
Abstract
Essential hypertension is a complex multifactorial disease process that involves the interaction of multiple genes at various loci throughout the genome, and the influence of environmental factors such as diet and lifestyle, to ultimately determine long-term arterial pressure. These factors converge with physiological signaling pathways to regulate the set-point of long-term blood pressure. In hypertension, structural changes in arteries occur and show differences within and between vascular beds, between species, models and sexes. Such changes can also reflect the development of hypertension, and the levels of circulating humoral and vasoactive compounds. The role of perivascular adipose tissue in the modulation of vascular structure under various disease states such as hypertension, obesity and metabolic syndrome is an emerging area of research, and is likely to contribute to the heterogeneity described in this review. Diversity in structure and related function is the norm, with morphological changes being causative in some beds and states, and in others, a consequence of hypertension. Specific animal models of hypertension have advantages and limitations, each with factors influencing the relevance of the model to the human hypertensive state/s. However, understanding the fundamental properties of artery function and how these relate to signalling mechanisms in real (intact) tissues is key for translating isolated cell and model data to have an impact and relevance in human disease etiology. Indeed, the ultimate aim of developing new treatments to correct vascular dysfunction requires understanding and recognition of the limitations of the methodologies used.
Similar content being viewed by others

Introduction
The structure of the arterial tree and its influence on blood vessel function is an important determinant of long-term blood pressure (BP) regulation. In this review, the relationship between changes in vascular structure and function, and their association with causality in hypertension is clarified. The data examined are predominantly from in vivo studies of animal models of hypertension, as they relate to that in humans. Primary structural changes that contribute to hypertension development are separated from secondary adaptive changes in response to hypertension (summarized, Table 1). Indeed, on this point the definition of ‘in vivo’ is critical, and here it refers to data from vessels of hypertensive animals and those treated with anti-hypertensives; as well as those subjected to surgical interventions to alter pressure or flow. Data from in vitro studies using cell or organ culture are not expressly considered here, because such methods often induce states that do not reflect normal physiology.1 Further, comparison of vascular structure between groups needs to be conducted under a defined standard state. For this reason, only results from vessels fixed under maximal relaxation are considered. By setting this standard, a critical appraisal of real changes that occur in vascular structure-function relationships can be made.
To be considered primary changes, the following criteria apply, whereby the;
-
i
Change precedes hypertension development.
-
ii
Treatment affecting these changes also alters BP, such that after treatment withdrawal, BP remains lower.
To be considered secondary adaptive changes, the following criteria apply, whereby the;
-
i
Change occurs after hypertension development.
-
ii
Prevention of hypertension development with drug therapy, or intervention to lower local BP (for example, selective ligation of blood vessels or vascular beds) prevents such changes.
Arteries are classified according to their location and branch point, size and structural characteristics; the latter showing variability within and between beds, between sexes and species, and in development, ageing and disease (for example refs 2, 3, 4; Tables 2,3).
Structural changes in different artery types
Conduit or elastic arteries
Conduit or elastic arteries are characterized by the presence of distinct internal and external elastic lamina (IEL/EEL, respectively), and of alternating medial elastin and smooth muscle cell (SMC) layers; with such vessels having a relatively thick adventitia compared with other arteries. These vessels include aorta, hepatic, renal, iliac, femoral, brachial and superior mesenteric arteries, and provide a low resistance path for blood supply to the limbs and visceral organs.5 The vasa vasorum provides a distinct blood supply to the outer media and adventitia of such thick-walled vessels, and is present in the aortic media of large mammals such as humans, cows and horses; but absent in vessels with <29 lamellar units in the media,6 where it can have a role in large artery disease.7 Thus, vasa vasorum is absent in the aorta of mice, rats and rabbits; animals commonly used as models of human hypertension. The total number of aortic medial lamellar units is increased with age during growth, although the increase is less for small (for example, 10% in guinea pigs), over larger animals (for example, 100% in humans6).
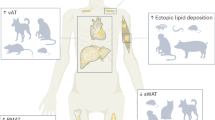
In rat superior mesenteric arteries, lumen diameter is 500–900 μm,8 with 6.5 SMC layers in 3–12 week Wistar Kyoto rats (WKY);9 where the media occupies 62 and 54% of the vessel wall in young compared older animals, respectively.10 The media contains 20% and 14% collagen and elastin, respectively, whereas the adventitia consists of 71% and 12% collagen and elastin, respectively; with fibroblasts (16%) and nerves (<1%) occupying the remaining volume.6 In human thoracic internal artery, perivascular adipose tissue (PVAT) is composed of white fat, compared with rat aorta, which is composed of mostly brown fat.11 Rat vena cava PVAT contains a mixture of white and brown fat cells,12 whereas Wistar rat mesenteric and main renal arteries are surrounded by white fat cells (see Figure 113). The ratio of cross-section area (CSA) of PVAT to CSA of vessel wall is 11:1 in the thoracic aorta (Lee, RMKW and Gao, YJ, unpublished observations).

Critical key interactions between hypertension and vascular remodeling.
