
Abstract
Endogenous damage-associated molecular patterns (DAMPs) are released during tissue damage and have increasingly recognized roles in the etiology of many human diseases. The inflammatory bowel diseases (IBD), ulcerative colitis (UC) and Crohn’s disease (CD), are immune-mediated conditions where high levels of DAMPs are observed. DAMPs such as calprotectin (S100A8/9) have an established clinical role as a biomarker in IBD. In this review, we use IBD as an archetypal common chronic inflammatory disease to focus on the conceptual and evidential importance of DAMPs in pathogenesis and why DAMPs represent an entirely new class of targets for clinical translation.
Similar content being viewed by others

Introduction
The inflammatory bowel diseases (IBD), ulcerative colitis (UC) and Crohn’s disease (CD), affect an estimated 4 million people in the United States and Europe and have a rising incidence in the developing world.1, 2, 3, 4 Both conditions are incurable, often diagnosed at a young age and are associated with significant morbidity and socioeconomic costs.5, 6 UC is characterized by confluent superficial inflammation affecting only the colon; in CD, deep patchy ulcerations can affect any part of the gastrointestinal tract. In UC, 15% will develop acute severe colitis where the failure rate of medical therapy is high (∼30% requiring surgical removal of the colon).7 In CD, most patients will encounter a disabling disease course and approximately half will require surgery within 10 years of diagnosis.8, 9
The past decade has seen remarkable progress in understanding the pathogenesis of IBD, with notable advances in the contribution of genetic susceptibility, microbial flora, and environmental factors.4, 10, 11, 12 There are clear differences between UC and CD (Table 1). However, failure to resolve mucosal inflammation (which commonly reactivates upon withdrawal of anti-inflammatory treatments such as glucocorticoids) is a notable shared clinical feature. Complete mucosal healing, the strongest predictive factor for long-lasting remission, is difficult to achieve. Here, we review the relatively underexplored but potentially critical contribution of immunogenic endogenous “damage-associated molecular patterns” (DAMPs) as distinct stimuli that maintain the state of abnormal mucosal inflammation in IBD. We focus on their roles in initiating, perpetuating, and amplifying inflammation in IBD and cover key areas namely: (i) DAMPs implicated in IBD; (ii) their roles in modulating the abnormal inflammatory response; (iii) factors governing specific DAMP release; and finally (iv) why DAMPs represent attractive targets for clinical translation in IBD.
DAMPS: Alerting the host to danger and promoting inflammation
The inflammatory response is an essential component of host defense, primarily ensuring containment and clearance of pathogens. This sentinel function of the innate immune system rapidly and precisely distinguishes between “self” and “non-self” by recognizing microbial invariant molecular patterns (pattern-associated molecular patterns (PAMPs)) through a system of germline-encoded pattern recognition receptors (PRRs).13 PRR activation leads to intracellular signaling cascades, transcriptional upregulation of inflammatory genes, production of proinflammatory cytokines, chemokines, and type I interferons, and recruitment of inflammatory cells such as neutrophils.
Similar strong immune responses are seen in the absence of invasive pathogens (“sterile inflammation”) such as in autoimmunity, trauma, and ischemia. This phenomenon is explained by Matzinger’s “danger hypothesis” in which immune responses are geared toward recognizing danger whether these signals arise endogenously or exogenously.14 In this context, PRRs are activated by both non-self (PAMPs) and endogenous molecules released at times of danger to the host (DAMPs)15, 16, 17 (Figure 1). The major classes of PRRs are cell surface or endosomal Toll-like receptors (TLRs), cytoplasmic nucleotide binding and oligomerization domain (NOD)-like receptors (NLRs) and inflammasomes, C-type leptin receptors, RIG-1 like receptors (RLRs), and absence in melanoma 2 (AIM2)-like receptors.18, 19 In addition, the more DAMP-specific receptor for advanced glycation end-products (RAGE) is also categorized as a PRR.20, 21

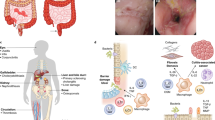
Danger recognition by the innate immune system. PRRs such as TLR, NLR, and RAGE sense danger associated with infection via recognition of evolutionarily conserved PAMPs on pathogens or sterile injury via recognition of DAMPs. Activation of cell surface or intracellular PRRs leads to intracellular signaling and inflammatory responses. DAMP cellular mechanisms. Cellular stress may also lead to damaged cellular components such as ROS generating mitochondria. Increased ROS production and oxidative stress may have multiple effects including increased translocation and active release of DAMPs and further cellular stress, leading to a vicious cycle. Defects in homeostatic pathways such as autophagy leads to escape of DAMPs such as mtDNA. Intranuclear DAMPs require translocation into the cytosol before active release. Active release (“secretion”) occurs through nonclassical pathways and cellular membrane rupture after necrosis or necroptosis results in passive release of DAMPs. ER stress contributes to the functional activity of DAMPs, e.g., through increased translocation and contributing to its role as an adjuvant; DAMPs can directly lead to increased ER stress. APC, antigen-presenting cell; DAMP, damage-associated molecular pattern; ER stress, endoplasmic reticulum stress; IBD, inflammatory bowel disease; IEC, intestinal epithelial cell; mtDNA, mitochondrial DNA; NLR, nucleotide binding oligomerization domain-like receptor; PAMP, pathogen-associated molecular pattern; PRR, pattern recognition receptor; RAGE, receptor for advanced glycation end-products; ROS, reactive oxygen species; TLR, Toll-like receptor.
DAMPs comprise structurally diverse nonpathogen-derived molecules that share a number of characteristics: (i) they bind to and activate PRRs; (ii) are passively leaked after plasma membrane rupture following various forms of cell death including necrosis, necroptosis, and secondary necrosis; (iii) may be actively secreted by stressed cells via nonclassical pathways independent of the endoplasmic reticulum (ER)/Golgi apparatus; and (iv) may change from a physiological to a proinflammatory function when released into the extracellular milieu.22 Extracellular DAMPs may activate cell surface PRRs or intracellular PRRs after phagocytosis, endocytosis, or other mechanisms of internalization.23 DAMPs may originate from any compartment of stressed cells and include intracellular proteins, extracellular matrix-derived proteins, and purinergic molecules. The list of recognized DAMPs is growing rapidly—a list of putative DAMPs and their receptors is provided in Table 2 (references are provided in Supplementary Table S1 online).
DAMPS in acute and chronic inflammation
Under physiological conditions, DAMPs reside intracellularly or are sequestered in the extracellular matrix, and are thus hidden from recognition by innate immune cells bearing PRRs. In response to perceived danger such as tissue damage, DAMPs are liberated extracellularly, serving to signal danger to the host and promoting inflammation and repair processes that are initially beneficial and protective.23 However, in the setting of significant and persistent DAMP release, their downstream effects may result in deleterious “collateral damage” and therefore have a central role in disease pathogenesis (Figure 2). The clearest example is in acute gout, where uric acid crystals directly trigger the NLRP3 inflammasome, leading to overwhelming inflammation and, if uncontrolled, joint destruction.24

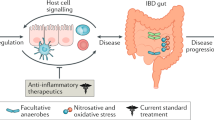
Contribution of DAMPs to inflammatory response in IBD. In health, IECs undergo constant shedding and apoptosis. Tissue damage releases danger signals that initiate a protective inflammatory response to restore tissue homeostasis. In IBD, nonapoptotic cell death, mucosal oxidative stress, and deregulation of homeostatic pathways lead to overwhelming release of DAMPs, creating a proinflammatory milieu. These DAMPs lead to an inflammatory response through a variety of pathways, leading to further tissue damage and ongoing IEC death. APC, antigen-presenting cell; DAMP, damage-associated molecular pattern; DC, dendritic cell; HMGB1, high-mobility group box 1; IBD, inflammatory bowel disease; IEC, intestinal epithelial cell; IL, interleukin; PRR, pattern recognition receptor; RAGE, receptor for advanced glycation end-products; TREM-1, triggering receptor expressed on myeloid cells 1; UPR, unfolded protein response.
The role of DAMPs has been explored in disease models using direct administration of purified or recombinant DAMPs and/or depletion via antagonists or antibodies.25 DAMP genetic knockout (KO) studies have limitations as they are unable to discriminate between the physiological intracellular and proinflammatory extracellular functions of DAMPs. In the first study to demonstrate how DAMP administration can cause inflammation in vivo, Johnson et al.26 observed features resembling the systemic inflammatory response syndrome after administration of the DAMP soluble heparan sulfate. Systemic administration of a recombinant form of the DAMP high-mobility group box 1 protein (HMGB1) in mice is lethal,27 with gut epithelial barrier dysfunction being a notable feature.28 In a study of trauma patients, mitochondrial DAMPs released at the time of injury led to systemic inflammatory response syndrome mediated via TLR9 and formyl peptide receptor-1 (FPR1) activation.29 In sepsis, initial PAMP-mediated cellular damage may lead to further DAMP release and subsequent DAMP–PRR inflammatory signaling. In a study illustrating this concept, lethal anthrax challenge in baboons was associated with only transiently elevated bacterial DNA, whereas mitochondrial DAMP levels remained elevated until death.30 When DAMP release was indirectly suppressed by activated protein C treatment in this study, an increased rate of survival was noted. This suggests that endogenous DAMPs may potentiate disease severity in conditions where PAMPs have an initial triggering role.
Levels of DAMPS are increased in IBD
Although the importance of DAMPs in acute inflammation is well documented, their precise role in chronic inflammatory diseases is less clear. High levels of various DAMPs have been observed in active inflammatory autoimmune, skin, cardiovascular, renal, allergic, and metabolic conditions.31, 32, 33, 34, 35, 36 In IBD, the chronic and extensively inflamed gut mucosa represents an enriched source of local and systemic DAMPs. It rationally follows, and unsurprisingly, several DAMPs are found in abundance during active IBD including the S100A calgranulins (S100A8/9 complex or calgranulin A/B or MRP8/14 or calprotectin; and S100A12), HMGB1, and interleukin (IL)-1α and IL-33. The latter group DAMPs are regarded as “alarmins,”37 molecules that possess cytokine-like functions that are stored in cells and released upon uncontrolled cell death.
