergoes several developmental changes until reaching adult state. The neonatal mucosa is characterized by a lack of crypts and crypt-residing Paneth cells, a major source of antimicrobial substances in the adult tissue. The neonatal enterocytes, however, express cathelicidin-related antimicrobial peptide (CRAMP). Birth initiates the colonization of the intestinal mucosa and microbial density in the neonate reaches plateau levels fast. The neonatal microbiota is dominated by Lactobacilli, Streptococci, and Bifidobacteria, whereas species of the Bacteriodetes phylum become prevalent only in adults. Microbial diversity in the postnatal gut is reduced about 3-fold compared with the adult situation. Despite the presence of goblet cells, the expression of the mucins muc2, muc3, and muc5ac is reduced in the neonate resulting in a thinner mucus layer. αβ T (including thymus derived regulatory T cells (tTregs) and B lymphocytes begin to populate the intestine shortly after birth, however, they display a discrete homing pattern only to the Peyer’s patches and remain naive throughout the neonatal phase. Only after weaning, the occurrence of germinal centers and activated lymphocytes is noted in the Peyer’s patches. The maturation of M cells, which are major routes of antigen uptake in the adult, also occurs after the second week of life. The effector sites lamina propria and the intraepithelial compartment are populated by lymphocytes only after weaning. Formation of cryptopatches is noted after birth and their further maturation into isolated lymphoid follicles (ILFs) is dependent on the presence of a microbiota. During the postnatal phase, many bioactive factors are supplied by maternal breast milk including cytokines, growth factors, as well as secretory immunoglobulin A (SIgA). Endogenous SIgA is only produced by plasma cells in the adult mucosal tissue.
During ontogeny, anlagen of the mesenteric lymph nodes (MLN) appear at embryonic day (E) 9–10, whereas Peyer’s patch (PP) anlagen develop up until birth.10 Mucosal lymphoid tissue development follows the same general program and requires interactions between lymphoid tissue inducer (LTi) cells and lymphoid tissue organizer or stromal cells.11 LTi cells are type 3 innate lymphoid cells (ILC3) that depend on the expression of the transcription factors RAR-related orphan receptor γt (RORγt) and Id2 and express the chemokine receptors CXCR5 and CCR7.12, 13, 14 Maturation of the MLN and PPs occurs in the sterile environment in utero and is assumed to follow a developmental program independent of environmental cues. However, a recent study has shown that the numbers of LTi cells and later the size of the secondary lymphoid organs are markedly determined by the uptake of dietary retinoic acid by the mother during gestation.15
Moreover, exposure to maternally derived microbial constituents during gestation impact fetal/neonatal intestinal immune development and induce sustained transcriptional changes in the intestinal tissue of the offspring.16 For example, aryl hydrocarbon ligands stemming from maternal microbiota are detected in the placenta and breast milk and stimulate the development of ILC3 in the intestine of neonates.16
Tissue morphogenesis and innate immune mechanisms in the prenatal lung
Mammalian lung organogenesis occurs in four distinct phases (pseudoglandular stage, canalicular stage, saccular stage, and alveolar stage), out of which only three are close to completion at birth,17 leaving an organ under constant reconstruction during the first weeks of life (Figure 2). Although low in absolute number, the first innate immune cells reach the lungs very early during development18, 19. Embryonic macrophages and dendritic cells (DCs) are first found during the pseudoglandular stage (embryonic day (E) 35–119 in humans, E9.5–E16.5 in mouse),20, 21 when a primitive respiratory structure is built by epithelial cell branching.17 It has been shown that these fetal lung-resident DCs are less potent in inducing T-cell proliferation than their adult counterparts.21 Embryonic macrophages have been assigned an important role during early fetal development in that they largely orchestrate fetal lung morphogenesis. Through the release and deposit of a variety of mediators (e.g., cytokines, proteoglycans, and collagen) needed in tissue remodeling, these embryonic macrophages are involved in processes including vascularization and airway branching.22, 23 However, activation of these embryonic macrophages can lead to aberrant lung morphogenesis and preterm birth, or perinatal death, via the inhibition of multiple developmental genes in the fetal lung such as fibroblast growth factor 10 and integrin α8β1.20 On the other hand, it has been shown that fetal lung inflammation, which is likely to cause activation of immune cells inside the tissue, leads to accelerated lung maturation.24 At the checkpoint towards the canalicular phase (around E16.5 in mouse), when the airway epithelium differentiates and vascularization and angiogenesis begins, embryonic macrophages and DCs are quickly outnumbered by liver-derived fetal monocytes, which further differentiate into alveolar macrophages (AM) immediately after birth.22, 25 In healthy fetuses, these monocytes show an immature phenotype; however, intra-amniotic exposure to endotoxin or other inflammatory mediators can induce rapid maturation and differentiation of these monocytes already before birth.26 Thus, innate immune cells colonize the lung very early during fetal development and can react to intrauterine inflammation. In addition to DCs and macrophages, natural killer (NK) cells represent a large proportion of the innate immune cells found in the fetal lung.27 A proportion of these fetal lung NK cells show a differentiated phenotype, including perforin and granzyme B production as seen in adult NK cells. Moreover, these fetal NK cells have been demonstrated to be highly responsive to cytokine stimulation,27 thus indicating that NK cells could respond to in utero infection and inflammation. Other important components of the innate immune response in the fetal lung are pulmonary surfactants, especially surfactant protein A. These mixtures of lipids and proteins are expressed by alveolar type-II cells and are important to reduce surface tension at the air–liquid interface of the alveolus, thus preventing it from collapsing during exhalation.28 They are also involved in the innate immune response against pathogens by acting as antimicrobial peptides, directly inhibiting the growth of bacteria,29 or by opsonizing pathogens and, hence, capturing them for phagocytosis by macrophages.30 Surfactant protein A is associated with lung maturation and its expression increases during the course of gestation.31 Its increased release into the amniotic fluid has been identified as a signal for parturition;32 however, in the context of Toll-like receptor (TLR) ligand-induced inflammation, it has been shown that surfactant protein A can prevent preterm delivery by the suppression of pro-inflammatory mediators.33 Mucins are an additional key innate defense mechanism in the airways. Muc5b is essential for macrophage function as these cells display impaired phagocytic capacity and decreased anti-bacterial cytokine (e.g., interleukin (IL) 23) production in the lungs of Muc5b−/− mice.34 Muc5b is detectable in the lung as early as E14.5, well before other mucins such as Muc5ac and Muc2.35 Hence, early Muc5b production during the fetal period could be a central mediator priming lung-infiltrating macrophages and monocytes for responsiveness against microbes after parturition.

Immune cell development in the pre- and perinatal lung. Before birth, during the pseudoglandular stage of lung morphogenesis, embryonic macrophages, fetal dendritic cells (DCs), and natural killer (NK) cells are the first cell types that infiltrate the fetal lung. At the same time, Muc5b is produced by goblet cells likely influencing the function of embryonic macrophages. During the canicular phase, surfactant proteins (SP), especially SP-A, are produced and can serve as antimicrobial peptides. Fetal liver-derived monocytes start replacing the embryonic macrophages. Immediately after birth, these fetal liver-derived monocytes differentiate into alveolar macrophages, a process orchestrated by epithelial cell-derived granulocyte-macrophage colony-stimulating factor (GM-CSF). Other innate and adaptive immune cell types (including invariant natural killer T (iNKT) cells, B and T cells, natural T-regulatory (nTreg) cells) slowly start to infiltrate the lung. B and T cells exhibit an immature/naive phenotype during the neonatal period and are present in low numbers, whereas nTreg cells peak in frequencies shortly after birth. Simultaneously with the nTregs, CD11b+ conventional DCs are high in proportion, expressing their highest levels of programmed cell death ligand (PD-L) 1, an inhibitory marker induced by the microbiota. During this early-life period, the microbiota consists of Firmicutes and Proteobacteria γ. Later during development, Bacteroidetes also colonize the lung, forming a significant proportion of the bacteria present in the adult organ. CD11b+ conventional DCs decrease in frequency, as does their expression of PD-L1. On the contrary, adaptive immune cells increase steadily during postnatal development, displaying a more mature phenotype with age. Although the nTreg compartment is reduced in the adult compared with the neonatal organ, a significant increase can be found in the frequencies of inducible Treg (iTreg) cells in the lung, which are absent early after birth.