Resistance arteries
Resistance arteries are also known as muscular or reactive arteries, due to the presence of mostly SMC in the media. Fragments of EEL are present in some larger resistance vessels, but these are usually visible only with electron microscopy. In adult rats, vessel diameter ranges from 120 to 280 μm in the first/second order mesenteric artery branch (large muscular artery) to 70–110 μm in the second/third order branch (‘small’ muscular artery7). In both large and small muscular arteries from WKY, media occupies ~60–65% of the total vessel wall, and no age-related changes in this proportion occur.10 In muscular arteries and arterioles, endothelial cells (ECs) can communicate directly with the SMC through holes (fenestrae) in the IEL by forming close contacts, some of which contain gap junctions (with an area of ⩽0.08 μm214, 15), at a proportion of EC-SM projections.16, 17 Gap junctions of similar size to those between EC and SMC are also present among the medial SMCs, whereas significantly larger gap junctions are present between ECs.18 The ratio of CSA of PVAT to CSA of vessel wall is 126:1 in the third order branch of rat mesenteric arteries (Lee, RMKW and Gao YJ, unpublished observations).
Arterioles
Arterioles are vessels with 1–2 SMC layers as the most distal segments of the vascular supply; linking to the capillary and subsequent venous networks. Like larger arteries, arteriole properties vary in a vascular bed-specific manner. The IEL can consist of an intermittent mesh-like elastin network, or as a continuous elastin sheet; the latter with intermittent perforating holes, in the same manner as that in arteries. In mature rodents, arterioles are from ~20 to 70 μm in diameter, with variability relating to specific vascular beds. In rat iris and mesenteric arterioles of ~40 μm diameter, the IEL is intermittent in the former and a continuous sheet in the latter19 (and Sandow, SL, unpublished observations). Proximal (‘1A’) arterioles in the cremaster muscle of the rat are up to ~170 μm in diameter and have a continuous IEL (albeit with holes),20 whereas the distal end of this vessel of <~80 μm in diameter has a discontinuous-intermittent IEL (Sandow, SL, unpublished observations); with arterioles of the hamster cheek pouch of ~80 μm diameter having an intermittent IEL.18 Relative to overall vessel diameter, the arteriolar adventitia is usually thin; but can also be sparse to thick, depending on the vascular bed, species (see comparative citations above) and developmental state; at least in arterioles of the rat iris (Sandow, SL, unpublished observations).
Site of vascular resistance and relation to hypertension
Around 70–90% of vascular resistance is modulated by muscular arteries and arterioles; with a significantly lesser contribution from conduit vessels (~10–30%). Notably however, in the cerebral circulation, large arteries play a major role in the regulation of blood flow.21 This is especially relevant in studies of hypertension because microcirculation studies of different vascular beds of spontaneously hypertensive rats (SHR) show that small arterioles control only 10–15% of the total normal resistance, as compared with 60-70% controlled by small arteries and large arterioles.22 In the cremaster muscle of SHR and WKY, ~65% of the pre-capillary pressure fall occurs in arteries with a diameter >100 μm.23 Direct measurement of BP in conscious adult rats show that 31% of systemic BP was dissipated proximal to the base of the mesenteric arcade, and that arcade arteries can contribute substantially to the control of peripheral resistance.24 Furthermore, in the mesenteric8 and renal25 vascular beds of adult SHR, increased SMC layers occur only in arteries/arterioles >70 μm in diameter. Among the intestinal arterioles in SHR, SMC hypertrophy underlies significant changes in the wall.26 These results indicate that in hypertension, most structural changes occur in muscular resistance arteries. Thus, such vessels have a critical role in the control of vascular resistance in hypertension, simply because constriction of these arteries will increase BP, irrespective of whether changes occur in downstream vessels or not. In this review, in small animals such as rats, arteries with a diameter of 100–300 μm are considered resistance vessels; irrespective of the evidence supporting this being mainly circumstantial.27
Changes in conduit arteries
Much of the early work on structural changes in hypertension focused on large elastic vessels such as aorta. In the SHR aorta, it was surmised that increased BP facilitated the secondary adaptive structural changes; given that these are absent in young SHR prior to increased BP,28 and treatment that lowered BP also prevented wall thickening.29 The increased medial mass of SHR aorta was largely owing to SMC hypertrophy associated with polyploidy.30, 31 Chronic treatment of SHR from gestation to 28 weeks of age with nadolol, a β-adrenoceptor blocker, did not prevent hypertension development, but reduced polyploidy SMC incidence in the aorta, indicating that polyploidy SMC development in hypertension is mediated by β-adrenoceptors.32
In the superior mesenteric artery, a thickened medial wall was found only in SHR with established hypertension,33 suggesting that as in the aorta, the change was likely also an adaptive response. However, in SHR treated in utero with vasodilator hydralazine, which prevented hypertension development, medial hypertrophy progress was unaffected, indicating that other intrinsic factors besides elevated BP underlie vascular remodeling in this vessel.34 In the carotid artery, the medial wall is already thickened in 135 and 15 day old36 SHR. In a similar manner, in the main renal artery, a thicker medial wall is present in pre-hypertensive SHR, compared with age-matched WKY.25 These results suggest that changes in the carotid and main renal artery are likely primary changes related to hypertension development in SHR. However, these arteries are not typical of large elastic vessels such as the aorta. The media of the carotid artery consists mostly of SM, and the main renal artery resembles the superior mesenteric moreso than the aorta. Notably, no structural change occurs in the femoral artery of female SHR as compared with normotensive male Sprague-Dawley rats.37