It is salutary to note that the use of DAMPs as biomarkers in IBD is established. Fecal calprotectin testing has revolutionized IBD clinical practice with roles in differentiating IBD from functional gut disorders;38, 39, 40 as a marker of disease activity41 and to predict subsequent course of disease.42 Calprotectin is now also a measurable outcome in current clinical IBD therapeutic trials. Calprotectin is a major cytosolic protein found in neutrophils and other inflammatory cells and is released by stressed cells during intestinal inflammation. Elevated serum and/or plasma levels of calprotectin have been found in numerous inflammatory diseases including IBD,43 psoriasis,44 vasculitis,45 and rheumatoid arthritis.46, 47 Lactoferrin, a marker of neutrophil degranulation that acts as an alarmin,48 is also detectable in the stool and can be used to differentiate IBD from functional disorders.49 High levels of serum and fecal S100A12 is found in active IBD, although existing studies are limited by size and most relate to the pediatric cohort.50, 51, 52, 53, 54, 55, 56 Similarly, fecal HMGB1 is raised in intestinal inflammation associated with IBD.57, 58 Serum59, 60 and mucosa epithelial-derived IL-33 expressions are increased in active IBD;59, 60, 61, 62, 63, 64 high levels of IL-1α are found in cultured colonic biopsies65 and lamina propria mononuclear cells66 of IBD patients. A comprehensive list of DAMPs implicated in IBD and experimental colitis is provided in Table 3, although it is noteworthy that many DAMPs have yet to be studied in the context of intestinal inflammation.
The functional consequence of DAMP release in IBD
Direct proinflammatory role of DAMPs
PRR signaling and activation of downstream transcription factors such as nuclear factor-κB is essential to maintain intestinal mucosal host defense and barrier function.11, 67 However, excessive or persistent PRR signaling can result in chronic intestinal inflammation, when this balance is lost.11 Despite their structural heterogeneity, PAMPs and DAMPs are often recognized by the same PRRs, although the structural biology underlying DAMP–PRR interaction remains poorly understood. As evident in the examples below, it is an oversimplification to suggest that all gut-released DAMPs are proinflammatory. In general, the nature and extent of the inflammatory response after DAMP–PRR interaction is likely to depend on the setting and the specific DAMP(s) involved.
HMGB1, the prototypic DAMP, provides a model of the impact of DAMPs when released after injury. HMGB1 is an abundant nuclear chromatin-binding protein expressed in almost all cell types.68 Once extracellular, HMGB1 can bind to one of several PRRs including RAGE, TLR2, TLR4, and TLR969, 70, 71, 72 or form complexes with DNA, lipopolysaccharide, cytokines, or lipids.73 Under physiological conditions, nuclear HMGB1 binds double-stranded DNA and facilitates chromatin bending that supports gene transcription.74 HMGB1 translocates to the cytoplasm in response to cellular stress; cytoplasmic but not nuclear HMGB1 expression is significantly enhanced in the biopsies of inflamed gut tissues.57 Passive release of cytoplasmic HMGB1 occurs after necrosis and associated loss of cell membrane integrity. Active extracellular secretion of HMGB1 may occur by a variety of immune cells (predominantly macrophages and monocytes but also natural killer cells, dendritic cells (DCs), neutrophils, eosinophils, and platelets) in response to plasma membrane receptor activation by extracellular components such as lipopolysaccharide and proinflammatory cytokines, endogenous inflammatory stimuli, or apoptotic cells.27, 74, 75
In intestinal inflammation, high HMGB1 levels are found in the feces58, 76, 77 and serum.78 In dextran sulfate sodium (DSS) colitis, cytoplasmic expression of epithelial and macrophage HMGB1 are associated with areas of necrosis, indicating translocation from its physiological nuclear compartment.78 Inhibition of HMGB1 appears to be protective in acute DSS colitis.76, 78 Constitutive deletion of HMGB1 is not compatible with survival.79 Of interest however, gut epithelial-specific HMGB1-KOs exacerbates DSS colitis, highlighting the additional physiological role of intracellular HMGB1.80 Other tissue-specific conditional KOs of HMGB1 have found conflicting survival outcomes, underlining its divergent intracellular and extracellular roles.81, 82, 83, 84 Here myeloid-, hepatocyte-, or pancreas-specific KOs of HMGB1 did not ameliorate but instead exacerbated lipopolysaccharide- or injury-induced damage and inflammation. This again may reflect on the homeostatic role of HMGB1 in maintaining the genome and cell survival, and preventing histone release.
Calprotectin, the most clinically relevant DAMP in IBD, is primarily expressed in neutrophils and macrophages with intracellular functions including calcium binding, regulation of microtubules, and modulation of the cytoskeleton.85 Like HMGB1, calprotectin may be passively released extracellularly after cellular rupture or actively secreted by inflamed endothelium-primed phagocytes.47 Calprotectin can bind to TLR4, RAGE, and surface heparan sulfate proteoglycan and carboxylated N-glycans on endothelial cells, resulting in downstream nuclear factor-κB activation.86, 87, 88 In certain vasculitides, the sites of inflammation are characterized by infiltration of leukocytes,45 higher overall circulating serum calprotectin levels, and higher cell surface calprotectin expression on macrophages.89
The case for calprotectin as a strictly proinflammatory DAMP appears more complex as it also functions as an antimicrobial protein.90 In this study, the name “calprotectin” was first suggested because of its calcium-binding properties and the finding that the protein inhibited the growth of various fungi and bacteria. Furthermore, when liberated in high quantities in the feces, calprotectin sequesters essential micronutrients metals such as zinc, thereby limiting their availability to microbes, a process termed nutritional immunity.91 During the release of calprotectin following uncontrolled cell death, human neutrophils also contain high concentrations of anti-inflammatory defensins.92 Furthermore, extracellular traps produced by dying neutrophils sequester calprotectin that may limit its proinflammatory effect.93 Most biomarker studies in IBD have focused on fecal calprotectin. As will be discussed later, calprotectin released into the local and systemic circulation may have different functional consequence to that released into the gut lumen.
The alarmins IL-1α and IL-33 are DAMPs implicated in IBD and experimental colitis (Table 3). Full-length IL-1α and IL-33 (pro-IL-1α and pro-IL-33) are constitutively expressed in resting cells, including epithelial cells, under normal conditions and retain intracellular function as transcription factors.94, 95 They do not require proteolytic processing for activity and can therefore exert their biological activity when released into the extracellular milieu,96, 97, 98, 99 a characteristic that ensures quick action at the time of initial tissue injury to act as effective alarm signals. IL-1α and IL-33 bind with high affinity to specific receptors of the Toll–IL-1 receptor (TIR) superfamily (IL-1 receptor type I (IL-1RI) for IL-1α; ST2 (also known as IL1RL1) for IL-33). Although these receptors are not classic PRRs, they perform PRR-like functions in recognizing endogenous alarmins to activate proinflammatory pathways. IL-1RI shares a common cytoplasmic TIR domain with TLRs;100 a key study showed that IL-1α-dependent activation of IL-1R by dead cells was an important trigger of the inflammatory response.101 In addition, release of IL-1α induces the recruitment of neutrophils during sterile inflammation.102
In colitis, stressed or necrotic intestinal epithelial cells (IECs) initially release extracellular full-length IL-33 that engages the ST2 receptor, leading to the release of proinflammatory cytokines via a MyD88-dependent pathway.103 Oboki et al.103 found that colitis was less severe in IL-33−/− mice during early stages of DSS challenge, fitting with a DAMP pattern of contribution to innate injury-driven colitis. Later, IL-33 is secreted by a variety of lamina propria cells in response to inflammatory cytokines104 and can engage T helper type 2 (Th2), as well as T helper type 1/17 (Th1/Th17) immune responses.105, 106 Interestingly, in healthy colons, ST2 expression appears to be abundantly expressed in colonic epithelial cells, whereas this expression is lost during inflammation, at which time it is upregulated in the lamina propria.60 Hence, the picture is different in chronic inflammatory settings (to be discussed later). This pathway is clinically relevant to IBD as Latiano et al.107 found a significant association between IL-33/ST2 single-nucleotide polymorphisms with both UC and CD, implicating IL-33 as a novel IBD susceptibility gene. In the case of IL-1α, high levels of mRNA are detectable early in the course of immune complex-induced colitis in rabbits with a high degree of correlation with necrosis and inflammation.108 Bersudsky et al.109 recently used IL-1α-deficient mice and neutralization experiments to show that IEC-derived IL-1α initiates and propagates DSS colitis, raising the possibility that IL-1α acting as a DAMP has an important triggering role early in IBD-associated inflammation.
DAMP pathways in IBD
Some aspects of PRR signaling relevant to IBD may be at least partially DAMP specific. One such example is activation of RAGE, a member of the immunoglobulin superfamily of cell surface molecules that recognizes a variety of ligands including HMGB1, S100 proteins, advanced glycation end-products, B2 integrins, amyloid-β, and amyloid fibrils but not PAMPs.110 RAGE expression is upregulated when its ligands are abundant;111 it follows that RAGE expression is increased in inflamed CD gut tissue where high levels of its ligands has been demonstrated.112, 113 Several studies have shown a major role for neutrophil recruitment and migration.81, 112, 114 Huebener et al.81 recently suggested that HMGB1-activating RAGE may have a dominant role in this context. The in vitro studies show that anti-RAGE antibodies inhibit neutrophil migration and cytokine release in intestinal epithelial cells.112, 114 The in vivo administration of soluble RAGE, which acts as a decoy receptor, suppresses inflammation in IL-10-deficient mouse model of colitis.115 A number of small studies have attempted to correlate blood soluble RAGE levels with the presence and activity of IBD with conflicting results.55, 116, 117, 118, 119
In addition to RAGE, DAMP regulatory pathways may play a role in IBD. The TREM-1 (triggering receptor expressed on myeloid cells 1) is an immunoglobulin present on monocytes and neutrophils that upregulates DAMP–PRR-mediated signaling.120 TREM-1 expression is upregulated in IBD and its expression correlates with endoscopic assessment of disease activity.121 Furthermore, TREM-1 blockade with small molecules attenuates mouse DSS colitis.122 In an in vitro study, TREM-1 inhibition with a recombinant chimeric protein attenuated the HMGB1 and heat shock protein 70 (HSP70)-induced proinflammatory response.120 In contrast to the upregulating effects of TREM-1, CD24-Siglec signaling (Siglec-G in mice; Siglec-10 in humans) has been shown to suppress DAMP-related but not PAMP-related inflammation.123 Siglecs (sialic acid-binding immunoglobulin-like lectins) are members of the Ig superfamily that bind with CD24 and selectively repress DAMP-mediated inflammation, possibly via phosphatases acting on PRRs.124 CD24-Siglec signaling has an anti-inflammatory role in models of acetaminophen-related hepatic injury123 and sepsis,125 but has not yet been investigated in vivo in colitis.