Prenatal adaptive immunity
In contrary to innate immune mechanisms, adaptive immunity has not been studied in great detail in the developing fetal lung as resident T and B cells are not detectable during this period.19 The murine fetal intestine is also mostly devoid of adaptive immune cells. Only γδ T lymphocytes that are generated in the thymus before the αβ T cells are already present.36 Those γδ T cells are likely to be IL-17 producers and are generated exclusively during the fetal phase. They reside in the lamina propria as the murine small intestine does not contain epithelium-associated lymphocytes until weaning.37, 38 Thus, unlike human neonates,39 mice do not seem to harbor “natural” intraepithelial lymphocytes.40
Prenatal microbial exposure
Dietary and microbial constituents are able to cross the placenta and influence the development of the fetal mucosal lymphoid tissues.15, 16 In addition, bacterial DNA has been detected in healthy human placenta and amniotic fluid and the existence of a placental microbiome has been suggested.41, 42 This low abundance, low diversity microbiota appears to be predominated by Proteobacteria and displays the closest resemblance to the oral microbiota.41 Transient asymptomatic bacteremia following manipulation of the mucosal surface, e.g., during tooth brushing is well known and may explain the presence of the detected material. Culture-based data are, however, rare. Jimenez et al.43, 44 were able to recover a genetically labelled Enterococcus strain in amniotic fluid and meconium following oral administration to the dam. However, future work is required to establish the presence of viable and diverse bacterial communities at the placental-fetal interface.
Postnatal microbial exposure and tissue maturation
Postnatal microbial colonization of mucosal surfaces
Rupture of membranes at birth, mark a drastic change in environmental exposures, most notably being the progressive colonization of mucosal sites by microbes. Following vaginal delivery, the first neonatal intestinal microbiota is derived from the urogenital microbial community of the mother that is transmitted to the newborn during birth.45 A number of studies that compared the microbiota of neonates and adults have outlined significant differences and an overall increase in the microbial density and diversity.46, 47 Although microbial density reaches plateau levels shortly after birth in humans and mice,46 diversity and thus a competitive microbiota is only reached after several years in humans and after weaning in mice.47, 48, 49 Nutritional factors appear to have a major role as, for example, the dominance of Lactobacilli, Streptococci, and Bifidobacteria in the neonatal intestine is lost after cessation of breast milk.
Although the lung is a functional gas-exchange organ at birth, its structure is far from mature. During the first years in humans (first 4 weeks in mice), alveolarization and microvascular maturation are ongoing,17 subjecting the organ to a process of constant tissue remodeling. Furthermore, like all other body surfaces, starting at birth the lung is constantly exposed to a variety of environmental exposures of a pathogenic and nonpathogenic nature. At the same time, the airways start to be colonized by bacteria and fungi, which form the microbiome and mycobiome.50 Few studies have thus far focused on how the airway microbiota develops during the neonatal period and information concerning early-life fungal colonization is still lacking. However, it has been shown in mice that similar to the intestine, the airway microbiota increases in load and diversity over the first weeks of life.51 The early colonizers belong mainly to the Proteobacteria and Firmicutes phyla, whereas later during the development, Bacteroidetes are largely represented within the airway microbiota.51 In humans, to date, only one study has directly addressed the airway microbiota of newborns.52 It was conducted on preterm infants that underwent endotracheal intubation for up to 1 month. In all infants enrolled in the study, Acinetobacter was the predominant genus at birth. In “healthy” infants that did not develop bronchopulmonary dysplasia, Pseudomonas and Sneathia started dominating from day 3 of life onwards, thus, diminishing the proportion of Acinetobacter.52 However, as all these infants had been born premature and intubated for up to 1 month, it is unknown whether the microbial profile detected in these infants will correlate with that of healthy full-term infants.
Innate immunity in the neonatal intestine
Intestinal crypts with stem cells and rapidly proliferating transit amplifying cells develop in mice around 10–12 days after birth.3, 4 This initiates constant epithelial renewal characterized by migration along the crypt–villus axis and exfoliation at the villus tip. With the appearance of crypts, also crypt-based Paneth cells mature and the spectrum of antimicrobial peptides expands from epithelial Cramp to a variety of Paneth cell-derived α-defensins and cryptdin-related sequence peptides.7, 53, 54 In addition, expression of the c-type lectin regenerating islet derived protein 3γ (Reg3γ) shown to significantly contribute to host–microbial homeostasis increases markedly (own unpublished results).55 In the adult animal, the epithelial surface of the colon is covered by a thick, bacteria-free mucus layer composed of highly structured glycoproteins that segregates luminal bacteria from the epithelial surface.56 A similar but less pronounced mucus layer is also observed in the small intestine.57 Although the mucus-producing goblet cells are present in the neonatal intestine, the expression of the major mucins Muc2, 3, and 5a is strongly reduced during the postnatal period.5 The mucus layer is composed of glycoproteins and thus provides an important nutrient source for certain commensal bacteria. This is expected to have a major influence on host–microbial homeostasis.58, 59
Developmental and adaptive mechanisms dampen the sensitivity of the intestinal epithelial surface to microbial exposure during the critical period after birth. In mice, postnatal exposure to bacterial endotoxin was shown to induce endotoxin tolerance by microRNA 146a (mir146a)-mediated posttranscriptional downregulation of the innate immune signaling molecule interleukin 1-associated kinase (Irak) 1.60, 61 A similar adaptive process by enhanced expression of the regulatory secretory leukocyte protease inhibitor was recently also observed in the upper gastrointestinal tract in humans.62 Moreover, developmentally regulated low expression of the receptor for double-stranded RNA, TLR3, in the murine neonatal intestinal epithelium was observed.63 A reduced sensitivity to TLR stimulation was also reported in fetal as compared with adult human intestinal epithelial cells mediated by enhanced IκB expression suggesting that similar mechanisms are also active in humans.64
Tissue resident intestinal macrophages (CX3CR1hiCD64+MHCII+ cells) are already present before birth and derive from fetal hematopoiesis in liver and yolk sac. Recently, it was shown that these cells, however, do not persist throughout life but are rather substituted by macrophages originating from adult hematopoiesis in the bone marrow. Therefore, during the postnatal period, macrophages represent a mixed population originating from fetal as well as adult hematopoiesis, with cells derived from adult hematopoiesis becoming progressively dominant and finally after weaning the only detectable population.65, 66 Intestinal DCs are also present in the postnatal intestine;67 however, it is not known whether the different subsets (i.e., CD103+, CD103+CD11b+, and CD11b+) seen in the adult are functional and present in comparable numbers.