In most other non-SHR hypertensive models, a thickening of the aortic medial wall also occurs, but the specific changes differ. In the deoxycorticosterone acetate model, medial enlargement owing to increased elastin and collagen occurs in the thoracic aorta in one,38 but not in another study.39 Instead, an increase in medial CSA occurs in the abdominal aorta, owing to SM hyperplasia.39 Hyperplasia of SMC is involved in the enlargement of the medial wall in deoxycorticosterone acetate/ NaCl hypertensive rats,40 and aortic wall in rabbits made hypertensive by partial abdominal aortic constriction above both kidneys.41 In renal hypertensive rats, an increase in medial SMC mass occurs, but the specific type of SMC change (that is, hyperplasia or hypertrophy) was not reported.42 In rats made hypertensive by surgical constriction of the sub-diaphragmatic aorta, medial enlargement was due to SMC hypertrophy,43 which was also involved in increased medial mass in the superior mesenteric of Dahl salt-sensitive hypertensive rats.44
Thus, specific changes in the medial wall of elastic vessels in hypertension are highly dependent on the vessel type and animal model examined. In the SHR, changes such as hypertrophy of aortic SM are adaptive, following in response to hypertension, whereas those in carotid and the main renal artery are primary and may contribute to hypertension development. In other animal models, changes associated with SM hyperplasia or hypertrophy are likely associated with the types of stimulus in the respective models. For instance, medial SMC hypertrophy in animal models may be induced by increased Angiotensin II (Ang II) production; as suggested from studies showing that Ang II induced SMC hypertrophy in culture,43, 44 and reversal of aortic medial hypertrophy by Ang II block.45 The Goldblatt hypertensive rat is an example of where SMC hypertrophy contributes to the increased medial mass in the aorta, which was accompanied by an increase in DNA ploidy.46
The changes in conduit arteries are summarized in Table 1.
Changes in resistance arteries
In most hypertension models, consistent changes are observed in large arterioles and muscular arteries. As above, these vessels are of primary importance for the control of blood flow and pressure, and thus for the control of vascular resistance. In rat mesenteric arteries (diameter 120–280 μm), medial wall hypertrophy occurs in SHR at the pre-hypertensive phase of hypertension development,47 indicating changes in this type of vessel are primary, thus contributing to such development. Medial wall hypertrophy persists in SHR during the early and established phases of hypertension development.10 In SHR treated in utero with hydralazine, which prevented hypertension development, medial hypertrophy still occurred;34 and treatment withdrawal after 26 weeks resulted in increased BP within 2 days;48 supporting the primary nature of the vascular remodeling in these mesenteric arteries. Similar results in such vessels also occur in SHR treated with hydralazine from 4 weeks of age.49 Notably, the wall of the relaxed hypertrophied vessels did not encroach upon the mesenteric artery lumen in male SHR where most of the studies were done. In the female SHR, however, the superior and muscular mesenteric artery lumen is smaller with no change in the CSA of the total vessel wall, compared with WKY,50 thus illustrating a gender-based difference in structural change in SHR. In the deoxycorticosterone acetate–NaCl40 and Dahl salt-sensitive genetic hypertensive rats,44 medial wall hypertrophy again occurs without a change in lumen diameter under maximal relaxation. Based on the increased SM layers and SMC length,51 the medial hypertrophy in these animal models of human hypertension is attributed to both SMC hypertrophy and hyperplasia.
In the cerebral circulation, vessel wall hypertrophy occurs in the basilar (lumen diameter, 200–345 μm) and superior cerebellar arteries (lumen diameter, 115–270 μm) from SHR and stroke-prone SHR (SP-SHR), characterized by an increase in SMC layer numbers.52 No difference in basilar artery lumen size occurs among SHR, SP-SHR and WKY, but in the superior cerebellar, lumen was smaller in SP-SHR than SHR. In the posterior cerebral artery (lumen diameter, 90–230 μm), even though SMC layer numbers were higher in SHR and SP-SHR than WKY, medial wall hypertrophy occurs only in SP-SHR; mainly because these vessels are larger in the SP-SHR than SHR and WKY; given the larger lumen size. Nevertheless, media-to-lumen ratio was higher in SHR and SP-SHR than WKY.52 These results show that even among the hypertensives, there are differences in the specific changes between animal strains and vascular beds.
In the renal vasculature, with the exception of pre-glomerular arterioles, all renal arteries of pre-hypertensive SHR show an increased medial wall; the CSA in most instances being due to increased SMC layers, indicating that a primary change contributed to hypertension development.25 A similar change also occurs in adult SHR, with no change in the lumen size in the renal arteries.25 Treatment of SHR in utero with hydralazine to prevent hypertension development did not change the development of vascular remodeling in the renal arteries of SHR, showing that these are not secondary adaptive changes due to hypertension development.47 Withdrawal of hydralazine treatment results in a rapid BP rise to hypertensive levels within 1–2 days, showing the importance of these changes in the development of hypertension.48
The changes in muscular arteries are summarized in Table 1.