Modulation of the adaptive immune response
Beyond simply behaving as immunogenic molecules for the innate immune system, DAMPs have an increasingly recognized role as adjuvants, directly or indirectly interacting with the adaptive immune system. In IBD, the inflammatory milieu enriched with DAMPs is fertile ground for shaping adaptive immune responses. In general, and consistent with Matzinger’s danger hypothesis, necrotic cells appear to activate DCs and augment the generation of CD4+ and CD8+ T-cell responses.126, 127, 128 This mechanism was postulated to explain how T-cell responses are generated in conditions such as cancer, transplants, and autoimmunity in the absence of microbial infection.25 Subsequently, several studies in related fields have provided strong evidence that DAMPs have effects on T-cell function and are capable of modulating antigen-presenting cell/T-cell interaction. A number of DAMPs, including HMGB1,129, 130, 131 HSP60,132 and HSP70,133, 134 appear to assist with T-cell priming by indirect stimulation of DC maturation. Genomic DNA and uric acid released by necrotic cells also have a similar effect.135, 136 Furthermore, culture of DCs in the presence of HMGB1 (ref. 137) or HSP60 (ref. 132) result in a Th1-type cytokine response, demonstrating a role for DAMPs in driving particular adaptive immune responses.
DAMPs have been shown to act as adjuvants promoting antigen-specific T-cell responses. After coinjection with antigen in vivo, uric acid enhanced CD8+ T-cell responses and uric acid depletion led to reduced adjuvant activity.136, 138Vaccination with hyaluronan as an adjuvant leads to increased cytokine responses in mice after antigen rechallenge.139 Similarly, lactoferrin augments the efficacy of the BCG (Bacillus Calmette–Guérin) vaccine through the generation of a T helper response140 and defensins promote T cell-dependent cellular immunity and antigen-specific Ig production in mice.141 The evolutionary basis of this as a protective mechanism against microbes is clear. However, in the context of exacerbated T-cell responses such as in IBD, this adjuvant role of DAMPs may in fact be harmful. This hypothesis has not yet been fully investigated. In a different setting, DAMP release from dying cancer cells has received considerable recent attention because of the possibility of DAMP-mediated activation of antitumorigenic T-cell immunity with implications for immunotherapy.142
Calprotectin is important for the induction of autoreactive CD8+ T cells and the development of systemic autoimmunity.143 In a T cell-mediated autoimmune mouse model of transgenic mice overexpressing the CD40 ligand (CD40lg), Loser et al.143 found that disease onset and severity was delayed and reduced respectively when Mrp14 was deleted. The authors suggested that Mrp8/14 functions as a TLR4 ligand on autoreactive CD8+ T cells that upregulate IL-17 expression and induce autoimmunity in mice and humans. This has yet to be studied in detail in mouse colitis models and may be more complex when considered in different disease settings. For example, in a T cell-mediated model of allergic contact dermatitis, Mrp14 deletion led to more severe disease.144 Here, it is suggested that loss of Mrp8 and Mrp14 resulted in enhanced DC differentiation and antigen presentation accounted for this finding.
More recently, Schiering et al.145 showed that IL-33 also has an immunoregulatory role in the intestine, where it enhances transforming growth factor-β-mediated differentiation of T regulatory cells and provides the necessary signal for T regulatory cell accumulation in inflamed mucosa. Here, ST2 appears to be preferentially expressed on colonic T regulatory cells. IL-23, an important proinflammatory cytokine in IBD, is shown to limit IL-33 effect. Hence, in this context, IL-33 plays an anti-inflammatory role; as discussed earlier, the role of IL-33, and indeed for DAMPs in general, is likely to be context dependent and, in this instance, dependent on the stage of colitis. This is further supported by the finding that IL-33, when administered to DSS-treated mice, led to an aggravation of acute colitis but a significant improvement in chronic colitis.146
DAMPs and epithelial barrier function
Intestinal epithelial dysfunction has an important contributory role in IBD where disruption of any components of this strategic barrier can lead to pathogenic interaction between luminal contents and resident immune cells within the underlying lamina propria.147, 148 A number of studies show how DAMPs can affect epithelial barrier function.28, 149, 150 Several mechanisms have been proposed: IL-33 administration impairs epithelial barrier in experimental colitis;149 HMGB1 has similar effects via an inducible nitric oxide synthase-dependent pathway in mice;28 and calprotectin causes epithelial barrier dysfunction in endothelial cells by engaging TLR4 and RAGE thereon influencing the endothelial cytoskeleton and tight junction proteins.151 The effects of HMGB1 may be potentiated via an autocrine feedback loop in immunostimulated enterocytes that further release HMGB1.152 Anti-HMGB1 neutralizing antibodies ameliorate gut barrier dysfunction in a hemorrhagic shock model.150 In humans, calprotectin and S100A12 from biopsies of active IBD areas upregulated adhesion molecules and chemokines in normal colonic endothelial cells in vitro.153 Furthermore, calprotectin increases vascular permeability via downregulation of cell junction-associated proteins and subsequent effects on endothelial monolayer integrity.154
Although activation of the inflammasomes by DAMPs is strongly proinflammatory,155 inflammasome activation also has important effects on epithelial barrier homeostasis. Like TLR activation, IL-18 has a compartmentalized effect on the epithelium. Upon activation within IECs, IL-18 induces IEC proliferation and regeneration, whereas its effect via lamina propria-resident immune cells aggravates gut barrier dysfunction through production of proinflammatory mediators and chemoattractants.156 Several studies show that mice deficient in NLRP3 are highly susceptible to gut epithelial injurious stimuli and death.157, 158, 159 Furthermore, NLRP6 inflammasome regulates colonic mucus production and microbiota, which are key components to maintain epithelial health.160, 161
Mechanisms regulating DAMP activity and clearance relevant to IBD
As discussed, current evidence suggests the load and composition of DAMPs may determine whether their effects become pathogenic, hence reemphasizing the delicate balance between the protective and pathologic roles of DAMPs. Here we further review the different factors that may influence this balance in the context of IBD.
The manner of cell death affects DAMP release
In health, the intestinal epithelium is replaced every 5–7 days; epithelial cells are either shed or die by apoptosis. In active IBD, nonapoptotic cell death, for example epithelial necrosis, occurs more commonly.162 More recently, necroptosis or programmed necrosis is increasingly appreciated as an alternative mechanism163 that appears to contribute to intestinal inflammation similar to that found in IBD.164, 165 The factors that determine whether a cell commits to necroptosis as opposed to apoptosis are complex and not yet fully understood.166 A key step in necroptosis is caspase-8 inhibition that results in RIPK1 and RIPK3 accumulation, phosphorylation, and RIPK1/RIPK3 complex IIb (“necrosome”) assembly.167, 168 Necrosome formation leads to RIPK3-dependent phosphorylation of mixed-lineage kinase domain-like protein (MLKL)169 that promotes an orderly form of necrotic cell death district from caspase-dependent apoptosis. RIPK1 also appears to have a kinase-independent role in regulating intestinal homeostasis where IEC-specific RIPK1 KO mice develop severe intestinal inflammation associated with IEC apoptosis.170, 171 Necrostatins such as necrostatin-1 inhibit necroptosis through inhibition of RIPK1 and have been used to investigate the functional role of necroptosis in animal models.172
Of interest, relevant KO mouse models suggest a role for necroptosis in IBD.164, 165, 173 IEC-specific FADD KO164 results in spontaneous enteritis/colitis and IEC-specific caspase-8 KO165 leads to reduced goblet cells, terminal ileum inflammation, and increased susceptibility to colitis. Intriguingly, both these necroptosis models exhibited Paneth cell depletion that is a feature of IBD; Paneth cells have an important role in the maintenance of epithelial barrier function including secretion of antimicrobial peptides. Furthermore, acute systemic deletion of caspase-8 (tamoxifen induced-Cre recombinase in floxed caspase-8) resulted in marked weight loss and lethality, with a predominant picture of gut enterocyte death and inflammation.173 Both FADD and caspase-8 KO is rescued by RIPK3 ablation.164, 173 These findings collectively show that that IEC necroptosis is a major factor that can trigger gut inflammation. It remains possible that these clinical phenotypes are primarily driven by loss of barrier and specialized enterocyte function (Paneth cells in this case) rather than mucosal DAMP release. Some limited evidence in human studies links necroptosis to IBD. Paneth cell loss in ileal biopsies is triggered by tumor necrosis factor but necrostatin-1 reversed this phenomenon.165, 174 High levels of RIPK3, MLKL, and lower caspase-8 are observed in IBD intestinal biopsies;174 in CD, increased necroptosis and decreased Paneth cell numbers are observed in affected ileal sections.165
Necroptosis lacks the massive caspase activation seen in apoptosis and this leads to comparative DAMP activation. For example, the lack of caspase-activated DNase means genomic DNA is not cleaved, leading to higher molecular weight DNA with greater proinflammatory potential.175 Similarly, full-length IL-33 is released in necroptosis compared with the nonimmunological IL-33 in apoptosis because of caspase-dependent proteolysis.98 HMGB1 is oxidized into its nonimmunological form during apoptosis by caspase-mediated reactive oxygen species (ROS) with irreversible binding to chromatin, but this does not occur in necroptosis.176 The DAMP–necroptosis link has been illustrated in several experimental models of necroptosis in skin, brain, and systemic inflammation that have shown higher levels of various DAMPs such as S100A9, IL-33, mitochondrial DNA (mtDNA), and HMGB1.163
The influence of the mucosal milieu on the inflammatory properties of DAMPs
Increased mucosal oxidative stress is another key feature of active IBD that can enhance the proinflammatory effects of DAMPs. An oxidative milieu modifies various proteins and lipids such as cholesteryl ester hydroperoxides and oxidized phospholipids, activating their role as potent DAMPs causing further inflammation.177, 178 There are several important examples. HMGB1 is redox sensitive and high levels of oxidative stress modulates its inflammatory potential.73 Purified HMGB1 only has weak proinflammatory activity.179 Low levels of ROS generation leads to cytosolic translocation of acetylated HMGB1 and autophagy-assisted secretion of the reduced, all-thiol form extracellularly that has chemotactic but no immunostimulatory properties.180, 181 Increasing oxidative stress initially leads to activation of the caspase cascade and oxidation of HMGB1 that is immunologically inactive when released extracellularly.182 At a critical level, excessive ROS results in uncontrolled cell death with subsequent passive, immunologically active HMGB1 release.73, 183 Similarly, oxidized mtDNA also becomes significantly more inflammatogenic. Shimada et al.184 found that cytosolic oxidized mtDNA rather than its nonoxidised form directly activates the NLRP3 inflammasome and IL-1β production. Pazmandi et al.185 further showed the increased immunogenicity of oxidatively modified mtDNA on plasmacytoid DCs compared with native mtDNA. Other DAMPs such as calreticulin and uric acid have been postulated to be susceptible to oxidative stress modification because of their regulatory protein and antioxidant properties.182
Deregulation of mucosal homeostatic pathways prime the inflammatory potential of DAMPs
Defective autophagy and the unfolded protein response (UPR) regulating ER stress are important in the pathogenesis of IBD.186 A meta-analysis of genome-wide association studies has identified the autophagy genes ATG16L1 and IRGM as key susceptibility genes particularly in CD.10 The T300A genetic mutation in ATG16L1 (a single-nucleotide polymorphism conferring a twofold risk for CD) sensitizes the gene to caspase-3-mediated degradation and consequent loss of autophagy function in response to cellular stress.187 ER stress-related genes have been implicated in IBD by genome-wide association studies (ORMDL3)10 and candidate gene approaches (XBP1 and AGR2).186, 188 The importance of autophagy in endogenous DAMP-mediated inflammation is increasingly appreciated, although its role in the clearance of intracellular pathogens (“xenophagy”) is established.