Innate immunity in the neonatal lung
Immediately after birth, fetal monocytes that infiltrated the lungs during the fetal period start differentiating into AMs (Figure 2).25 This process is dependent on the expression of granulocyte–macrophage colony-stimulating factor by lung epithelial cells, which is upregulated during the first days of life.25 The stimulus leading to increased granulocyte–macrophage colony-stimulating factor production shortly after birth is still unknown. However, it has been shown that maturation of AMs and other CD11c+ cells can be achieved by intranasal administration of a mixture of microbial extracts,68 indicating that the exposure to an airway microbiota could assist in the maturation of these cell types. AMs have a crucial role in the first-line defense against a variety of inhaled antigens. Although newborns have a higher risk of establishing pulmonary infections, AMs from neonatal rats have been reported to exhibit a higher phagocytic capacity than those of adults.69 They are also able to induce an adult-like response towards LPS challenge.69 CD11b+ conventional DCs constitute a large proportion of cells during the first 2 weeks of life in mouse lungs51 and they exhibit a unique activation profile compared with adults. A variety of activation markers are highly expressed and it has been shown that these neonatal CD11b+ DCs are potent in processing antigens and inducing T helper (Th) 2-type immune responses.51 Moreover, it has been demonstrated that the activation profile of these DCs during the neonatal period is partially altered by the presence of a microbiota. Microbial colonization induces a transient expression of PD-L1 (programmed cell-death ligand 1) on these cells during the first 2 weeks of life, which allows them to induce regulatory T (Treg) cells.51 Similar activation profiles have been reported for the CD103+ conventional DC subset;51 however, their relative abundance in the lungs of neonates is controversial.51, 70 The functionality of neonatal pulmonary DCs has been tested in a variety of animal models. Different murine and an ovine model described a similar capacity of neonatal and adult pulmonary DCs to take up and process antigens and to stimulate T cell responses;51, 70, 71 however, it has been shown in rats that the antigen-presenting capacity of neonatal DCs in response to granulocyte–macrophage colony-stimulating factor is significantly reduced in comparison with adult DCs.72 Another lung-resident DC subset, plasmacytoid DCs, is found in very low absolute numbers and frequency in the neonatal lung.70 Their functionality in the newborn has yet to be addressed in detail. Invariant natural killer T (iNKT) cells are also very low in frequency during the neonatal period.73 Accumulation of these cells in mucosal tissues, including the lung, throughout life is prevented by the induction of CXCL16 via signals obtained from the microbiota.73
Adaptive immunity in the neonatal intestine
Around birth, αβ T cells and B cells mature and begin to populate the anlagen of the secondary lymphoid organs that have developed prenatally.74 Interestingly, until weaning, T cells in the small intestine are only detected in the gut-associated lymphoid tissue structures (PPs), but not at effector sites, and despite the presence of food and microbial antigens early after birth, display a naive phenotype in stark contrast to the adult situation (Figure 1).67 Germinal center formation in PPs, a reflection of constant exposure to microbiota and food-derived antigens, does not occur in the neonate.38 The effector sites lamina propria and the epithelium-associated compartment are populated with (activated) T cells starting around weaning. A major fraction of intestinal regulatory T cells (Tregs) in the adult intestine are negative for Neuropilin 1 and therefore induced in the periphery likely being responsive to microbiota- and food-derived antigen in the colon and small intestine, respectively.75, 76 Similar to the emergence of activated T cells at intestinal effector sites, these peripheral (p)Tregs appear after weaning.76 However, Tregs are already detected when first T cells populate the neonatal small intestine soon after birth (own unpublished observations). Those Tregs are thymus-derived and enforce peripheral tolerance to self-antigens already shortly after birth.77 The intestinal T-cell immunity of the neonate appears to resemble the central rather than the mucosal state in the adult. Human neonatal intestines are already populated with T cells at the effector compartments at birth but also here, a postnatal maturation can be seen. The proportion of naive T cells in pediatric patients is significantly higher than in young adults.78
B cells readily populate the intestine during the postnatal phase in mice;67 however, the occurrence of functional plasma cells and secretory immunoglobulin A (SIgA) production are only noted after weaning and a stable immunoglobulin (Ig) A repertoire formation is completed around 8–10 weeks of age in mice (Figure 1).79, 80, 81 Also in human neonates, total serum IgA rises to adult levels only after the first year of life.82 During the postnatal phase, breast milk is the major source of SIgA in the intestinal lumen of the neonate.80 The absence of maternal SIgA leads to premature maturation of T and B cells in the neonate.79 It has been a matter of a long debate whether the intestine can serve as a primary lymphoid organ generating adaptive lymphocytes. A recent study using Rag reporter mice has shown that in weanling mice, the lamina propria harbors nascent B cells where their development is shaped by the presence of the microbiota.83
Although MLN and PP anlagen develop prenatally, solitary intestinal lymphoid tissue structures are not seen before 2 weeks post parturition.84 Cryptopatches develop independently of the presence of live bacteria. Microbial constituents such as the NOD1 ligand peptidoglycan then induce maturation of cryptopatches into isolated lymphoid follicles where IgA producing plasma cells are induced independently of T cell help.85, 86
Exposure of the neonatal intestinal mucosa to commensal bacteria does not remain unseen by the host’s adaptive immune system. Suppressive mechanisms are in place to protect against the hazard posed by an overt stimulation during the initial exposure of the intestine to the outside world. T-cell activation within the PPs is dampened by the presence of regulatory T cells and maternal SIgA.67 Erythroid CD71+ cells that are present abundantly in the neonate suppress myeloid and lymphoid cell activation in the intestine following bacterial colonization.87
Adaptive immunity in the neonatal lung
As reported for systemic immunity, adaptive immune cells in the lung of neonates are scarce and exhibit limited functionality (Figure 2). T cells are found in low frequencies in the neonatal lung,70 and are predominantly comprised of naive T cells,78 which show an intrinsic bias towards the production of Th2 cytokines and transcription factors when stimulated in vitro.70 Thus, susceptibility to viral lung infections in neonates could be largely due to insufficient, or inappropriate, T-cell responses. However, it has also been reported that in the context of adult Pneumocystis carinii infection, adoptive transfer of neonatal T cells was as effective in resolving the infection as adult T cells.88 Hence, the authors conclude that there is no intrinsic defect of neonatal T cells inside the lungs, but that the neonatal lung environment during homeostasis is not able to sufficiently prime these T cells.88 Only Treg cells have been reported to substantially accumulate inside the neonatal lung51, 78 and their accumulation is linked to microbial exposure.51 Importantly, the Treg compartment in neonates is only comprised of natural thymus-derived Treg (nTreg) cells.51 Peripherally induced Treg (iTreg) cells can only be found in the lung later in life and their appearance is initiated by the microbiota in a PD-L1-dependent manner.51 Taken together, during the postnatal period, immune cells slowly accumulate in the lung while the lung tissue is remodeling and the airways are colonized by microbes. Out of all the cell types investigated, only CD11b+ conventional DCs and Treg cells are present in higher frequencies in the neonatal lung than in the lung of adults.51 The phenotype and function of these two cell types is strongly influenced by microbial colonization.
Contribution of breast milk to intestinal immunity
Major differences exist between the neonate and adult mucosa. This concerns the microbiota composition, the chemical and metabolic milieu, the cellular composition and functional maturation of the epithelial barrier, as well as multiple aspects of innate and adaptive immune recognition and effector function. Concerning at least the intestine, maternal breast milk might partially compensate for these differences providing a number of important antimicrobial and immunomodulatory constituents.89 Beside macronutrients, a large number of non-nutritive bioactive factors such as immunoglobulins (SIgA, IgG), growth factors, hormones, maternal cells, cytokines, antimicrobial peptides, and complex oligosaccharides modulate innate immune signaling and influence the developing host–microbial interaction.90, 91 Maternal immunoglobulins transferred via placenta and breast milk provide an effective protection against common infectious diseases such as rotavirus.92, 93 Complex human milk oligosaccharides, lysozyme, lactoferrin as well as mucins might compensate for the reduced mucus production and the smaller spectrum of secreted antimicrobial peptides in the neonate.5, 7 Endothelial growth factor and transforming growth factor β modulate inflammatory signaling and might reduce the immunostimulatory potential of microbial exposure after birth.91 The spectrum of SIgA provided by breast milk reflects the maternal intestinal SIgA.94 The combined transfer of members of the maternal microbiota with the corresponding maternal SIgA might provide the optimal strategy to rapidly establish a stable host–microbial homeostasis.