Vascular changes in arterioles
Arteriolar medial and adventitial area and media-to-lumen ratio can be altered in hypertension. For example, in the cremaster muscle arteriole of the hypertensive (and hyper-insulinemic, -lipidemic and -glycemic) diet-induced obese rat, media-to-lumen ratio and IEL thickness is increased and adventitia thickened compared with non-obese control.53 Of note, few studies have examined anatomical arteriolar remodeling in sufficient resolution (such as with electron microscopy) to clarify whether such changes are typical of hypertension; or indeed, causative to hypertension.
Vascular changes in veins
Medial hypertrophy in the portal vein and vena cava of 6-month old SHR has been reported,54 and based on parabiotic pair experiments, a circulating factor was suggested as causative.55 However, in small mesenteric veins (lumen diameter, 366–377 μm56) and the portal vein57 of adult SHR, no change in vessel wall structure occurs. Further clarifying studies are needed to examine whether structural hypertension-related changes occur in the venous system.
Hyperplasia or hypertrophy of SMC in hypertension?
The general medial hypertrophy associated with hypertension in arteries is due to SM hypertrophy and/or hyperplasia, with the specific cause of medial enlargement varying between forms of hypertension (as well as between models) and between vascular beds. Some of the controversy on this issue is owing to methodology. Suggested hyperplastic changes in SM were partly based on cultured SMC data from SHR and WKY, and these cells were often derived from thoracic aorta.58, 59, 60, 61, 62 This is in spite of the findings from studies of intact tissues that medial hypertrophy in SHR thoracic aorta is a secondary adaptive response, owing to hypertrophy and polyploidy of SMC, as discussed above (per, ‘Changes in conduit arteries’). These studies also failed to show that the multi-nucleated and polyploid nature of some SMC in SHR aorta occurred or was maintained in their culture systems. Polyploid SMC proliferate at a much slower rate than diploid SMC, and a need to consider ploidy changes when evaluating in vitro SMC growth kinetics is required.63 It is possible that the SMCs derived from the thoracic aorta of SHR and WKY were diploid; and yet studies using cultured aortic SMC from SHR and WKY made conclusions that were applied to the entire vasculature generally; when this is an inherently invalid premise. Cell isolation and culture have significant effects on SMC growth kinetics; as a further example of cell isolation- and stress-induced phenotypic drift (see ref. 1). Indeed, SMC derived from mesenteric arteries grow at a much slower rate than that from aorta.64, 65 Unfortunately, to date, systematic studies comparing the growth characteristics of isolated SMC from mesenteric arteries (and resistance vessels in general) normotensive and hypertensive models has not been conducted.
In studies of SMC growth, several limitations occur when using isolated such cells. Primarily, these involve the phenotypic changes that arise as a result of the isolation and culture process; being stress- or experimental/condition-specific. Indeed, even with the same agonist, such as Ang II, the SMC growth response can be due to hypertrophy or hyperplasia, depending on the cell isolation and culture methods, and the presence other growth co-factors,66 including chemokines.67, 68 In a similar manner, the lack of modulating influence of other vascular cells such as the innervation, endothelium and adventitial mediator cells in an isolated cell system can limit the relevance of data obtained. Further, unlike in isolated SMC, in intact cultured segments or strips of human, pig, rabbit or rat arteries,69 only a small fraction of the medial cells of an intact vessel can be stimulated to synthesize DNA. In a similar manner to isolated and cultured cell states, the use of organ culture to reportedly simulate in vivo conditions is also of limited relevance given that wall properties alter with culture conditions.70 Thus, considerable caution needs to be exercised in drawing conclusions relating data obtained from isolated cells or tissues to that from intact vessels.