From a DAMP perspective, failure to clear proinflammatory damaged mitochondria is a key consequence of defective autophagy. Dysfunctional, ROS-generating mitochondria,189 and specifically oxidized mtDNA184 can activate the NLRP3 inflammasome. Other DAMPs such as extracellular matrix components biglycan and hyaluronic acid can additionally prime inflammasome activation in this context.190 Nakahira et al.155 showed that defective autophagy promotes the accumulation of mitochondrial DAMPs leading to NLRP3 activation. Indeed, in ATG16L1 deficiency there is an increased susceptibility to inflammasome-mediated release of IL-1β and IL-18.191 A further study showed that defective autophagy can lead to the release of DAMPs and subsequently contribute directly to inflammatory pathology in vivo.192 Here, Oka et al.192 showed that mice deficient in DNase leaked mtDNA and developed a TLR9-mediated proinflammatory state, cardiomyopathy, and heart failure. These studies point to a failure in autophagy resulting in a higher load of inflammatory intracellular DAMPs. It is noteworthy that in vivo mouse models of ATG16L1 deficiency (chimeric,191 hypomorphic,193 human IBD ATG16L1 polymorphism T300A knock-in,194 and epithelial specific ATG16L1-deficiency195, 196) do not develop spontaneous colitis but are very susceptible to gut inflammation when subjected to additional injurious stimuli (DSS, murine norovirus, or genetic deficiency of ER stress). This suggests a possible potentiating rather than initiating role in gut inflammation.
In terms of ER stress, there is some evidence to show that DAMPs can directly result in increased ER stress.197, 198 Endothelial cells exposed to HMGB1 led to higher expression of the ER stress sensors PERK and IREI that were markedly reduced after pretreatment with anti-RAGE antibodies.197 Furthermore, protein and mRNA levels of the ER stress marker GRP78 were elevated in HMGB1-treated DCs.198 Intriguingly, HMGB1 coculture enhanced the T-cell proliferation capabilities of DCs but this was not seen when XBP-1 was silenced, implicating the ER stress response and the UPR in the maturation and activation of DCs activated by DAMPs. In addition, high levels of ER stress may modify the inflammatory potential of DAMPs. In a study by Garg et al.,199 high levels of ROS-mediated ER stress before cell death increased calreticulin expression and adenosine triphosphate secretion.
Targeting DAMP-mediated inflammation and clinical translation
The role of DAMPs as functionally active mediators of inflammation makes this class a highly novel and exciting therapeutic target in IBD that has already shown promise in related inflammatory diseases (summarized in Supplementary Table S2). Presently, most potential DAMP therapeutics have yet to be studied in clinical trials. A number of challenges exist and these include: understanding complex disease-specific DAMP biology with their diverse often competing effects; how to localize therapeutic effects to the site of inflammation; deciphering DAMP–PRR and DAMP–DAMP interactions; understanding the triggers for DAMP release; and how DAMP-mediated signaling varies depending on context.
The list of DAMPs is rapidly growing and here we provide brief overviews of the potential strategies of translation in IBD: (i) targeting the mechanism or pathways mediating DAMP release; (ii) direct inhibition of DAMP action and its downstream interactions; (iii) modulation of factors that shape the pathogenicity of DAMP; and finally (iv) as potential functional biomarkers of disease activity. We envisage the clinical position for such approaches to be therefore complementary to current anti-inflammatory treatments (e.g., corticosteroids, anti-tumor necrosis factors) to reduce the severity and promote complete resolution of inflammation.
In (i), specific DAMP pathways as described earlier are relevant in IBD, namely necroptosis and autophagy. In the former, necrostatin-1, a necroptosis suppressor, improves the outcome of a number of inflammatory experimental mouse models200, 201 with lower levels of HMGB1, IL-23, IL-17A, and ROS.202 RIPK1, RIPK3, and MLKL203 may be plausible targets for therapy in addition to upstream (e.g., FADD/caspase-8) mechanisms. For example, the small-molecule necrosulfonamide inhibits MLKL and arrests necroptosis in human cells.169 This approach however may be an oversimplification as the biological processes of inflammation vis-a-vis with apoptosis and necroptosis remain complex and requires further thought. For example, RIPK1 plays a key role at the crossroads of nuclear factor-κB-mediated cell survival, caspase-8-dependent apoptosis, and RIPK3-dependent necroptosis. Such consideration is also noteworthy in autophagy, given its diverse biological roles in cellular homeostasis. There is some evidence to show that pharmacological activation of autophagy (sirolimus or everolimus)204 are effective at ameliorating murine models of colitis.205, 206 Sirolimus has been used successfully to treat CD in a case report,207 but clinical trials in everolimus have been negative in CD.208
In (ii), HMGB1 provides a good example of direct therapeutic targeting of DAMPs via small molecules or antibodies. There are several compounds (including anti-HMGB1 neutralizing antibodies, steroid derivatives, ethyl pyruvate, ghrelin, and others) that block HGMB1 cytoplasmic translocation and cellular release and demonstrate protective effects in mouse models of inflammation (Supplementary Table S2). The downstream DAMP–PRR interaction also offers opportunities, specifically via targeting PRRs (as in the case of ST2 or RAGE) or factors that modify this signaling (e.g., TREM-1). In the case of IL-33, which is elevated in active IBD,60 inhibition of ST2 has been successful in experimental models of colitis and arthritis.149, 209 Targeting of RAGE, which is a receptor for multiple DAMPs, has also been successful.115, 210, 211, 212, 213, 214 A recent study suggests that some of the anti-inflammatory activity of methotrexate may be because of inhibition of HMGB1/RAGE signaling via attachment to the RAGE-binding region of HMGB1.215 TREM-1, which upregulates DAMP–PRR signaling, is already highly expressed in human IBD121, 122 and its potential role as a target is supported by mouse models.121, 122, 216 DAMP–inflammasome signaling also offers a potential target, although most research thus far has focused on the downstream effects, e.g., IL-1β and IL-18.
Targeting calprotectin as a functional biomarker is of interest given its established biological actions. S100A9-deficient mice lack both S100A8 and S100A9 proteins because of S100A8 instability in the absence of S100A9.217, 218 In this way, a number of studies have targeted calprotectin via S100A9 in animal models. The quinolone-3-carboxamide ABR-215757 binds to S100A9 and the S100A8/S100A9 complex thus blocking interaction with TLR4 and RAGE.219 Quinoline-3-carboxamides are compounds with anti-inflammatory actions in inflammatory models.220, 221, 222, 223 Quinoline-3-carboxamides have been used in humans with encouraging results in type 1 diabetes,224 systemic lupus erythematosus,220 and multiple sclerosis.225 More specific calprotectin targeting may be possible via antibodies, and topical blockade at the level of the intestinal mucosa in IBD could be an effective strategy with increased efficacy and decreased toxicity. This approach was successful in an atherosclerosis model where nanoparticles displaying antibodies against S100A9 were designed for preferential uptake and retention within atherosclerotic plaques.226
In (iii), specific antioxidant approaches (e.g., targeting xanthine oxidase, NADPH oxidases [Nox enzymes], mitochondrial ROS or endothelial nitric oxide synthase) and/or targeted delivery (e.g., at the mitochondria or gut epithelium) may be more advantageous compared to general antioxidant therapies that are generally in effective.227 ER stress may also be amenable to pharmacological intervention either by suppressing ER stress or enhancing the UPR—animal models exist for type 2 diabetes and small bowel inflammation.228, 229, 230
Finally, in (iv), DAMPs offer great potential as biomarkers in disease diagnosis, prediction of outcome, monitoring of progression, and response to treatment. We have discussed calprotectin as an established IBD biomarker; other DAMPs found in high levels in serum, feces, or at the mucosal level in IBD (Table 3) may similarly find important clinical roles in the future. At a broader level, investigating whether respective IBD subphenotypes have specific DAMP signatures offers an opportunity to stratify patients for therapy and clinical trials.
In conclusion, our review highlights the emerging role of DAMPs in mediating abnormal inflammation in IBD and also many exciting potential prospects in clinical translation in the wider human inflammatory disease setting. Our mechanistic understanding of DAMPs, although far from complete, is rapidly expanding particularly in relation to novel areas such as autophagy and necroptosis. A number of DAMPs have already been implicated in IBD and others are currently under investigation, although the exact role of these DAMPs needs further clarification. There remain a number of unanswered questions and unexplored areas that are potentially fertile fields of research given the role of DAMPs as functional mediators of inflammation.
References
Molodecky, N.A. et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 142, 46–54.e42 (2012).
Danese, S. & Fiocchi, C. Ulcerative Colitis. N. Engl. J. Med. 365, 1713–1725 (2011).
Abraham, C. & Cho, J.H. Inflammatory bowel disease. N. Engl. J. Med. 361, 2066–2078 (2009).
Boyapati, R., Satsangi, J. & Ho, G.T. Pathogenesis of Crohn's disease. F1000Prime Rep. 7, 44 (2015).
Baumgart, D.C. & Sandborn, W.J. Crohn's disease. Lancet 380, 1590–1605 (2012).
Ordas, I., Eckmann, L., Talamini, M., Baumgart, D.C. & Sandborn, W.J. Ulcerative colitis. Lancet 380, 1606–1619 (2012).
Turner, D., Walsh, C.M., Steinhart, A.H. & Griffiths, A.M. Response to corticosteroids in severe ulcerative colitis: a systematic review of the literature and a meta-regression. Clin. Gastroenterol. Hepatol. 5, 103–110 (2007).
Peyrin-Biroulet, L., Loftus, E.V. Jr, Colombel, J.F. & Sandborn, W.J. The natural history of adult Crohn's disease in population-based cohorts. Am. J. Gastroenterol. 105, 289–297 (2010).
Beaugerie, L., Seksik, P., Nion-Larmurier, I., Gendre, J.P. & Cosnes, J. Predictors of Crohn's disease. Gastroenterology 130, 650–656 (2006).
Jostins, L. et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119–124 (2012).
Maloy, K.J. & Powrie, F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature 474, 298–306 (2011).
Wlodarska, M., Kostic, A.D. & Xavier, R.J. An integrative view of microbiome-host interactions in inflammatory bowel diseases. Cell Host Microbe 17, 577–591.
Akira, S., Uematsu, S. & Takeuchi, O. Pathogen recognition and innate immunity. Cell 124, 783–801 (2006).
Matzinger, P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 12, 991–1045 (1994).