Immunity and disease susceptibility
Early intestinal colonization in health and disease
Developmental and adaptive mechanisms that facilitate fetal postnatal transition must provide a robust regulatory framework to cope with the sudden onset and extent of environmental and microbial exposure.46 It may therefore not come as a surprise that the transition between fetal and postnatal life can fail leading to organ dysfunction and clinical disease. Three clinical examples might illustrate this scenario. First, genetic predisposition can cause chronic inflammatory diseases. For example, a genetic defect in IL-10 receptor expression leads to early-onset colitis manifesting within the first year of life and most likely due to the uncontrolled secretion of proinflammatory mediators such as TNF in response to microbial exposure.95 As both IL-10 production and the IL-10-targeted cell population reside within the hematopoietic lineage, bone marrow transplantation provides an effective treatment option.96 This is consistent with animal studies that report the development of mucosal inflammation under similar conditions of an impaired immune homeostasis.97, 98 Another example is necrotizing enterocolitis of the newborn.99 This condition is characterized by a devastating inflammatory response of the intestinal tissue in preterm-born human neonates leading to high morbidity and mortality. Disease onset is typically observed 1–2 weeks after birth, consistent with the current hypothesis that an inappropriate innate immune response of the mucosal tissue to postnatal bacterial colonization drives this inflammatory response.100 Animal studies suggest that lipopolysaccharide recognition by TLR4 on somatic cells and in particular intestinal epithelial cells and endothelial cells has a major role in the etiology of the disease.101, 102 Several mechanisms such as breast milk-derived epidermal growth factor or intestinal alkaline phosphatase, secretory leukocyte protease inhibitor, or the acquisition of tolerance were identified to dampen inflammation in mature neonatal mice and may function insufficiently in the preterm intestine.60, 61, 62, 91 Interestingly, a recent meta-analysis reported a significant benefit of postnatal oral administration of probiotic bacteria to preterm human neonates with both reduced necrotizing enterocolitis incidence and mortality.103 Thus, alterations in the enteric microbiota in preterm neonates might promote the etiology of necrotizing enterocolitis, consistent with the reported temporal clustering of necrotizing enterocolitis cases in individual institutions resembling the epidemiology of an infectious disease. The third example is somewhat speculative and addresses a possible association of cesarean section delivery with the development of immune-mediated, mainly atopic, diseases.104, 105 A direct link between the mode of delivery and the early enteric microbiota has been established. The early microbial composition in turn has significant and lasting impact on immune maturation.45, 73, 106 The high rate of cesarean sections in industrialized countries and the continuing rise of allergic diseases such as asthma, allergic rhinitis and celiac disease have provoked a discussion on a possible causal relationship. Although more research is needed to confirm this link and identify the underlying mechanisms, attempts are being made to reconstitute the vaginal maternal microbiota in cesarean-section-born human neonates directly after birth.107
On the other hand, the postnatal phase also provides a unique window of opportunity to shape the host immune homeostasis towards tolerance that is effective throughout life.108 Microbial exposure during the postnatal development constrains the expansion of iNKT cells and IgE-producing plasma cells.73, 106 As a consequence, the lack of microbial exposure during the postnatal phase leads to an increased susceptibility of the adult individual to inflammatory bowel and allergic diseases. In contrast, enhanced exposure to microbial stimuli during early postnatal life, e.g., by growing up on a farm appears to protect against atopic diseases in humans.109
Neonatal susceptibility to intestinal infection
The need for an enhanced mucosal immunoregulatory control in the neonate might impair an efficient host response to infection. Indeed, infections represent the leading cause of death in this age group worldwide and gastrointestinal and pulmonary infections account for a major fraction of all cases.110 In addition, not only the incidence of infectious diseases but also the spectrum of pathogens greatly varies with age. For example, orally acquired bacteria such as group B streptococci (S. agalactiae), Listeria monocytogenes, and E. coli K1 represent major causal agents of sepsis and meningitis in neonates, whereas they are rarely found in adults.111 This also accounts for the intestine where rotavirus, enterohemorrhagic and enteropathogenic E. coli represent major human pathogens in children but not adults.112, 113 Although environmental exposure might in part explain this difference, it is likely that particularities of the neonatal innate and adaptive immune system contribute as well.
Indeed, animal studies have identified specific age-dependent differences in the host–microbial interaction that explain the susceptibility of the neonatal host to certain pathogens (Table 1). First, the early microbiota shows low diversity and thus low colonization resistance. Even low numbers of pathogens such as E. coli K1 and Salmonella efficiently colonize the neonatal murine intestine.5, 114 This is in contrast to the situation in adult individuals, where enteropathogens require mechanisms to overcome their commensal neighbors and generate an ecological niche. Not surprisingly, large inocula or antibiotic pretreatment are required in many animal models to allow colonization, a prerequisite of infection.115 Second, functional differences in innate immune recognition and effector molecule production might account for enhanced susceptibility. In neonatal mice, epithelial endotoxin tolerance and low postnatal expression of the innate immune receptor for double-stranded RNA, TLR3, were observed.60, 63 Although delayed responsiveness to bacterial lipopolysaccharide might increase the susceptibility to infection with gram-negative pathogens such as Salmonella, low postnatal expression of TLR3, confers enhanced susceptibility to infection with rotavirus.5, 60, 63 Decreased epithelial TLR3 expression was also found in human infants, which might contribute to the enhanced susceptibility in this age group.63 Rotavirus displays a marked tropism for the intestinal epithelium and represents one of the most frequent causative agents of gastroenteritis in human infants worldwide.113
In humans, bacillary dysentery caused by Shigella spp. primarily affects children under low sanitary conditions in developing countries and causes a severe colitis. The altered spectrum of antimicrobial substances in the neonatal mouse intestine was identified to allow Shigella flexneri to infect newborn animals.116 Moreover, the small intestine in neonatal mice lacks mature M cells, an important entry portal for bacterial enteropathogens.5 As a consequence, Salmonella requires epithelial invasion to penetrate the mucosal barrier. Enterocyte invasion leads to the generation of large intraepithelial microcolonies, most likely due to the reduced epithelial turnover in the neonatal host.5 Finally, the altered milieu with increased serum levels of IL-10 and reduced IL-12 and IFN-γ favor infection by intracellular pathogens such as Listeria monocytogenes.117, 118, 119, 120 L. monocytogenes translocates through the intestinal mucosa and spreads systemically causing abortion but also sepsis and meningitis in human neonates. Reduced IFN-γ secretion in combination with the above mentioned intestinal epithelial endotoxin tolerance might also enhance the susceptibility to infection with non-typhoidal Salmonella and cryptosporidia.60, 121, 122, 123 Cryptosporidium parvum typically causes watery diarrhea in human infants,124 whereas non-typhoidal Salmonella represents an important cause of sepsis and meningitis in human neonates in many African countries.111
Despite these examples, neonates are able to mount protective innate and adaptive mucosal immune responses. Several studies from the mid-1990s demonstrated that under certain conditions, e.g., the use of an adjusted inoculum of the infectious agent or of a stronger adjuvant for vaccination, neonates were able to mount adult-like immune responses.125, 126, 127 Subsequent analyses showed that neonatal immunity exhibits a wide variety of responses that range from immunodeficient through immunodeviant to fully immunocompentent.118, 128 For example, a potent T-cell response mediates viral clearance during rotavirus infection in neonatal mice.129 In addition, the neonatal host even shows enhanced resistance to oral Yersinia enterocolitica infection. Strong IFN-γ and IL-17 production within the MLN leading to the recruitment of activated phagocytes and a protective CD4+ T-cell response was identified to protect the neonatal host.130, 131 The neonate might thus compensate for a less-efficient mucosal response with a heightened immune activation in the protective firewall, the lymphoid tissue. Another example is the resistance of human neonates to infection with Clostridium difficile, the most common etiological agent of hospital-acquired diarrhea in adults. Many small children are colonized by toxin-producing C. difficile, but remain asymptomatic.132 The underlying mechanisms have not been resolved yet.
Neonatal susceptibility to pulmonary allergy and infection
Unsurprisingly, also in the case of the lung, the difference in immune cell numbers and phenotypes between newborns and adults leads to distinct immune responses against the same agents (Table 1). Once exposed to an allergen such as house dust mite, neonates show increased allergic airway inflammation compared with adults, which has been correlated to the missing iTreg cells in the lungs of neonates.51 In line with this, lung T cells show an intrinsic bias towards Th2 during the neonatal period,70 likely contributing to the exaggerated inflammatory response against allergens. Pulmonary viral infections are very common in young infants and have a tendency to be more severe than in adults. A significant proportion of these infections are caused by respiratory syncytial virus (RSV).133 It has been shown that, although viral kinetics are similar between neonatal and adult RSV infection,134, 135 the primary response towards RSV in neonates differs significantly from that in adults. IFN-γ production is attenuated and delayed in neonatal compared with adult mice,134, 136 whereas IL-13 is increased during the course of infection in the newborn.135 Epitope-specificity of CD8+ T cells is distinct between neonatal and adult infection,137 which is in part due to altered precursor frequencies. The neonatal CD8+ T cell repertoire towards RSV is restricted and possesses a lower functional avidity.137 In addition, an altered composition of the pulmonary DC subsets in response to RSV, which results in a domination of CD103+ DCs with limited co-stimulatory capacity, contributes to the generation of functionally different CD8+ T cells.138 Thus, there are strong age-dependent differences in the response to RSV. This has also inevitable consequences during reinfection. Reinfection following primary neonatal RSV infection leads to elevated production of inflammatory mediators139 and enhanced inflammatory cell infiltration,134 resulting in increased weight loss and immunopathology.134 It has also been demonstrated that primary neonatal RSV infection leads to airway hyperresponsiveness, enhanced mucus production and airway eosinophilia upon reinfection.135 This phenomenon has been attributed to the presence of RSV-specific IgE and the production of IL-13 upon initial infection.135, 140 Moreover, the amount of IFN-γ produced during primary infection, which is barely detectable in newborns,134, 136 largely determines the extent of airway hyperresponsiveness, mucus production and airway eosinophilia during reinfection.141 Whether the immunological findings obtained with RSV also hold true in the case of other pulmonary viral infections (e.g., with influenza A or Rhinovirus) has yet to be determined. Moreover, although bacterial pneumonia is a complication that can be seen more often during the immediate postnatal period compared with later during childhood,142 immunological studies giving mechanistic insights into the cause and consequences are still scarce.