Earlier morphometric studies also contributed to some of the abovementioned confusion. Based on the reported absence of change in SMC length in SHR mesenteric arteries compared with WKY,71, 72 an assumption was made in the protocol for the measurement of vascular changes in hypertension that the length of SMC in the SHR mesenteric arteries was similar to WKY.10 This led to the conclusion that the increased SMC layers in the mesenteric arteries of SHR was due to SMC hyperplasia10 (see also ref. 73). Other studies also arrived at the same conclusion for mesenteric arteries26, 74 and renal vasculature25 of WKY and SHR. Such data supported the concept that during hypertension development, SMC hyperplasia is a major contributor to medial hypertrophy. In time, this concept became so entrenched such that authors of reviews generally accepted it a priori. With the advent of confocal microscopy and appropriate staining techniques for serial sectioning and serial reconstruction, two fixation methods were applied, in vitro and in situ, to the mesenteric vascular bed of young SHR and WKY, and the volume of the medial wall and lumen, numerical density of SMC nuclei, SMC and nuclear length were measured.51 Both methods of fixation yielded similar results showing that increased SMC length is responsible for vascular hypertrophy in the mesenteric arteries of SHR. There are several explanations of why earlier studies suggested that SMC hyperplasia is a major contributor to medial wall hypertrophy in hypertension, as clarified above. Furthermore, three-dimensional reconstruction (two paragraphs, below) via confocal microscopy allowed vascular structure to be assessed directly and without the bias that came with estimating cell number from single histological sections.75
Cell proliferation and apoptosis
The structure of superior and large mesenteric arteries from newborn SHR is similar to that of WKY, indicating that structural changes in these SHR vessels occur after birth either through differentiation of existing myofibroblasts, or SMC proliferation.76 Autoradiographic studies showed that SMC density was similar between superior mesenteric artery from SHR and WKY at 1, 2 and 4 weeks after birth, whereas in the primary and secondary mesenteric branches, SMC density was higher in SHR than WKY at 1 week after birth; but became similar in older age groups.77 These results suggest that increased SMC DNA synthesis may have a role in hypertension development in SHR. Given that medial hypertrophy in these vessels was due to SMC hypertrophy,51 the increased DNA synthesis resulted from larger SMCs rather than an increased cell number. At ~6 weeks of age when SHR BP differentiates and becomes higher than WKY, a higher SMC density occurs in large conduit vessels such as aorta, renal and femoral artery of SHR compared with WKY, but no difference occurs in resistance arteries from mesenteric and renal vascular beds.78 These data suggest that the elevated DNA synthesis in large arteries of 6-week old SHR represents a secondary adaptive response to the marginally elevated BP.78
Another potential contributor to medial hypertrophy in SHR vessels relates to apoptosis. In first order mesenteric arteries from young 1–2-week old SHR, apoptotic SMC incidence is lower than in WKY;79 although at ~1% of total medial SMCs,79 the impact of this change was unlikely to be significant. Notably, in second order mesenteric arteries of 8 and 12, but not young 4-week old SHR, medial SMC apoptosis was significantly increased compared with WKY.80 Thus, apoptosis may exert a role in resistance artery remodeling during hypertension development79, 80 (for review, see refs 47, 81). Indeed, given that this development begins ~6 weeks of age in SHR,47, 82 these data are consistent with SMC apoptosis exerting a role in resistance artery remodeling during hypertension development79, 80 (for review, see refs 47, 81). Thus, in SHR mesenteric artery, vascular remodeling associated with increased medial thickness and BP is potentially developmentally related to increased cell size and an altered apoptotic SMC incidence.
Three-dimensional confocal reconstructions
Three-dimensional confocal reconstruction of tissues ex vivo allows for statistically valid measurement of blood vessel characteristics such as medial, luminal and adventitial volume and SMC nuclei and layer numbers. This method thereby facilitates the evaluation of vessel structure and its effect on BP development, via eliminating the distortion associated with embedding, sectioning and potential investigator bias;83 although fixation remains a (minor) distortion issue. Regardless, confocal microscopy is optimal as thin noninvasive sections, similar to those used for electron microscopy, can be produced and the distance between these optical sections, in terms of tissue volume, can be precisely known.83 Such confocal methods have shown that medial volume of site-matched mesenteric resistance vessels in SHR at 4 weeks of age is significantly larger than in WKY (~16 800 vs. 11 200 μm3 μm−1 length of artery, respectively) when only a small difference in BP between these strains exist at the young age.83 Further, SM layer numbers in the media of these vessels are increased in SHR compared with WKY (4.1 vs 2.7, respectively) and correlation analysis revealed that the increase in SMC layers accounted for 81% of the medial volume increase.83 Confocal microscopy also shows that increased medial volume in pre-hypertensive SHR mesenteric arteries is due to increased SMC length;50 whereby medial volume expansion occurs in SHR, and not WKY, without a lumen change in the maximally relaxed state. Further, SMC density is less in SHR compared with WKY, whereas SMCs (and their nuclei) are longer in SHR than that WKY.50 Interestingly, in basilar artery of SP-SHR, SMCs have an abnormal orientation, with discrete regions of SMC at an altered angle of orientation in the XY plane84 (for review, see ref. 85), suggesting abnormal SMC and adventitial cell interaction (for review, see ref. 85).