Matzinger, P. The danger model: a renewed sense of self. Science 296, 301–305 (2002).
Chen, G.Y. & Nunez, G. Sterile inflammation: sensing and reacting to damage. Nat. Rev. Immunol. 10, 826–837 (2010).
Bianchi, M.E. DAMPs, PAMPs and alarmins: all we need to know about danger. J. Leukoc. Biol. 81, 1–5 (2007).
Takeuchi, O. & Akira, S. Pattern recognition receptors and inflammation. Cell 140, 805–820 (2010).
Blander, J.M. & Sander, L.E. Beyond pattern recognition: five immune checkpoints for scaling the microbial threat. Nat. Rev. Immunol. 12, 215–225 (2012).
Xie, J. et al. Structural Basis for Pattern Recognition by the Receptor for Advanced Glycation End Products (RAGE). J. Biol. Chem. 283, 27255–27269 (2008).
Schmidt, A.M., Yan, S.D., Yan, S.F. & Stern, D.M. The biology of the receptor for advanced glycation end products and its ligands. Biochim. Biophys. 1498, 99–111 (2000).
Rock, K.L. & Kono, H. The inflammatory response to cell death. Annu. Rev. Pathol. 3, 99–126 (2008).
Schaefer, L. Complexity of danger: the diverse nature of damage-associated molecular patterns. J. Biol. Chem. 289, 35237–35245 (2014).
Martinon, F., Petrilli, V., Mayor, A., Tardivel, A. & Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241 (2006).
Kono, H. & Rock, K.L. How dying cells alert the immune system to danger. Nat. Rev. Immunol. 8, 279–289 (2008).
Johnson, G.B., Brunn, G.J. & Platt, J.L. Cutting edge: an endogenous pathway to systemic inflammatory response syndrome (SIRS)-like reactions through Toll-like receptor 4. J. Immunol. 172, 20–24 (2004).
Wang, H. et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science 285, 248–251 (1999).
Sappington, P.L. et al. HMGB1 B box increases the permeability of Caco-2 enterocytic monolayers and impairs intestinal barrier function in mice. Gastroenterology 123, 790–802 (2002).
Zhang, Q. et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464, 104–107 (2010).
Sursal, T. et al. Plasma bacterial and mitochondrial DNA distinguish bacterial sepsis from sterile SIRS and quantify inflammatory tissue injury in nonhuman primates. Shock 39, 55–62 (2013).
Piccinini, A.M. & Midwood, K.S. DAMPening inflammation by modulating TLR signalling. Mediators Inflamm Article ID: 672395 2010 (2010).
Ehrchen, J.M., Sunderkötter, C., Foell, D., Vogl, T. & Roth, J. The endogenous Toll-like receptor 4 agonist S100A8/S100A9 (calprotectin) as innate amplifier of infection, autoimmunity, and cancer. J. Leukoc. Biol. 86, 557–566 (2009).
Cayrol, C. & Girard, J.-P. IL-33: an alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Cur. Opin. Immunol. 31, 31–37 (2014).
Harris, H.E., Andersson, U. & Pisetsky, D.S. HMGB1: a multifunctional alarmin driving autoimmune and inflammatory disease. Nat. Rev. Rheumatol. 8, 195–202 (2012).
Liew, F.Y., Pitman, N.I. & McInnes, I.B. Disease-associated functions of IL-33: the new kid in the IL-1 family. Nat. Rev. Immunol. 10, 103–110 (2010).
Rosin, D.L. & Okusa, M.D. Dangers within: DAMP responses to damage and cell death in kidney disease. J. Am. Soc. Nephrol. 22, 416–425 (2011).
Garlanda, C., Dinarello, C.A. & Mantovani, A. The interleukin-1 family: back to the future. Immunity 39, 1003–1018 (2013).
Tibble, J. et al. A simple method for assessing intestinal inflammation in Crohn's disease. Gut 47, 506–513 (2000).
Schoepfer, A.M., Trummler, M., Seeholzer, P., Seibold-Schmid, B. & Seibold, F. Discriminating IBD from IBS: comparison of the test performance of fecal markers, blood leukocytes, CRP, and IBD antibodies. Inflamm. Bowel Dis. 14, 32–39 (2008).
Henderson, P., Anderson, N.H. & Wilson, D.C. The diagnostic accuracy of fecal calprotectin during the investigation of suspected pediatric inflammatory bowel disease: a systematic review and meta-analysis. Am. J. Gastroenterol. 109, 637–645 (2014).
Lin, J.F. et al. Meta-analysis: fecal calprotectin for assessment of inflammatory bowel disease activity. Inflamm. Bowel Dis. 20, 1407–1415 (2014).
Ho, G.T. et al. Fecal calprotectin predicts the clinical course of acute severe ulcerative colitis. Am. J. Gastroenterol. 104, 673–678 (2009).
Meuwis, M.A. et al. Serum calprotectin as a biomarker for Crohn's disease. J. Crohns Colitis 7, e678–e683 (2013).
Benoit, S. et al. Elevated serum levels of calcium-binding S100 proteins A8 and A9 reflect disease activity and abnormal differentiation of keratinocytes in psoriasis. Br. J. Dermatol. 155, 62–66 (2006).
Pepper, R.J. et al. Leukocyte and serum S100A8/S100A9 expression reflects disease activity in ANCA-associated vasculitis and glomerulonephritis. Kidney Int. 83, 1150–1158 (2013).
Brun, J.G., Jonsson, R. & Haga, H.J. Measurement of plasma calprotectin as an indicator of arthritis and disease activity in patients with inflammatory rheumatic diseases. J. Rheumatol. 21, 733–738 (1994).
Frosch, M. et al. Myeloid-related proteins 8 and 14 are specifically secreted during interaction of phagocytes and activated endothelium and are useful markers for monitoring disease activity in pauciarticular-onset juvenile rheumatoid arthritis. Arthritis Rheum. 43, 628–637 (2000).
de la Rosa, G., Yang, D., Tewary, P., Varadhachary, A. & Oppenheim, J.J. Lactoferrin acts as an alarmin to promote the recruitment and activation of APCs and antigen-specific immune responses. J. Immunol. 180, 6868–6876 (2008).
Lewis, J.D. The utility of biomarkers in the diagnosis and therapy of inflammatory bowel disease. Gastroenterology 140, 1817–1826.e2 (2011).
de Jong, N.S., Leach, S.T. & Day, A.S. Fecal S100A12: a novel noninvasive marker in children with Crohn's disease. Inflamm. Bowel Dis. 12, 566–572 (2006).
Kaiser, T. et al. Faecal S100A12 as a non-invasive marker distinguishing inflammatory bowel disease from irritable bowel syndrome. Gut 56, 1706–1713 (2007).
Sidler, M.A., Leach, S.T. & Day, A.S. Fecal S100A12 and fecal calprotectin as noninvasive markers for inflammatory bowel disease in children. Inflamm. Bowel Dis. 14, 359–366 (2008).
Dabritz, J. et al. Improving relapse prediction in inflammatory bowel disease by neutrophil-derived S100A12. Inflamm. Bowel Dis. 19, 1130–1138 (2013).
Foell, D. et al. Neutrophil derived human S100A12 (EN-RAGE) is strongly expressed during chronic active inflammatory bowel disease. Gut 52, 847–853 (2003).
Leach, S.T. et al. Serum and mucosal S100 proteins, calprotectin (S100A8/S100A9) and S100A12, are elevated at diagnosis in children with inflammatory bowel disease. Scand. J. Gastroenterol. 42, 1321–1331 (2007).
Manolakis, A.C. et al. Moderate performance of serum S100A12, in distinguishing inflammatory bowel disease from irritable bowel syndrome. BMC Gastroenterol. 10, 118 (2010).
Vitali, R. et al. Fecal HMGB1 is a novel marker of intestinal mucosal inflammation in pediatric inflammatory bowel disease. Am. J. Gastroenterol. 106, 2029–2040 (2011).
Palone, F. et al. Role of HMGB1 as a suitable biomarker of subclinical intestinal inflammation and mucosal healing in patients with inflammatory bowel disease. Inflamm. Bowel Dis. 20, 1448–1457 (2014).
Beltran, C.J. et al. Characterization of the novel ST2/IL-33 system in patients with inflammatory bowel disease. Inflamm. Bowel Dis. 16, 1097–1107 (2010).
Pastorelli, L. et al. Epithelial-derived IL-33 and its receptor ST2 are dysregulated in ulcerative colitis and in experimental Th1/Th2 driven enteritis. Proc. Natl. Acad. Sci. USA 107, 8017–8022 (2010).
Kobori, A. et al. Interleukin-33 expression is specifically enhanced in inflamed mucosa of ulcerative colitis. J. Gastroenterol. 45, 999–1007 (2010).
Seidelin, J.B. et al. IL-33 is upregulated in colonocytes of ulcerative colitis. Immunol. Lett. 128, 80–85 (2010).
Sponheim, J. et al. Inflammatory bowel disease-associated interleukin-33 is preferentially expressed in ulceration-associated myofibroblasts. Am. J. Pathol. 177, 2804–2815 (2010).
Wakahara, K. et al. Human basophils interact with memory T cells to augment Th17 responses. Blood 120, 4761–4771 (2012).
Ludwiczek, O. et al. Imbalance between interleukin-1 agonists and antagonists: relationship to severity of inflammatory bowel disease. Clin. Exp. Immunol. 138, 323–329 (2004).
Youngman, K.R. et al. Localization of intestinal interleukin 1 activity and protein and gene expression to lamina propria cells. Gastroenterology 104, 749–758 (1993).
Nenci, A. et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature 446, 557–561 (2007).
Lotze, M.T. & Tracey, K.J. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 5, 331–342 (2005).
Park, J.S. et al. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J. Biol. Chem. 279, 7370–7377 (2004).
Yu, M. et al. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock 26, 174–179 (2006).
Tian, J. et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat. Immunol. 8, 487–496 (2007).
Dumitriu, I.E. et al. Release of high mobility group box 1 by dendritic cells controls T cell activation via the receptor for advanced glycation end products. J. Immunol. 174, 7506–7515 (2005).
Li, G., Tang, D. & Lotze, M.T. Ménage à Trois in stress: DAMPs, redox and autophagy. Semin. Cancer Biol. 23, 380–390 (2013).
Andersson, U., Erlandsson-Harris, H., Yang, H. & Tracey, K.J. HMGB1 as a DNA-binding cytokine. J. Leukoc. Biol. 72, 1084–1091 (2002).
Qin, S. et al. Role of HMGB1 in apoptosis-mediated sepsis lethality. J. Exp. Med. 203, 1637–1642 (2006).
Dave, S.H. et al. Ethyl pyruvate decreases HMGB1 release and ameliorates murine colitis. J. Leukoc. Biol. 86, 633–643 (2009).