Neonatal predisposition to chronic lung disorders
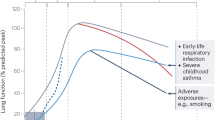
Chronic lung disorders, such as asthma, are widespread among the population and account for a tremendous number of hospitalizations and deaths every year. Epidemiologically, a strong link between fetal and postnatal exposures and the predisposition to chronic lung diseases, especially asthma, has been described.143, 144 The most common risk factors are pre- and perinatal tobacco exposure, antibiotic treatment and neonatal severe lower respiratory tract infections.145 Murine studies have started to shed light on the immunological basis behind the phenomenon of this “environmentally induced” early-life predisposition towards asthma. It has, for example, been shown that antibiotic treatment throughout the perinatal period increases baseline serum IgE levels and reduces the number of Treg cells,146 thus, leading to increased allergic airway inflammation upon allergen exposure. Infection with RSV during the neonatal period also has profound consequences for Treg cell responses, enhancing allergic airway inflammation during adulthood.147 In comparison with adult RSV infection, neonatal infection induces a Th2-bias in Treg cells by promoting GATA-3 expression and the production of Th2-type cytokines in these cells.147 A study in humans also made a link between maternal tobacco smoking and the number of Treg cells found in cord blood, further associating smoking and lower Treg cell counts with the development of allergy.148 In addition, environmental influences protecting against the development of asthma have been linked to an increase in the Treg cell pool in neonates.149, 150 Breast-feeding150 (by allergen-sensitized mothers) and maternal farming149 are two of these protective exposures. Thus, Treg cells seem to be a crucial cell type in the prevention of asthma as a variety of early-life risk and protective factors influence Treg cell numbers or alter their function in the newborn.146, 147, 148, 149, 150 Another common point of nearly all these factors is their influence on the microbiota composition.151, 152, 153 The mechanisms by which they can alter microbial load and composition have not been elucidated in great detail yet. However, a link between neonatal microbial colonization of the lungs, pulmonary Treg numbers/phenotypes, and protection against allergic airway inflammation during adulthood has been established in mice,51 thus, providing evidence for one of potentially a variety of mechanisms by which early-life exposures could influence predisposition to asthma.
Conclusions
Taken together, in the context of mucosal immunity of the newborn, it might be more appropriate to speak of a distinct rather than immature mucosal immune system. This conclusion has important consequences. Only a better knowledge of age-dependent differences of host mucosal immunity and the neonatal immune system might allow us to understand the establishment of host–microbial homeostasis. In addition, the pathogenesis of infectious and inflammatory diseases in neonates might significantly differ from what we know from the adult host. Neonates and small infants might therefore require a different therapeutic approach and clinical management as compared to the established regimen applied to adult patients. Finally, environmental and especially microbial exposure during the postnatal period have a major long-term impact on the host’s immune system and health. This again stresses the need to better understand the neonatal immune system and identify exogenous factors that prime this system. In the future, this will allow the avoidance of detrimental environmental signals but also identify stimuli that facilitate the development of a balanced and robust immune response with reduced predisposition to diseases in later life.
References
Noah, T. K., Donahue, B. & Shroyer, N. F. Intestinal development and differentiation. Exp. Cell Res. 317, 2702–2710 (2011).
Drozdowski, L. A., Clandinin, T. & Thomson, A. B. Ontogeny, growth and development of the small intestine: understanding pediatric gastroenterology. World J. Gastroenterol. 16, 787–799 (2010).
de Santa Barbara, P., van den Brink, G. R. & Roberts, D. J. Development and differentiation of the intestinal epithelium. Cell. Mol. Life Sci. 60, 1322–1332 (2003).
Hirano, S. & Kataoka, K. Histogenesis of the mouse jejunal mucosa, with special reference to proliferative cells and absorptive cells. Arch. Histol. Jpn. 49, 333–348 (1986).
Zhang, K. et al. Age-dependent enterocyte invasion and microcolony formation by Salmonella. PLoS Pathog. 10, e1004385 (2014).
Salzman, N. H. et al. Enteric defensins are essential regulators of intestinal microbial ecology. Nat. Immunol. 11, 76–83 (2010).
Menard, S. et al. Developmental switch of intestinal antimicrobial peptide expression. J. Exp. Med. 205, 183–193 (2008).
Dorschner, R. A., Lin, K. H., Murakami, M. & Gallo, R. L. Neonatal skin in mice and humans expresses increased levels of antimicrobial peptides: innate immunity during development of the adaptive response. Pediatr. Res. 53, 566–572 (2003).
Starner, T. D., Agerberth, B., Gudmundsson, G. H. & McCray, P. B. Jr. Expression and activity of beta-defensins and LL-37 in the developing human lung. J. Immunol. 174, 1608–1615 (2005).
Rennert, P. D., Browning, J. L., Mebius, R., Mackay, F. & Hochman, P. S. Surface lymphotoxin alpha/beta complex is required for the development of peripheral lymphoid organs. J. Exp. Med. 184, 1999–2006 (1996).
Honda, K. et al. Molecular basis for hematopoietic/mesenchymal interaction during initiation of Peyer's patch organogenesis. J. Exp. Med. 193, 621–630 (2001).
Cherrier, M., Sawa, S. & Eberl, G. Notch, Id2, and RORgammat sequentially orchestrate the fetal development of lymphoid tissue inducer cells. J. Exp. Med. 209, 729–740 (2012).
Eberl, G. et al. An essential function for the nuclear receptor RORgamma(t) in the generation of fetal lymphoid tissue inducer cells. Nat. Immunol. 5, 64–73 (2004).
van de Pavert, S. A. et al. Chemokine CXCL13 is essential for lymph node initiation and is induced by retinoic acid and neuronal stimulation. Nat. Immunol. 10, 1193–1199 (2009).
van de Pavert, S. A. et al. Maternal retinoids control type 3 innate lymphoid cells and set the offspring immunity. Nature 508, 123–127 (2014).
Gomez de Aguero, M. et al. The maternal microbiota drives early postnatal innate immune development. Science 351, 1296–1302 (2016).
Warburton, D. et al. Lung organogenesis. Curr. Top. Dev. Biol. 90, 73–158 (2010).
Holt, P. G. & Jones, C. A. The development of the immune system during pregnancy and early life. Allergy 55, 688–697 (2000).
Stoltenberg, L., Thrane, P. S. & Rognum, T. O. Development of immune response markers in the trachea in the fetal period and the first year of life. Pediatr. Allergy Immunol. 4, 13–19 (1993).
Blackwell, T. S. et al. NF-kappaB signaling in fetal lung macrophages disrupts airway morphogenesis. J. Immunol. 187, 2740–2747 (2011).
McCarthy, K. M., Gong, J. L., Telford, J. R. & Schneeberger, E. E. Ontogeny of Ia+ accessory cells in fetal and newborn rat lung. Am. J. Respir. Cell Mol. Biol. 6, 349–356 (1992).
De Kleer, I., Willems, F., Lambrecht, B. & Goriely, S. Ontogeny of myeloid cells. Front. Immunol. 5, 423 (2014).
Jones, C. V. et al. M2 macrophage polarisation is associated with alveolar formation during postnatal lung development. Respir. Res. 14, 41 (2013).
Kramer, B. W., Kallapur, S., Newnham, J. & Jobe, A. H. Prenatal inflammation and lung development. Semin. Fetal Neonatal Med. 14, 2–7 (2009).
Guilliams, M. et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J. Exp. Med. 210, 1977–1992 (2013).