In the adventitia of rat mesenteric and basilar artery, confocal imaging shows a higher cell density and heterogeneous morphology in SP-SHR and L-NAME-induced hypertensive rat models over control84, 86 (for review, see ref. 85), consistent with the increased role of sympathetic constrictor and sensory dilator innervation in hypertension (see following section, below); and increased adventitial stiffening with elevated pressure load and stress.87 Confocal data further show that Ang II-infused rats display significantly increased media-to-lumen ratio and a significantly higher expression of vascular cell adhesion molecule 1 in mesenteric arteries compared with untreated control, with these effects being abolished by peroxisome proliferator-activated receptor α docosahexaenoic acid.88 Thus, these data show that lowering BP by docosahexaenoic acid is associated with improved endothelium-dependent relaxation (EDR) and altered vascular structure in Ang II-infused rats.88
Confocal studies also show that drug-mediated correction of hypertension has implications for vessel structure. A combined treatment with the angiotensin-converting enzyme inhibitor (ACEI; see also ‘Renin–angiotensin section’, below) quinapril and HMG-CoA reductase inhibitor (and cholesterol medication) atorvastatin lowered systolic BP in both SHR and WKY rats.89 Confocal myography found a decrease in medial thickness and volume and SMC layer number in mesenteric arteries; in addition to a decrease in the media-to-lumen ratio in the interlobular arteries from quinapril and atorvastatin treated SHR, but not in WKY rats.89
Mechanisms of vascular remodeling in vivo associated with hypertension development
Sympathetic nervous system
In humans, sympathetic activity is increased in a significant number of patients with hypertension, and the increase in sympathetic drive is widespread across many organs (summarized in Table 2).90 A similar state occurs in rat models, where vessel function is altered in sympathectomy (for example, ref. 91) potentially and likely reflecting anatomical observations; per examples, below. In the SHR, tachycardia in pre-hypertensive young rats is a good predictor of eventual BP outcome, and this correlation occurs even among normotensive rats.92 In order to demonstrate the essential role of the sympathetic nervous system in the development of hypertension multiple methods have been used to induce sympathectomy. However, traditional methods using sympatholytic agents such as guanethidine, or anti-nerve growth factor, were ineffective in the SHR, partly because of the resistance of the SHR to these agents.93 The failure to induce sympathectomy using the common sympatholytic agents in neonates is also related to the sympathetic perivascular plexus developing earlier in vessels of SHR than WKY.91 In cerebral arteries, a higher innervation density occurs in SHR compared with WKY 1 day after birth. Sympathectomy with guanethidine and anti-nerve growth factor after birth reduced cerebral artery nerve density in SHR and SP-SHR, but did not cause complete sympathectomy, as is the case in WKY.94 Mesenteric arteries were not innervated at birth, and a higher innervation density was present by 10 days after birth in SHR compared with WKY.94 The only effective method to achieve complete sympathectomy in SHR is a combination of guanethidine and anti-nerve growth factor after birth, which eliminates sympathetic nerves in the mesenteric arteries of SHR and WKY.94, 95 Such sympathectomy prevents hypertension development and structural and functional changes in SHR mesenteric arteries;95 thus showing the trophic influence of sympathetic nerves on vascular changes and hypertension development in SHR. Sympathetic nerves also have a trophic influence on cerebral artery remodeling during hypertension development in the SHR and SP-SHR.52 In sympathectomized SHR, circulating levels of catecholamines from the adrenal medulla still cause a higher BP than in WKY.95 Adrenal demedullation caused a further decrease in BP to WKY levels, which is associated with increased lumen size in muscular arteries.96 Sympathectomy alone or in combination with demedullation increased adventitia size in both hypertensive and normotensive animals.95, 96, 97
The effects of other perivascular nerve types, such as the sensory population, on vascular remodeling and hypertension development are unclear. Neonatal sympathectomy with anti-nerve growth factor and guanethidine did not destroy all the nerves surrounding the blood vessels of SHR and WKY.94 Vasoactive intestinal peptide is 50–100 times more potent than acetylcholine as a vasodilator98 through its endothelium-mediated nitric oxide (NO) activity.99 Vasoactive intestinal peptide-containing fiber density was higher in veins and superior mesenteric artery of SP-SHR than WKY, and lower in cerebral arteries of SP-SHR than of WKY; although no difference occurs in mesenteric resistance arteries of these strains. Sympathectomy reduced nerve density in all the peripheral vessels, but had little effect on cerebral arteries.100 Substance P-containing nerves are involved in the control of the mesenteric artery myogenic response.101 Neonatal SHR treatment with capsaicin, which destroyed substance P-containing sensory nerves also prevented hypertension development;102 although via an unknown mechanism. The density of substance P-containing nerves is similar between SP-SHR and WKY in the peripheral vessels, but higher in cerebral arteries of WKY than SP-SHR. Sympathectomy reduced the density of these nerves in the peripheral vessels, but increased the density in some cerebral arteries of SP-SHR.100 Thus, it is unlikely that substance P is involved in hypertension development, at least not in the SHR. The density of neuropeptide Y containing nerves was higher in the peripheral vessels of SP-SHR than WKY, and sympathectomy caused a near-complete removal of these nerves,100 which may be related to the association of sympathetic and sensory innervations.