Vitali, R. et al. Dipotassium glycyrrhizate inhibits HMGB1-dependent inflammation and ameliorates colitis in mice. PLoS One 8, e66527 (2013).
Maeda, S. et al. Essential roles of high-mobility group box 1 in the development of murine colitis and colitis-associated cancer. Biochem. Biophys. Res. Commun. 360, 394–400 (2007).
Calogero, S. et al. The lack of chromosomal protein Hmg1 does not disrupt cell growth but causes lethal hypoglycaemia in newborn mice. Nat. Genet. 22, 276–280 (1999).
Zhu, X. et al. Cytosolic HMGB1 controls the cellular autophagy/apoptosis checkpoint during inflammation. J. Clin. Invest. 125, 1098–1110 (2015).
Huebener, P. et al. The HMGB1/RAGE axis triggers neutrophil-mediated injury amplification following necrosis. J. Clin. Invest. 125, 539–550 (2015).
Huang, H. et al. Hepatocyte-specific high-mobility group box 1 deletion worsens the injury in liver ischemia/reperfusion: a role for intracellular high-mobility group box 1 in cellular protection. Hepatology 59, 1984–1997 (2014).
Kang, R. et al. Intracellular Hmgb1 inhibits inflammatory nucleosome release and limits acute pancreatitis in mice. Gastroenterology 146, 1097–1107 (2014).
Yanai, H. et al. Conditional ablation of HMGB1 in mice reveals its protective function against endotoxemia and bacterial infection. Proc. Natl. Acad. Sci. USA 110, 20699–20704 (2013).
Foell, D., Wittkowski, H., Vogl, T. & Roth, J. S100 proteins expressed in phagocytes: a novel group of damage-associated molecular pattern molecules. J. Leukoc. Biol. 81, 28–37 (2007).
Robinson, M.J., Tessier, P., Poulsom, R. & Hogg, N. The S100 family heterodimer, MRP-8/14, binds with high affinity to heparin and heparan sulfate glycosaminoglycans on endothelial cells. J. Biol. Chem. 277, 3658–3665 (2001).
Vogl, T. et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat. Med. 13, 1042–1049 (2007).
Srikrishna, G. et al. Two proteins modulating transendothelial migration of leukocytes recognize novel carboxylated glycans on endothelial cells. J. Immunol. 166, 4678–4688 (2001).
Frosch, M. et al. Expression of MRP8 and MRP14 by macrophages is a marker for severe forms of glomerulonephritis. J. Leukoc. Biol. 75, 198–206 (2004).
Steinbakk, M. et al. Antimicrobial actions of calcium binding leucocyte L1 protein, calprotectin. Lancet 336, 763–765 (1990).
Liu, J.Z. et al. Zinc sequestration by the neutrophil protein calprotectin enhances Salmonella growth in the inflamed gut. Cell Host Microbe 11, 227–239 (2012).
Miles, K. et al. Dying and necrotic neutrophils are anti-inflammatory secondary to the release of alpha-defensins. J. Immunol. 183, 2122–2132 (2009).
Urban, C.F. et al. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 5, e1000639 (2009).
Rider, P., Carmi, Y., Voronov, E. & Apte, R.N. Interleukin-1α. Semin. Immunol. 25, 430–438 (2013).
Moussion, C., Ortega, N. & Girard, J.-P. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel ‘alarmin’? PLoS One 3, e3331 (2008).
Carta, S., Lavieri, R. & Rubartelli, A. Different members of the IL-1 family come out in different ways: DAMPs vs cytokines? Front. Immunol. 4, 123 (2013).
Lamkanfi, M. & Dixit, V.M. IL-33 raises alarm. Immunity 31, 5–7 (2009).
Lüthi, A.U. et al. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity 31, 84–98 (2009).
Cohen, I. et al. Differential release of chromatin-bound IL-1alpha discriminates between necrotic and apoptotic cell death by the ability to induce sterile inflammation. Proc. Natl. Acad. Sci. USA 107, 2574–2579 (2010).
Dinarello, C.A. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 27, 519–550 (2009).
Chun-Jen, C. et al. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat. Med. 13, 851–856 (2007).
Rider, P. et al. IL-1alpha and IL-1beta recruit different myeloid cells and promote different stages of sterile inflammation. J. Immunol. 187, 4835–4843 (2011).
Oboki, K. et al. IL-33 is a crucial amplifier of innate rather than acquired immunity. Proc. Natl. Acad. Sci. USA 107, 18581–18586 (2010).
Salas, A. The IL-33/ST2 axis: yet another therapeutic target in inflammatory bowel disease? Gut 62, 1392–1393 (2013).
Nunes, T., Bernardazzi, C. & de Souza, H.S. Interleukin-33 and inflammatory bowel diseases: lessons from human studies. Mediators Inflamm. 2014, 423957 (2014).
Baumann, C. et al. T-bet- and STAT4-dependent IL-33 receptor expression directly promotes antiviral Th1 cell responses. Proc. Natl. Acad. Sci. USA 112, 4056–4061 (2015).
Latiano, A. et al. Associations between genetic polymorphisms in IL-33, IL1R1 and risk for inflammatory bowel disease. PLoS One 8, e62144 (2013).
Cominelli, F. et al. Interleukin 1 (IL-1) gene expression, synthesis, and effect of specific IL-1 receptor blockade in rabbit immune complex colitis. J. Clin. Invest. 86, 972–980 (1990).
Bersudsky, M. et al. Non-redundant properties of IL-1α and IL-1β during acute colon inflammation in mice. Gut 63, 598–609 (2014).
Fritz, G. RAGE: a single receptor fits multiple ligands. Trends Biochem. Sci. 36, 625–632 (2011).
Stern, D., Yan, S.D., Yan, S.F. & Schmidt, A.M. Receptor for advanced glycation endproducts: a multiligand receptor magnifying cell stress in diverse pathologic settings. Adv. Drug Deliv. Rev. 54, 1615–1625 (2002).
Ciccocioppo, R. et al. Role of the advanced glycation end products receptor in Crohn's disease inflammation. World J. Gastroenterol. 19, 8269–8281 (2013).
Dabritz, J. et al. The functional -374 T/A polymorphism of the receptor for advanced glycation end products may modulate Crohn's disease. Am. J. Physiol. Gastrointest. Liver Physiol. 300, G823–G832 (2011).
Zen, K., Chen, C.X., Chen, Y.T., Wilton, R. & Liu, Y. Receptor for advanced glycation endproducts mediates neutrophil migration across intestinal epithelium. J. Immunol. 178, 2483–2490 (2007).
Hofmann, M.A. et al. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell 97, 889–901 (1999).
Malickova, K. et al. Anti-inflammatory effect of biological treatment in patients with inflammatory bowel diseases: calprotectin and IL-6 changes do not correspond to sRAGE changes. Scand. J. Clin. Lab. Invest. 70, 294–299 (2010).
Yilmaz, Y., Yonal, O., Eren, F., Atug, O. & Over Hamzaoglu, H. Serum levels of soluble receptor for advanced glycation endproducts (sRAGE) are higher in ulcerative colitis and correlate with disease activity. J. Crohns Colitis 5, 402–406 (2011).
Ciccocioppo, R. et al. The circulating level of soluble receptor for advanced glycation end products displays different patterns in ulcerative colitis and Crohn's disease: a cross-sectional study. Dig. Dis. Sci. 60, 2327–2337 (2015).
Meijer, B. et al. Total soluble and endogenous secretory receptor for advanced glycation endproducts (RAGE) in IBD. J. Crohns Colitis 8, 513–520 (2014).
El Mezayen, R. et al. Endogenous signals released from necrotic cells augment inflammatory responses to bacterial endotoxin. Immunol. Lett. 111, 36–44 (2007).
Saurer, L. et al. Elevated levels of serum-soluble triggering receptor expressed on myeloid cells-1 in patients with IBD do not correlate with intestinal TREM-1 mRNA expression and endoscopic disease activity. J. Crohns Colitis 6, 913–923 (2012).
Schenk, M., Bouchon, A., Seibold, F. & Mueller, C. TREM-1—expressing intestinal macrophages crucially amplify chronic inflammation in experimental colitis and inflammatory bowel diseases. J. Clin. Invest. 117, 3097–3106 (2007).
Chen, G.Y., Tang, J., Zheng, P. & Liu, Y. CD24 and Siglec-10 selectively repress tissue damage-induced immune responses. Science 323, 1722–1725 (2009).
Liu, Y., Chen, G.Y. & Zheng, P. CD24-Siglec G/10 discriminates danger- from pathogen-associated molecular patterns. Trends Immunol. 30, 557–561 (2009).
Chen, G.Y. et al. Amelioration of sepsis by inhibiting sialidase-mediated disruption of the CD24-SiglecG interaction. Nat. Biotechnol. 29, 428–435 (2011).
Shi, Y. & Rock, K.L. Cell death releases endogenous adjuvants that selectively enhance immune surveillance of particulate antigens. Eur. J. Immunol. 32, 155–162 (2002).
Gallucci, S., Lolkema, M. & Matzinger, P. Natural adjuvants: endogenous activators of dendritic cells. Nat. Med. 5, 1249–1255 (1999).
Shi, Y., Zheng, W. & Rock, K.L. Cell injury releases endogenous adjuvants that stimulate cytotoxic T cell responses. Proc. Natl. Acad. Sci. USA 97, 14590–14595 (2000).
Rovere-Querini, P. et al. HMGB1 is an endogenous immune adjuvant released by necrotic cells. EMBO Rep. 5, 825–830 (2004).
Yang, D. et al. High mobility group box-1 protein induces the migration and activation of human dendritic cells and acts as an alarmin. J. Leukoc. Biol. 81, 59–66 (2007).
Dumitriu, I.E., Bianchi, M.E., Bacci, M., Manfredi, A.A. & Rovere-Querini, P. The secretion of HMGB1 is required for the migration of maturing dendritic cells. J. Leukoc. Biol. 81, 84–91 (2007).
Flohe, S.B. et al. Human heat shock protein 60 induces maturation of dendritic cells versus a Th1-promoting phenotype. J. Immunol. 170, 2340–2348 (2003).
Somersan, S. et al. Primary tumor tissue lysates are enriched in heat shock proteins and induce the maturation of human dendritic cells. J. Immunol. 167, 4844–4852 (2001).
Chen, T., Guo, J., Han, C., Yang, M. & Cao, X. Heat shock protein 70, released from heat-stressed tumor cells, initiates antitumor immunity by inducing tumor cell chemokine production and activating dendritic cells via TLR4 pathway. J. Immunol. 182, 1449–1459 (2009).
Ishii, K.J. et al. Genomic DNA released by dying cells induces the maturation of APCs. J. Immunol. 167, 2602–2607 (2001).
Shi, Y., Evans, J.E. & Rock, K.L. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature 425, 516–521 (2003).