Kramer, B. W. et al. Endotoxin-induced maturation of monocytes in preterm fetal sheep lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 293, L345–L353 (2007).
Ivarsson, M. A. et al. Differentiation and functional regulation of human fetal NK cells. J. Clin. Invest. 123, 3889–3901 (2013).
Daniels, C. B. & Orgeig, S. Pulmonary surfactant: the key to the evolution of air breathing. News Physiol. Sci. 18, 151–157 (2003).
Wu, H. et al. Surfactant proteins A and D inhibit the growth of Gram-negative bacteria by increasing membrane permeability. J. Clin. Invest. 111, 1589–1602 (2003).
LeVine, A. M. et al. Distinct effects of surfactant protein A or D deficiency during bacterial infection on the lung. J. Immunol. 165, 3934–3940 (2000).
Hallman, M., Arjomaa, P., Mizumoto, M. & Akino, T. Surfactant proteins in the diagnosis of fetal lung maturity. I. Predictive accuracy of the 35 kD protein, the lecithin/sphingomyelin ratio, and phosphatidylglycerol. Am. J. Obstet. Gynecol. 158, 531–535 (1988).
Condon, J. C., Jeyasuria, P., Faust, J. M. & Mendelson, C. R. Surfactant protein secreted by the maturing mouse fetal lung acts as a hormone that signals the initiation of parturition. Proc. Natl. Acad. Sci. USA 101, 4978–4983 (2004).
Agrawal, V., Smart, K., Jilling, T. & Hirsch, E. Surfactant protein (SP)-A suppresses preterm delivery and inflammation via TLR2. PLoS One 8, e63990 (2013).
Roy, M. G. et al. Muc5b is required for airway defence. Nature 505, 412–416 (2014).
Roy, M. G. et al. Mucin production during prenatal and postnatal murine lung development. Am. J. Respir. Cell. Mol. Biol. 44, 755–760 (2011).
Ramond, C. et al. Two waves of distinct hematopoietic progenitor cells colonize the fetal thymus. Nat. Immunol. 15, 27–35 (2014).
Haas, J. D. et al. Development of interleukin-17-producing gammadelta T cells is restricted to a functional embryonic wave. Immunity 37, 48–59 (2012).
Ferguson, A. & Parrott, D. M. The effect of antigen deprivation on thymus-dependent and thymus-independent lymphocytes in the small intestine of the mouse. Clin. Exp. Immunol. 12, 477–488 (1972).
Latthe, M., Terry, L. & MacDonald, T. T. High frequency of CD8 alpha alpha homodimer-bearing T cells in human fetal intestine. Eur. J. Immunol. 24, 1703–1705 (1994).
Cheroutre, H., Lambolez, F. & Mucida, D. The light and dark sides of intestinal intraepithelial lymphocytes. Nat. Rev. Immunol. 11, 445–456 (2011).
Aagaard, K. et al. The placenta harbors a unique microbiome. Sci. Transl. Med. 6, 237ra265 (2014).
Collado, M. C., Rautava, S., Aakko, J., Isolauri, E. & Salminen, S. Human gut colonisation may be initiated in utero by distinct microbial communities in the placenta and amniotic fluid. Sci. Rep. 6, 23129 (2016).
Jimenez, E. et al. Isolation of commensal bacteria from umbilical cord blood of healthy neonates born by cesarean section. Curr. Microbiol. 51, 270–274 (2005).
Jimenez, E. et al. Is meconium from healthy newborns actually sterile? Res. Microbiol. 159, 187–193 (2008).
Dominguez-Bello, M. G. et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc. Natl. Acad. Sci. USA 107, 11971–11975 (2010).
Palmer, C., Bik, E. M., DiGiulio, D. B., Relman, D. A. & Brown, P. O. Development of the human infant intestinal microbiota. PLoS Biol. 5, e177 (2007).
Yatsunenko, T. et al. Human gut microbiome viewed across age and geography. Nature 486, 222–227 (2012).
Backhed, F. et al. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe 17, 690–703 (2015).
Pantoja-Feliciano, I. G. et al. Biphasic assembly of the murine intestinal microbiota during early development. ISME J. 7, 1112–1115 (2013).
Marsland, B. J. & Gollwitzer, E. S. Host-microorganism interactions in lung diseases. Nat. Rev. Immunol. 14, 827–835 (2014).
Gollwitzer, E. S. et al. Lung microbiota promotes tolerance to allergens in neonates via PD-L1. Nat. Med. 20, 642–647 (2014).
Lohmann, P. et al. The airway microbiome of intubated premature infants: characteristics and changes that predict the development of bronchopulmonary dysplasia. Pediatr. Res. 76, 294–301 (2014).
Bry, L. et al. Paneth cell differentiation in the developing intestine of normal and transgenic mice. Proc. Natl. Acad. Sci. USA 91, 10335–10339 (1994).
Hornef, M. W., Putsep, K., Karlsson, J., Refai, E. & Andersson, M. Increased diversity of intestinal antimicrobial peptides by covalent dimer formation. Nat. Immunol. 5, 836–843 (2004).
Vaishnava, S. et al. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science 334, 255–258 (2011).
Birchenough, G. M., Johansson, M. E., Gustafsson, J. K., Bergstrom, J. H. & Hansson, G. C. New developments in goblet cell mucus secretion and function. Mucosal Immunol. 8, 712–719 (2015).
Dupont, A. et al. Intestinal mucus affinity and biological activity of an orally administered antibacterial and anti-inflammatory peptide. Gut 64, 222–232 (2015).
Meyer-Hoffert, U. et al. Secreted enteric antimicrobial activity localises to the mucus surface layer. Gut 57, 764–771 (2008).
Pelaseyed, T. et al. The mucus and mucins of the goblet cells and enterocytes provide the first defense line of the gastrointestinal tract and interact with the immune system. Immunol. Rev. 260, 8–20 (2014).
Lotz, M. et al. Postnatal acquisition of endotoxin tolerance in intestinal epithelial cells. J. Exp. Med. 203, 973–984 (2006).
Chassin, C. et al. miR-146a mediates protective innate immune tolerance in the neonate intestine. Cell Host Microbe 8, 358–368 (2010).
Menckeberg, C. L. et al. Human buccal epithelium acquires microbial hyporesponsiveness at birth, a role for secretory leukocyte protease inhibitor. Gut 64, 884–893 (2015).
Pott, J. et al. Age-dependent TLR3 expression of the intestinal epithelium contributes to rotavirus susceptibility. PLoS Pathog. 8, e1002670 (2012).
Claud, E. C. et al. Developmentally regulated IkappaB expression in intestinal epithelium and susceptibility to flagellin-induced inflammation. Proc. Natl. Acad. Sci. USA 101, 7404–7408 (2004).
Bain, C. C. et al. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat. Immunol. 15, 929–937 (2014).
van de Laar, L. et al. Yolk sac macrophages, fetal liver, and adult monocytes can colonize an empty niche and develop into functional tissue-resident macrophages. Immunity 44, 755–768 (2016).
Torow, N. et al. Active suppression of intestinal CD4(+)TCRalphabeta(+) T-lymphocyte maturation during the postnatal period. Nat. Commun. 6, 7725 (2015).
Kasahara, K. et al. Intranasal priming of newborn mice with microbial extracts increases opsonic factors and mature CD11c+ cells in the airway. Am. J. Physiol. Lung Cell. Mol. Physiol. 303, L834–L843 (2012).
Lee, P. T., Holt, P. G. & McWilliam, A. S. Role of alveolar macrophages in innate immunity in neonates: evidence for selective lipopolysaccharide binding protein production by rat neonatal alveolar macrophages. Am. J. Respir. Cell. Mol. Biol. 23, 652–661 (2000).
Roux, X. et al. Neonatal lung immune responses show a shift of cytokines and transcription factors toward Th2 and a deficit in conventional and plasmacytoid dendritic cells. Eur. J. Immunol. 41, 2852–2861 (2011).
Fach, S. J., Brockmeier, S. L., Hobbs, L. A., Lehmkuhl, H. D. & Sacco, R. E. Pulmonary dendritic cells isolated from neonatal and adult ovine lung tissue. Vet. Immunol. Immunopathol. 112, 171–182 (2006).
Nelson, D. J. & Holt, P. G. Defective regional immunity in the respiratory tract of neonates is attributable to hyporesponsiveness of local dendritic cells to activation signals. J. Immunol. 155, 3517–3524 (1995).