Renin–angiotensin system
Infusion of Ang II has a significant effect on vascular morphology, hypertension development and BP; including a sensory innervation-mediated affect. In maximally relaxed vessels, Ang II infusion increased the polyamine concentration and medial area in rat mesenteric arteries.103 The increased medial wall area was mediated by angiotensin II type 1 (AT1) receptors,104 and Ang II infusion also caused changes in PVAT function in the aorta and mesenteric arteries, and structural and functional changes in the latter that are associated with hypertension development.105
Studies of antihypertensive treatment on artery structure have helped unravel the mechanisms underlying vascular changes in hypertension. Among the different types of anti-hypertensives, treatment with ACEI have significant effects on vascular structure. Treatment of SHR with the ACEI captopril, before and after birth prevented hypertension development, structural and functional changes of mesenteric arteries and cardiac hypertrophy.50 Smaller lumen size in female SHR was normalized to that in WKY. In superior and large mesenteric arteries, captopril treatment prevented medial hypertrophy, and the increased number of SMC layers.50 Treatment of SHR beginning at 3 weeks of age for 12 weeks with an AT1 receptor antagonist L-158-809, lowered BP and decreased wall thickness in mesenteric arteries, but did not prevent hypertension permanently, as treatment withdrawal caused an increase in BP to hypertensive levels, and an increase in wall thickness to the level of untreated control.106 Notably, ACE inhibition is effective in delaying the redevelopment of hypertension following inhibitor withdrawl or calcium channel blocker (nitrendipine) administration.107, 108 Interestingly, a permanent inhibition of the potentiating effects of Ang II on the nerve-mediated response was achieved with AT1 receptor blocker treatment.106 In contrast, treatment of adult (15-week old) SHR with the ACEI perindopril was more effective than treatment with an AT1 receptor blocker; since after 10 weeks of treatment and the switching of the drugs between the two treatment groups, BP was increased in the group switched from ACEI to AT1 blocker, whereas BP decreased in the group switched from AT1 blocker to ACEI.109 The better BP control achieved via treatment with an ACEI is likely due to its effects on the vascular wall; ACEI induced a thinner wall than did the AT1 blocker. Other effects of ACEI include the production of other vasoactive products such as angiotensin 1–7 (Ang-(1–7)) and an accumulation of bradykinin.109 Treatment of SHR with ACEI increased plasma levels of Ang-(1–7), 5–50-fold,110, 111 and infusion of Ang-(1–7) in SHR reduced BP.111, 112 This aspect is interesting in view of Ang-(1–7) being the only factor identified to date as the diffusible EDR factor released by PVAT,113 thereby providing a local source of Ang-(1–7) in the modulation of vascular structure and function. Of interest, other receptor-mediated mechanisms may also play a role in SM proliferation in hypertension. For example, in cultured aortic vascular SM the D4 dopamine receptor has an inhibitory effect on proliferation of, and is suggested to play a regulatory role in AT1a receptor expression, where its role is preserved in SHR over WKY.114
The effect of ACEI on vascular structure is in many cases likely related to its ability to induce SMC apoptosis. Treatment of 10-week old SHR with the AT1 blocker losartan or ACEI enalapril for 4 weeks increased the SMC apoptosis rate, resulting in a reduction in medial mass and SMC number.115 Treatment of adult (15-week old) SHR with quinapril for 10 weeks caused vascular remodeling through the induction of medial SMC apoptosis in rat mesenteric arteries, and reduced the augmented contractile response and improved EDR.116 These results suggest that Ang II has an inhibitory effect on SMC apoptosis. Interestingly, the response to ACEI differs between mesenteric and renal arteries. In mesenteric artery of adult SHR and WKY, quinapril reduced the media thickness-area and media-to-lumen ratio, but in renal vessels, only the media-to-lumen ratio of the interlobular arteries from SHR was reduced.116 In contrast, treatment of young SHR with enalapril for 10 weeks significantly reduced the media-to-lumen ratio of interlobar and interlobular arteries.117 These data show that in young SHR, treatment with enalapril prevented further development of vascular hypertrophy in the renal vessels, whereas in adult SHR with established hypertension, reversal of vascular hypertrophy was possible in the mesenteric, but only in some larger renal arteries.116 Treatment with ACEI also prolonged the lifespan of SHR after treatment withdrawal through reduced damage to the brain and kidneys.118 In addition to its effect on SMC growth and thereby vascular remodeling in hypertension,66 Ang II also affects stroke development.119, 120 In SP-SHR, the myogenic response of middle cerebral artery in response increased pressure was lost prior to stroke development.121 Treatment of SP-SHR with ACEI prevented or delayed the onset of stroke by preserving the cerebral artery myogenic response.120
Alteration in response to changes in flow and/or pressure
In the cerebral circulation, arteries respond to changes in pressure or flow by constriction or relaxation, as the autoregulatory mechanism. Thus, an increase in pressure or flow will induce constriction to protect small vessels downstream, whereas vessels will dilate in response to a decrease in pressure or flow, to maintain adequate perfusion to vessels downstream.
Hemodynamic changes are powerful inducers of arterial structural change. An elevated blood flow usually increases lumen size due to the increased fluid shear force applied on the vessel wall.122 Given that it occurs soon after the elevation of blood flow, the luminal expansion via an acute elevation of blood flow is mainly a functional adjustment, whereas permanent changes in vascular dimensions owing to chronic increased blood flow are associated with changes in cellular and extracellular components of vascular tissue.123, 124 Vasodilation associated with an acute elevation of blood flow involves the releases of vasodilators such as NO and/or prostaglandins125, 126 from the endothelium, and/or the activation of specific ion channels.127
Although mesenteric blood flow in SHR is similar to that of WKY,128, 129 arterioles from young SHR are subject to a significant increase in shear rate compared with WKY and older SHR.130 A higher blood flow velocity in mesenteric arterioles from SHR compared with WKY has been reported.131 Thus, arteries from SHR are subject to increased blood flow and pressure. By using the ligation method of Unthank et al.,132 responses to chronic increased blood flow in an in vivo environment can be examined; whereby small mesenteric arteries exposed to increased blood flow become thicker with larger diameter, due to hyperplasia and / or hypertrophy of SMC. The endothelium appears to be involved in this response as its removal prevented vascular changes associated with altered flow,122, 133 consistent with endothelium-mediated growth factor action.134, 135 Gao et al.,136 applied the ligation method in mesenteric arteries of 11–12-week old SHR and WKY for 7–10 days, and studied vascular changes with an emphasis on SMC growth and apoptosis. In WKY, increased blood flow resulted in a reduction of apoptotic SMC, no change in wall-to-lumen ratio, and an increase in lumen diameter, the number of SMC layers, and the proliferating cell nuclear antigen (PCNA)-positive SMC. In SHR, elevated blood flow increased vessel wall thickness and SMC layer numbers, and the wall-to-lumen ratio, and PCNA-positive SMC; but with no change in lumen size or the number of apoptotic SMC. These results show that mesenteric arteries from hypertensive and normotensive rats respond to an increase in blood flow differently. They also showed that an alteration in blood flow is a powerful modulator of vascular remodeling even in the presence of hypertension as in the case of SHR. The mechanism involved in causing vascular changes due to a chronic increase in blood flow is not known.