Messmer, D. et al. High mobility group box protein 1: an endogenous signal for dendritic cell maturation and Th1 polarization. J. Immunol. 173, 307–313 (2004).
Shi, Y., Galusha, S.A. & Rock, K.L. Cutting edge: elimination of an endogenous adjuvant reduces the activation of CD8 T lymphocytes to transplanted cells and in an autoimmune diabetes model. J. Immunol. 176, 3905–3908 (2006).
Scheibner, K.A. et al. Hyaluronan fragments act as an endogenous danger signal by engaging TLR2. J. Immunol. 177, 1272–1281 (2006).
Hwang, S.A., Kruzel, M.L. & Actor, J.K. Lactoferrin augments BCG vaccine efficacy to generate T helper response and subsequent protection against challenge with virulent Mycobacterium tuberculosis. Int. Immunopharmacol. 5, 591–599 (2005).
Kenji, T. et al. Defensins act as potent adjuvants that promote cellular and humoral immune responses in mice to a lymphoma idiotype and carrier antigens. Int. Immunol. 12, 691–700 (2000).
Garg, A.D., Martin, S., Golab, J. & Agostinis, P. Danger signalling during cancer cell death: origins, plasticity and regulation. Cell Death Differ. 21, 26–38 (2014).
Loser, K. et al. The Toll-like receptor 4 ligands Mrp8 and Mrp14 are crucial in the development of autoreactive CD8+ T cells. Nat. Med. 16, 713–717 (2010).
Petersen, B. et al. The alarmin Mrp8/14 as regulator of the adaptive immune response during allergic contact dermatitis. EMBO J. 32, 100–111 (2013).
Schiering, C. et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature 513, 564–568 (2014).
Groβ, P., Doser, K., Falk, W., Obermeier, F. & Hofmann, C. IL-33 attenuates development and perpetuation of chronic intestinal inflammation. Inflamm. Bowel Dis. 18, 1900–1909 (2012).
Salim, S.Y. & Soderholm, J.D. Importance of disrupted intestinal barrier in inflammatory bowel diseases. Inflamm. Bowel Dis. 17, 362–381 (2011).
Marchiando, A.M., Graham, W.V. & Turner, J.R. Epithelial barriers in homeostasis and disease. Annu. Rev. Pathol. 5, 119–144 (2010).
Sedhom, M.A.K. et al. Neutralisation of the interleukin-33/ST2 pathway ameliorates experimental colitis through enhancement of mucosal healing in mice. Gut 62, 1714–1723 (2012).
Yang, R. et al. Anti-HMGB1 neutralizing antibody ameliorates gut barrier dysfunction and improves survival after hemorrhagic shock. Mol. Med. 12, 105–114 (2006).
Wang, L., Luo, H., Chen, X., Jiang, Y. & Huang, Q. Functional characterization of S100A8 and S100A9 in altering monolayer permeability of human umbilical endothelial cells. PLoS One 9, e90472 (2014).
Liu, S. et al. HMGB1 is secreted by immunostimulated enterocytes and contributes to cytomix-induced hyperpermeability of Caco-2 monolayers. Am. J. Physiol. Cell Physiol. 290, C990–C999 (2006).
Foell, D. et al. Phagocyte-specific S100 proteins are released from affected mucosa and promote immune responses during inflammatory bowel disease. J. Pathol. 216, 183–192 (2008).
Viemann, D. et al. MRP8/MRP14 impairs endothelial integrity and induces a caspase-dependent and -independent cell death program. Blood 109, 2453–2460 (2007).
Nakahira, K. et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 12, 222–230 (2011).
Siegmund, B. Interleukin-18 in intestinal inflammation: friend and foe? Immunity 32, 300–302 (2010).
Zaki, M.H. et al. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 32, 379–391 (2010).
Bauer, C. et al. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut 59, 1192–1199 (2010).
Allen, I.C. et al. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J. Exp. Med. 207, 1045–1056 (2010).
Wlodarska, M. et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell 156, 1045–1059 (2014).
Elinav, E. et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145, 745–757 (2011).
Günther, C., Neumann, H., Neurath, M.F. & Becker, C. Apoptosis, necrosis and necroptosis: cell death regulation in the intestinal epithelium. Gut 62, 1062–1071 (2013).
Kaczmarek, A., Vandenabeele, P. & Krysko, D.V. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity 38, 209–223 (2013).
Welz, P.S. et al. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature 477, 330–334 (2011).
Gunther, C. et al. Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature 477, 335–339 (2011).
Pasparakis, M. & Vandenabeele, P. Necroptosis and its role in inflammation. Nature 517, 311–320 (2015).
Linkermann, A. & Green, D.R. Necroptosis. N. Engl. J. Med. 370, 455–465 (2014).
Vandenabeele, P., Galluzzi, L., Vanden Berghe, T. & Kroemer, G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 11, 700–714 (2010).
Sun, L. et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227 (2012).
Takahashi, N. et al. RIPK1 ensures intestinal homeostasis by protecting the epithelium against apoptosis. Nature 513, 95–99 (2014).
Dannappel, M. et al. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature 513, 90–94 (2014).
Degterev, A. et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 4, 313–321 (2008).
Weinlich, R. et al. Protective roles for caspase-8 and cFLIP in adult homeostasis. Cell Rep. 5, 340–348 (2013).
Pierdomenico, M. et al. Necroptosis is active in children with inflammatory bowel disease and contributes to heighten intestinal inflammation. Am. J. Gastroenterol. 109, 279–287 (2014).
Martin, S.J., Henry, C.M. & Cullen, S.P. A perspective on mammalian caspases as positive and negative regulators of inflammation. Mol. Cell 46, 387–397 (2012).
Taylor, R.C., Cullen, S.P. & Martin, S.J. Apoptosis: controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 9, 231–241 (2008).
Choi, S.H. et al. Lipoprotein accumulation in macrophages via toll-like receptor-4-dependent fluid phase uptake. Circ. Res. 104, 1355–1363 (2009).
Imai, Y. et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 133, 235–249 (2008).
Rouhiainen, A., Tumova, S., Valmu, L., Kalkkinen, N. & Rauvala, H. Pivotal advance: analysis of proinflammatory activity of highly purified eukaryotic recombinant HMGB1 (amphoterin). J. Leukoc. Biol. 81, 49–58 (2007).
Kang, R., Tang, D., Lotze, M.T. & Zeh, H.J. 3rd RAGE regulates autophagy and apoptosis following oxidative injury. Autophagy 7, 442–444 (2011).
Tang, D. et al. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene 29, 5299–5310 (2010).
Kazama, H. et al. Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity 29, 21–32 (2008).
Venereau, E. et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J. Exp. Med. 209, 1519–1528 (2012).
Shimada, K. et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36, 401–414 (2012).
Pazmandi, K. et al. Oxidative modification enhances the immunostimulatory effects of extracellular mitochondrial DNA on plasmacytoid dendritic cells. Free Radic. Biol. Med. 77, 281–290 (2014).
Kaser, A. et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 134, 743–756 (2008).
Murthy, A. et al. A Crohn's disease variant in Atg16l1 enhances its degradation by caspase 3. Nature 506, 456–462 (2014).
Zheng, W. et al. Evaluation of AGR2 and AGR3 as candidate genes for inflammatory bowel disease. Genes Immun. 7, 11–18 (2006).
Zhou, R., Yazdi, A.S., Menu, P. & Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225 (2011).
Iyer, S.S. et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc. Natl. Acad. Sci. 106, 20388–20393 (2009).
Saitoh, T. et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 456, 264–268 (2008).
Oka, T. et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 485, 251–255 (2012).
Cadwell, K. et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 456, 259–263 (2008).
Lassen, K.G. et al. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc. Natl. Acad. Sci. USA 111, 7741–7746 (2014).
Adolph, T.E. et al. Paneth cells as a site of origin for intestinal inflammation. Nature 503, 272–276 (2013).
Conway, K.L. et al. Atg16l1 is required for autophagy in intestinal epithelial cells and protection of mice from Salmonella infection. Gastroenterology 145, 1347–1357 (2013).
Luo, Y., Li, S.J., Yang, J., Qiu, Y.Z. & Chen, F.P. HMGB1 induces an inflammatory response in endothelial cells via the RAGE-dependent endoplasmic reticulum stress pathway. Biochem. Biophys. Res. Commun. 438, 732–738 (2013).
Zhu, X.M. et al. Endoplasmic reticulum stress and its regulator XBP-1 contributes to dendritic cell maturation and activation induced by high mobility group box-1 protein. Int. J. Biochem. Cell Biol. 44, 1097–1105 (2012).
Garg, A.D. et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J. 31, 1062–1079 (2012).
Wang, Y. et al. Necroptosis inhibitor necrostatin-1 promotes cell protection and physiological function in traumatic spinal cord injury. Neuroscience 266, 91–101 (2014).
Zhou, Y. et al. Protective effects of necrostatin-1 against concanavalin A-induced acute hepatic injury in mice. Mediators Inflamm. 2013, 706156–706156 (2013).
Zhang, A. et al. Necrostatin-1 inhibits Hmgb1-IL-23/IL-17 pathway and attenuates cardiac ischemia reperfusion injury. Transpl. Int. 27, 1077–1085 (2014).
Remijsen, Q. et al. Depletion of RIPK3 or MLKL blocks TNF-driven necroptosis and switches towards a delayed RIPK1 kinase-dependent apoptosis. Cell Death Dis. 5, e1004 (2014).
Rubinsztein, D.C., Gestwicki, J.E., Murphy, L.O. & Klionsky, D.J. Potential therapeutic applications of autophagy. Nat. Rev. Drug Discov. 6, 304–312 (2007).
Yin, H. et al. Sirolimus ameliorates inflammatory responses by switching the regulatory T/T helper type 17 profile in murine colitis. Immunology 139, 494–502 (2013).
Matsuda, C. et al. Therapeutic effect of a new immunosuppressive agent, everolimus, on interleukin-10 gene-deficient mice with colitis. Clin. Exp. Immunol. 148, 348–359 (2007).
Massey, D.C., Bredin, F. & Parkes, M. Use of sirolimus (rapamycin) to treat refractory Crohn's disease. Gut 57, 1294–1296 (2008).
Reinisch, W. et al. A multicenter, randomized, double-blind trial of everolimus versus azathioprine and placebo to maintain steroid-induced remission in patients with moderate-to-severe active Crohn's disease. Am. J. Gastroenterol. 103, 2284–2292 (2008).
Palmer, G. et al. Inhibition of interleukin-33 signaling attenuates the severity of experimental arthritis. Arthritis Rheum. 60, 738–749 (2009).
Sunahori, K. et al. Increased expression of receptor for advanced glycation end products by synovial tissue macrophages in rheumatoid arthritis. Arthritis Rheum. 54, 97–104 (2006).