Olszak, T. et al. Microbial exposure during early life has persistent effects on natural killer T cell function. Science 336, 489–493 (2012).
Garcia, A. M., Fadel, S. A., Cao, S. & Sarzotti, M. T cell immunity in neonates. Immunol. Res. 22, 177–190 (2000).
Lathrop, S. K. et al. Peripheral education of the immune system by colonic commensal microbiota. Nature 478, 250–254 (2011).
Kim, K. S. et al. Dietary antigens limit mucosal immunity by inducing regulatory T cells in the small intestine. Science 351, 858–863 (2016).
Yang, S., Fujikado, N., Kolodin, D., Benoist, C. & Mathis, D. Immune tolerance. Regulatory T cells generated early in life play a distinct role in maintaining self-tolerance. Science 348, 589–594 (2015).
Thome, J. J. et al. Early-life compartmentalization of human T cell differentiation and regulatory function in mucosal and lymphoid tissues. Nat. Med. 22, 72–77 (2016).
Harris, N. L. et al. Mechanisms of neonatal mucosal antibody protection. J. Immunol. 177, 6256–6262 (2006).
Rogier, E. W. et al. Secretory antibodies in breast milk promote long-term intestinal homeostasis by regulating the gut microbiota and host gene expression. Proc. Natl. Acad. Sci. USA 111, 3074–3079 (2014).
Lindner, C. et al. Age, microbiota, and T cells shape diverse individual IgA repertoires in the intestine. J. Exp. Med. 209, 365–377 (2012).
South, M. A., Warwick, W. J., Wolheim, F. A. & Good, R. A. The IgA system. 3. IgA levels in the serum and saliva of pediatric patients: evidence for a local immunological system. J. Pediatr. 71, 645–653 (1967).
Wesemann, D. R. et al. Microbial colonization influences early B-lineage development in the gut lamina propria. Nature 501, 112–115 (2013).
Kanamori, Y. et al. Identification of novel lymphoid tissues in murine intestinal mucosa where clusters of c-kit+ IL-7R+ Thy1+ lympho-hemopoietic progenitors develop. J. Exp. Med. 184, 1449–1459 (1996).
Bouskra, D. et al. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature 456, 507–510 (2008).
Knoop, K. A. & Newberry, R. D. Isolated lymphoid follicles are dynamic reservoirs for the induction of intestinal IgA. Front. Immunol. 3, 84 (2012).
Elahi, S. et al. Immunosuppressive CD71+ erythroid cells compromise neonatal host defence against infection. Nature 504, 158–162 (2013).
Qureshi, M. H. & Garvy, B. A. Neonatal T cells in an adult lung environment are competent to resolve Pneumocystis carinii pneumonia. J. Immunol. 166, 5704–5711 (2001).
Turfkruyer, M. & Verhasselt, V. Breast milk and its impact on maturation of the neonatal immune system. Curr. Opin. Infect. Dis. 28, 199–206 (2015).
Ballard, O. & Morrow, A. L. Human milk composition: nutrients and bioactive factors. Pediatr. Clin. North Am. 60, 49–74 (2013).
Good, M. et al. Breast milk protects against the development of necrotizing enterocolitis through inhibition of Toll-like receptor 4 in the intestinal epithelium via activation of the epidermal growth factor receptor. Mucosal Immunol. 8, 1166–1179 (2015).
Offit, P. A., Hoffenberg, E. J., Pia, E. S., Panackal, P. A. & Hill, N. L. Rotavirus-specific helper T cell responses in newborns, infants, children, and adults. J. Infect. Dis. 165, 1107–1111 (1992).
Tino De Franco, M. et al. Neutralizing activity and secretory IgA antibodies reactive with rotavirus SA-11 (serotype G3) in colostrum and milk from Brazilian women. Paediatr. Int. Child Health 33, 102–107 (2013).
Lindner, C. et al. Diversification of memory B cells drives the continuous adaptation of secretory antibodies to gut microbiota. Nat. Immunol. 16, 880–888 (2015).
Glocker, E. O. et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N. Engl. J. Med. 361, 2033–2045 (2009).
Engelhardt, K. R. et al. Clinical outcome in IL-10- and IL-10 receptor-deficient patients with or without hematopoietic stem cell transplantation. J. Allergy Clin. Immunol. 131, 825–830 (2013).
Kuhn, R., Lohler, J., Rennick, D., Rajewsky, K. & Muller, W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell 75, 263–274 (1993).
Kontoyiannis, D., Pasparakis, M., Pizarro, T. T., Cominelli, F. & Kollias, G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity 10, 387–398 (1999).
Berman, L. & Moss, R. L. Necrotizing enterocolitis: an update. Semin. Fetal Neonatal Med. 16, 145–150 (2011).
Hackam, D. J., Afrazi, A., Good, M. & Sodhi, C. P. Innate immune signaling in the pathogenesis of necrotizing enterocolitis. Clin. Dev. Immunol. 2013, 475415 (2013).
Sodhi, C. P. et al. Intestinal epithelial Toll-like receptor 4 regulates goblet cell development and is required for necrotizing enterocolitis in mice. Gastroenterology 143, 708–718.e1–5 (2012).
Yazji, I. et al. Endothelial TLR4 activation impairs intestinal microcirculatory perfusion in necrotizing enterocolitis via eNOS-NO-nitrite signaling. Proc. Natl. Acad. Sci. USA 110, 9451–9456 (2013).
Olsen, R., Greisen, G., Schroder, M. & Brok, J. Prophylactic probiotics for preterm infants: a systematic review and meta-analysis of observational studies. Neonatology 109, 105–112 (2016).
Thavagnanam, S., Fleming, J., Bromley, A., Shields, M. D. & Cardwell, C. R. A meta-analysis of the association between Caesarean section and childhood asthma. Clin. Exp. Allergy 38, 629–633 (2008).
Decker, E. et al. Cesarean delivery is associated with celiac disease but not inflammatory bowel disease in children. Pediatrics 125, e1433–e1440 (2010).
Cahenzli, J., Koller, Y., Wyss, M., Geuking, M. B. & McCoy, K. D. Intestinal microbial diversity during early-life colonization shapes long-term IgE levels. Cell Host Microbe 14, 559–570 (2013).
Dominguez-Bello, M. G. et al. Partial restoration of the microbiota of cesarean-born infants via vaginal microbial transfer. Nat. Med. 22, 250–253 (2016).
Hansen, C. H. et al. Patterns of early gut colonization shape future immune responses of the host. PLoS One 7, e34043 (2012).
Depner, M. et al. Atopic sensitization in the first year of life. J. Allergy Clin. Immunol. 131, 781–788 (2013).
Liu, L. et al. Global, regional, and national causes of child mortality: an updated systematic analysis for 2010 with time trends since 2000. Lancet 379, 2151–2161 (2012).
Ao, T. T. et al. Global burden of invasive nontyphoidal Salmonella disease, 2010(1). Emerg. Infect. Dis. 21 (2015).
Brando, R. J. et al. Renal damage and death in weaned mice after oral infection with Shiga toxin 2-producing Escherichia coli strains. Clin. Exp. Immunol. 153, 297–306 (2008).
Kotloff, K. L. et al. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet 382, 209–222 (2013).
Birchenough, G. M. et al. Altered innate defenses in the neonatal gastrointestinal tract in response to colonization by neuropathogenic Escherichia coli. Infect. Immun. 81, 3264–3275 (2013).
Barthel, M. et al. Pretreatment of mice with streptomycin provides a Salmonella enterica serovar Typhimurium colitis model that allows analysis of both pathogen and host. Infect. Immun. 71, 2839–2858 (2003).
Fernandez, M. I. et al. Maturation of paneth cells induces the refractory state of newborn mice to Shigella infection. J. Immunol. 180, 4924–4930 (2008).
Lee, H. H. et al. Delayed maturation of an IL-12-producing dendritic cell subset explains the early Th2 bias in neonatal immunity. J. Exp. Med. 205, 2269–2280 (2008).
Zaghouani, H., Hoeman, C. M. & Adkins, B. Neonatal immunity: faulty T-helpers and the shortcomings of dendritic cells. Trends Immunol. 30, 585–591 (2009).