Wendt, T.M. et al. RAGE drives the development of glomerulosclerosis and implicates podocyte activation in the pathogenesis of diabetic nephropathy. Am. J. Pathol. 162, 1123–1137 (2003).
Kislinger, T. et al. Receptor for advanced glycation end products mediates inflammation and enhanced expression of tissue factor in vasculature of diabetic apolipoprotein E-null mice. Arterioscler. Thromb. Vasc. Biol. 21, 905–910 (2001).
Myint, K.M. et al. Blockade of diabetic vascular injury by controlling of AGE-RAGE system. Curr. Drug Targets 6, 447–452 (2005).
Lutterloh, E.C. et al. Inhibition of the RAGE products increases survival in experimental models of severe sepsis and systemic infection. Crit. Care 11, R122 (2007).
Kuroiwa, Y. et al. Identification and characterization of the direct interaction between methotrexate (MTX) and high-mobility group box 1 (HMGB1) protein. PLoS One 8, e63073 (2013).
Zhou, J. et al. TREM-1 inhibition attenuates inflammation and tumor within the colon. Int. Immunopharmacol. 17, 155–161 (2013).
Hobbs, J.A. et al. Myeloid cell function in MRP-14 (S100A9) null mice. Mol. Cell. Biol. 23, 2564–2576 (2003).
Manitz, M.P. et al. Loss of S100A9 (MRP14) results in reduced interleukin-8-induced CD11b surface expression, a polarized microfilament system, and diminished responsiveness to chemoattractants in vitro. Mol. Cell. Biol. 23, 1034–1043 (2003).
Bjork, P. et al. Identification of human S100A9 as a novel target for treatment of autoimmune disease via binding to quinoline-3-carboxamides. PLoS Biol. 7, e97 (2009).
Bengtsson, A.A. et al. Pharmacokinetics, tolerability, and preliminary efficacy of paquinimod (ABR-215757), a new quinoline-3-carboxamide derivative: studies in lupus-prone mice and a multicenter, randomized, double-blind, placebo-controlled, repeat-dose, dose-ranging study in patients with systemic lupus erythematosus. Arthritis Rheum. 64, 1579–1588 (2012).
Karussis, D.M. et al. Treatment of chronic-relapsing experimental autoimmune encephalomyelitis with the synthetic immunomodulator linomide (quinoline-3-carboxamide). Proc. Natl. Acad. Sci. USA 90, 6400–6404 (1993).
Brunmark, C. et al. The new orally active immunoregulator laquinimod (ABR-215062) effectively inhibits development and relapses of experimental autoimmune encephalomyelitis. J. Neuroimmunol. 130, 163–172 (2002).
Bjork, J. & Kleinau, S. Paradoxical effects of LS-2616 (Linomide) treatment in the type II collagen arthritis model in mice. Agents Actions 27, 319–321 (1989).
Coutant, R. et al. Low dose linomide in Type I juvenile diabetes of recent onset: a randomised placebo-controlled double blind trial. Diabetologia 41, 1040–1046 (1998).
Polman, C. et al. Treatment with laquinimod reduces development of active MRI lesions in relapsing MS. Neurology 64, 987–991 (2005).
Maiseyeu, A. et al. In vivo targeting of inflammation-associated myeloid-related protein 8/14 via gadolinium immunonanoparticles. Arterioscler. Thromb. Vasc. Biol. 32, 962–970 (2012).
Spychalowicz, A., Wilk, G., Śliwa, T., Ludew, D. & Guzik, T.J. Novel therapeutic approaches in limiting oxidative stress and inflammation. Curr. Pharm. Biotechnol. 13, 2456–2466 (2012).
Ozcan, U. et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306, 457–461 (2004).
Ozcan, U. et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 313, 1137–1140 (2006).
Uchida, A., Yamada, T., Hayakawa, T. & Hoshino, M. Taurochenodeoxycholic acid ameliorates and ursodeoxycholic acid exacerbates small intestinal inflammation. Am. J. Physiol. 272, G1249–G1257 (1997).
Imaeda, H. et al. Interleukin-33 suppresses Notch ligand expression and prevents goblet cell depletion in dextran sulfate sodium-induced colitis. Int. J. Mol. Med. 28, 573–578 (2011).
Duan, L. et al. Interleukin-33 ameliorates experimental colitis through promoting Th2/Foxp3(+) regulatory T-cell responses in mice. Mol. Med. 18, 753–761 (2012).
Gross, P., Doser, K., Falk, W., Obermeier, F. & Hofmann, C. IL-33 attenuates development and perpetuation of chronic intestinal inflammation. Inflamm. Bowel Dis. 18, 1900–1909 (2012).
Peetermans, W.E., D'Haens, G.R., Ceuppens, J.L., Rutgeerts, P. & Geboes, K. Mucosal expression by B7-positive cells of the 60-kilodalton heat-shock protein in inflammatory bowel disease. Gastroenterology 108, 75–82 (1995).
Tomasello, G. et al. Hsp10, Hsp70, and Hsp90 immunohistochemical levels change in ulcerative colitis after therapy. Eur. J. Histochem. 55, e38–e38 (2011).
Ludwig, D. et al. Enhanced intestinal expression of heat shock protein 70 in patients with inflammatory bowel diseases. Dig. Dis. Sci. 44, 1440–1447 (1999).
Rodolico, V. et al. Hsp60 and Hsp10 increase in colon mucosa of Crohn's disease and ulcerative colitis. Cell Stress Chaperones 15, 877–884 (2010).
Hu, S. et al. Translational inhibition of colonic epithelial heat shock proteins by IFN-gamma and TNF-alpha in intestinal inflammation. Gastroenterology 133, 1893–1904 (2007).
Collins, C.B. et al. Targeted inhibition of heat shock protein 90 suppresses tumor necrosis factor-α and ameliorates murine intestinal inflammation. Inflamm. Bowel Dis. 20, 685–694 (2014).
Riedl, S. et al. Serum tenascin-C is an indicator of inflammatory bowel disease activity. Int. J. Colorectal Dis. 16, 285–291 (2001).
Riedl, S. et al. Mucosal tenascin C content in inflammatory and neoplastic diseases of the large bowel. Dis. Colon Rectum 41, 86–92 (1998).
Dueck, M. et al. Detection of tenascin-C isoforms in colorectal mucosa, ulcerative colitis, carcinomas and liver metastases. Int. J. Cancer 82, 477–483 (1999).
Magnusson, M.K. et al. Response to infliximab therapy in ulcerative colitis is associated with decreased monocyte activation, reduced CCL2 expression and downregulation of tenascin C. J. Crohns Colitis 9, 56–65 (2015).
Kessler, S. et al. Hyaluronan (HA) deposition precedes and promotes leukocyte recruitment in intestinal inflammation. Clin. Transl. Sci. 1, 57–61 (2008).
Zheng, Y., Humphry, M., Maguire, J.J., Bennett, M.R. & Clarke, M.C. Intracellular interleukin-1 receptor 2 binding prevents cleavage and activity of interleukin-1alpha, controlling necrosis-induced sterile inflammation. Immunity 38, 285–295 (2013).
Jensen-Jarolim, E. et al. The constitutive expression of galectin-3 is downregulated in the intestinal epithelia of Crohn's disease patients, and tumour necrosis factor alpha decreases the level of galectin-3-specific mRNA in HCT-8 cells. Eur. J. Gastroenterol. Hepatol. 14, 145–152 (2002).
Muller, S. et al. Galectin-3 modulates T cell activity and is reduced in the inflamed intestinal epithelium in IBD. Inflamm. Bowel Dis. 12, 588–597 (2006).
Frol'ova, L. et al. Detection of galectin-3 in patients with inflammatory bowel diseases: new serum marker of active forms of IBD? Inflamm. Res. 58, 503–512 (2009).
Santucci, L. et al. Galectin-1 suppresses experimental colitis in mice. Gastroenterology 124, 1381–1394 (2003).
Paclik, D. et al. Galectin-2 induces apoptosis of lamina propria T lymphocytes and ameliorates acute and chronic experimental colitis in mice. J. Mol. Med. (Berl) 86, 1395–1406 (2008).
Hokama, A. et al. Induced reactivity of intestinal CD4(+) T cells with an epithelial cell lectin, galectin-4, contributes to exacerbation of intestinal inflammation. Immunity 20, 681–693 (2004).
Neves, A.R. et al. Overexpression of ATP-activated P2X7 receptors in the intestinal mucosa is implicated in the pathogenesis of Crohn's disease. Inflamm. Bowel Dis. 20, 444–457 (2014).
Hofman, P. et al. Genetic and pharmacological inactivation of the purinergic P2RX7 receptor dampens inflammation but increases tumor incidence in a mouse model of colitis-associated cancer. Cancer Res 75, 1–11 (2015).
Marques, C.C. et al. Prophylactic systemic P2X7 receptor blockade prevents experimental colitis. Biochim. Biophys. Acta 1842, 65–78 (2014).
Kurashima, Y. et al. Extracellular ATP mediates mast cell-dependent intestinal inflammation through P2X7 purinoceptors. Nat. Commun. 3, 1034 (2012).
Broere, F., van der Zee, R. & van Eden, W. Heat shock proteins are no DAMPs, rather 'DAMPERs'. Nat. Rev. Immunol. 11, 565–565 (2011).
Chen, G.Y. & Nuñez, G. Are heat shock proteins DAMPs? Nat. Rev. Immunol. 11, 565–565 (2011).
Acknowledgements
This work is supported by Medical Research Council (to R.K.B. and G.-T.H.) and Edinburgh Gut Immunobiology and Gastroenterology Trustees Fund (to R.K.B.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest.
Additional information
SUPPLEMENTARY MATERIAL is linked to the online version of the paper
Supplementary information
Rights and permissions
About this article

Cite this article
Boyapati, R., Rossi, A., Satsangi, J. et al. Gut mucosal DAMPs in IBD: from mechanisms to therapeutic implications. Mucosal Immunol 9, 567–582 (2016). https://doi.org/10.1038/mi.2016.14
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2016.14
This article is cited by
-
Colon-targeted S100A8/A9-specific peptide systems ameliorate colitis and colitis-associated colorectal cancer in mouse models
Acta Pharmacologica Sinica (2023)
-
A subset of gut leukocytes has telomerase-dependent “hyper-long” telomeres and require telomerase for function in zebrafish
Immunity & Ageing (2022)
-
Hypo-osmotic stress induces the epithelial alarmin IL-33 in the colonic barrier of ulcerative colitis
Scientific Reports (2022)
-
Association of cell free mitochondrial DNA and caspase-1 expression with disease severity and ARTs efficacy in HIV infection
Molecular Biology Reports (2021)
-
HMGB1 released from GSDME-mediated pyroptotic epithelial cells participates in the tumorigenesis of colitis-associated colorectal cancer through the ERK1/2 pathway
Journal of Hematology & Oncology (2020)