Genovese, F. et al. Role of IL-10 in a neonatal mouse listeriosis model. J. Immunol. 163, 2777–2782 (1999).
Byun, H. J. et al. An evaluation of the neonatal immune system using a listeria infection model. Neonatology 92, 83–90 (2007).
Lantier, L. et al. Intestinal CD103+ dendritic cells are key players in the innate immune control of Cryptosporidium parvum infection in neonatal mice. PLoS Pathog. 9, e1003801 (2013).
Kupz, A. et al. Contribution of Thy1+ NK cells to protective IFN-gamma production during Salmonella typhimurium infections. Proc. Natl. Acad. Sci. USA 110, 2252–2257 (2013).
Zhou, R., Gong, A. Y., Eischeid, A. N. & Chen, X. M. miR-27b targets KSRP to coordinate TLR4-mediated epithelial defense against Cryptosporidium parvum infection. PLoS Pathog. 8, e1002702 (2012).
Bouzid, M., Hunter, P. R., Chalmers, R. M. & Tyler, K. M. Cryptosporidium pathogenicity and virulence. Clin. Microbiol. Rev. 26, 115–134 (2013).
Forsthuber, T., Yip, H. C. & Lehmann, P. V. Induction of TH1 and TH2 immunity in neonatal mice. Science 271, 1728–1730 (1996).
Ridge, J. P., Fuchs, E. J. & Matzinger, P. Neonatal tolerance revisited: turning on newborn T cells with dendritic cells. Science 271, 1723–1726 (1996).
Sarzotti, M., Robbins, D. S. & Hoffman, P. M. Induction of protective CTL responses in newborn mice by a murine retrovirus. Science 271, 1726–1728 (1996).
Adkins, B., Leclerc, C. & Marshall-Clarke, S. Neonatal adaptive immunity comes of age. Nat. Rev. Immunol. 4, 553–564 (2004).
Torow, N., Dittrich-Breiholz, O. & Hornef, M. W. Transcriptional profiling of intestinal CD4(+) T cells in the neonatal and adult mice. Genom. Data 5, 371–374 (2015).
Echeverry, A., Saijo, S., Schesser, K. & Adkins, B. Yersinia enterocolitica promotes robust mucosal inflammatory T-cell immunity in murine neonates. Infect. Immun. 78, 3595–3608 (2010).
Siefker, D. T. et al. Murine neonates infected with Yersinia enterocolitica develop rapid and robust proinflammatory responses in intestinal lymphoid tissues. Infect. Immun. 82, 762–772 (2014).
Adlerberth, I. et al. Toxin-producing Clostridium difficile strains as long-term gut colonizers in healthy infants. J. Clin. Microbiol. 52, 173–179 (2014).
Nair, H. et al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: a systematic review and meta-analysis. Lancet 375, 1545–1555 (2010).
Culley, F. J., Pollott, J. & Openshaw, P. J. Age at first viral infection determines the pattern of T cell-mediated disease during reinfection in adulthood. J. Exp. Med. 196, 1381–1386 (2002).
Dakhama, A. et al. The enhancement or prevention of airway hyperresponsiveness during reinfection with respiratory syncytial virus is critically dependent on the age at first infection and IL-13 production. J. Immunol. 175, 1876–1883 (2005).
Tregoning, J. S., Yamaguchi, Y., Harker, J., Wang, B. & Openshaw, P. J. The role of T cells in the enhancement of respiratory syncytial virus infection severity during adult reinfection of neonatally sensitized mice. J. Virol. 82, 4115–4124 (2008).
Ruckwardt, T. J. et al. Neonatal CD8 T-cell hierarchy is distinct from adults and is influenced by intrinsic T cell properties in respiratory syncytial virus infected mice. PLoS Pathog. 7, e1002377 (2011).
Ruckwardt, T. J., Malloy, A. M., Morabito, K. M. & Graham, B. S. Quantitative and qualitative deficits in neonatal lung-migratory dendritic cells impact the generation of the CD8+ T cell response. PLoS Pathog. 10, e1003934 (2014).
Tasker, L., Lindsay, R. W., Clarke, B. T., Cochrane, D. W. & Hou, S. Infection of mice with respiratory syncytial virus during neonatal life primes for enhanced antibody and T cell responses on secondary challenge. Clin. Exp. Immunol. 153, 277–288 (2008).
Dakhama, A. et al. Virus-specific IgE enhances airway responsiveness on reinfection with respiratory syncytial virus in newborn mice. J. Allergy Clin. Immunol. 123, 138–145 e135 (2009).
Lee, Y. M. et al. IFN-gamma production during initial infection determines the outcome of reinfection with respiratory syncytial virus. Am. J. Respir. Crit. Care Med. 177, 208–218 (2008).
Duke, T. Neonatal pneumonia in developing countries. Arch. Dis. Child Fetal Neonatal Ed. 90, F211–F219 (2005).
Martinez, F. D. The origins of asthma and chronic obstructive pulmonary disease in early life. Proc. Am. Thorac. Soc. 6, 272–277 (2009).
Yang, K. D. Perinatal programming of childhood asthma. Clin. Dev. Immunol. 2012, 438572 (2012).
Mukherjee, A. B. & Zhang, Z. Allergic asthma: influence of genetic and environmental factors. J. Biol. Chem. 286, 32883–32889 (2011).
Russell, S. L. et al. Perinatal antibiotic treatment affects murine microbiota, immune responses and allergic asthma. Gut Microbes 4, 158–164 (2013).
Krishnamoorthy, N. et al. Early infection with respiratory syncytial virus impairs regulatory T cell function and increases susceptibility to allergic asthma. Nat. Med. 18, 1525–1530 (2012).
Herberth, G. et al. Maternal and cord blood miR-223 expression associates with prenatal tobacco smoke exposure and low regulatory T-cell numbers. J. Allergy Clin. Immunol. 133, 543–550 (2014).
Schaub, B. et al. Maternal farm exposure modulates neonatal immune mechanisms through regulatory T cells. J. Allergy Clin. Immunol. 123, 774–782.e5 (2009).
Verhasselt, V. et al. Breast milk-mediated transfer of an antigen induces tolerance and protection from allergic asthma. Nat. Med. 14, 170–175 (2008).
Rinne, M., Kalliomaki, M., Arvilommi, H., Salminen, S. & Isolauri, E. Effect of probiotics and breastfeeding on the bifidobacterium and lactobacillus/enterococcus microbiota and humoral immune responses. J. Pediatr. 147, 186–191 (2005).
Dicksved, J. et al. Molecular fingerprinting of the fecal microbiota of children raised according to different lifestyles. Appl. Environ. Microbiol. 73, 2284–2289 (2007).
Tanaka, S. et al. Influence of antibiotic exposure in the early postnatal period on the development of intestinal microbiota. FEMS Immunol. Med. Microbiol. 56, 80–87 (2009).
Dominguez-Bello, M. G. et al. Partial restoration of the microbiota of cesarean-born infants via vaginal microbial transfer. Nat. Med. 22, 250–253 (2016).
Deshmukh, H. S. et al. The microbiota regulates neutrophil homeostasis and host resistance to Escherichia coli K1 sepsis in neonatal mice. Nat. Med. 20, 524–530 (2014).
Dupont, A. et al. Age-dependent susceptibility to enteropathogenic Escherichia coli (EPEC) infection in mice. PLoS Pathog 9, e1005616 (2016).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declared no conflict of interest.
PowerPoint slides
Rights and permissions
About this article

Cite this article
Torow, N., Marsland, B., Hornef, M. et al. Neonatal mucosal immunology. Mucosal Immunol 10, 5–17 (2017). https://doi.org/10.1038/mi.2016.81
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2016.81
This article is cited by
-
Neonatal intestinal mucus barrier changes in response to maturity, inflammation, and sodium decanoate supplementation
Scientific Reports (2024)
-
Early-life interactions between the microbiota and immune system: impact on immune system development and atopic disease
Nature Reviews Immunology (2023)
-
Questioning the fetal microbiome illustrates pitfalls of low-biomass microbial studies
Nature (2023)
-
Neonatal LTβR signaling is required for the accumulation of eosinophils in the inflamed adult mesenteric lymph node
Mucosal Immunology (2022)
-
Establishment of tissue-resident immune populations in the fetus
Seminars in Immunopathology (2022)