Endothelium-mediated mechanisms
Endothelium-dependent relaxation and constriction (EDC), and the balance therein are key mechanisms for the control of vascular tone and thus of blood flow, pressure and tissue perfusion. Defects in the mechanisms of EDR and EDC are associated with the etiology of hypertension (for example, for review, see refs 2, 137, 138). Indeed, extensive animal model data, supported by studies from human subjects showed that defects in all mechanisms of EDR and EDC have significant implications for hypertension. However, whether these are a cause or consequence of an elevated BP is not fully clarified. Here, a brief outline of the EDR-EDC mechanisms that contribute to hypertension, and their potential association with arterial remodeling is presented.
In the normal state, the mechanisms of EDR and EDC have different contributions in and between vascular beds, between species and sexes, and during development and ageing. This variability in specific mechanisms also applies in disease; including hypertension. The specific alterations in dilator and constrictor pathways are diverse and in part relate to the relative and differential contribution of specific mechanisms of EDR as endothelium-derived hyperpolarization (EDH), NO and cyclooxygenase; and of EDC action, including that of endothelin, superoxide and thromboxane. The characteristics of remodeling also differ in and between vascular beds, between species and sexes, and during development and ageing; but do not appear to be directly related to the mechanism of EDR that are altered in hypertension (Table 3). Indeed, typical conduit and resistance arteries and arterioles show diversity in EDR and specific structural and remodeling characteristics with hypertension associated with a rat model of diet-induced obesity (Table 3). Given the apparent lack of association of remodeling and altered EDR mechanisms, alterations in EDC may also not be directly linked to arterial remodeling. However, ultrastructural data on this latter point are not available.
Does arterial remodeling and hypertension arise as a cause or consequence of altered EDR and EDC? As above, whereas this is unknown, there are correlative ultrastructural and functional data that allude to whether or not this is the case. However, a significant caveat to this is that the morphological and ultrastructural characteristics of remodeling show significant diversity between different vascular beds, even within the same hypertensive animal model (Table 3) and do not change in a consistent manner in different vascular beds. Indeed, these data may be considered to be consistent with the generalized dogma of reduced lumen diameter and increased medial area underlying essential hypertension in resistance vessels, such as mesenteric and cremaster muscle arteries, as compared with conduit vessels such as the saphenous and femoral arteries and aorta.91, 139
Of note, EDH-dependent myoendothelial gap junctional coupling was absent in the saphenous artery (as a conduit vessel) of the adult normotensive control rat, but present in the hypertensive diet-induced obese rat, which also exhibited no change in medial area or media-to-lumen ratio compared with control;140 but also exhibited impaired NO-mediated EDR and increased endothelial caveolae density.141 Interestingly, EDH-dependent myoendothelial gap junctional coupling was present in the saphenous artery of the juvenile 14-day-old rat in contrast to the saphenous from the normal 16-week-old adult animal.142 Indeed, developmental BP in normotensive rats peaks at ~6–10 weeks of age.82 In combination, assuming that myoendothelial gap junction presence reflects the same overall function in the saphenous artery from young and hypertensive animals (being primarily a dilator mechanism; but also related to differentiation134, 135), these data suggest that the remodeling associated with the EDH-dependent myoendothelial coupling mechanism is not directly or solely related to elevated BP. However, given the additional significant functional and anatomical changes that occur in development and disease, altered myoendothelial coupling will likely have implications for the conduction of endothelium-dependent responses, in development and disease,137, 143 with future studies being required to address this issue.
As above, the specific signalling mechanisms associated with EDH are diverse within and between vascular beds, species, ageing, development and disease. Regardless, as a component of EDR a fundamental underlying mechanism of EDH involves small and/or intermediate conductance calcium-activated potassium channel (S/IKCa) activation. However, other signalling mechanisms associated with EDH can be both conserved and diverse within and between vascular beds. For example, the contribution of (in some cases, microdomain-related) inward rectifying potassium channels (Kir), Na-K-ATPase, and inositol-1,3,5-trisphosphate-mediated Ca2+ release3, 144, 145 varies in and between different arteries, as above, to be associated with both similar and different underlying signalling pathways, depending on the artery. In this regard, changes in EDH-related S/IKCa-Kir-Na-K-ATPase expression and activity have been reported in different arteries in the same model of obesity-related hypertension. In contrast to the conduit saphenous artery, in 4th order mesenteric (resistance) arteries from the hypertensive diet-induced obese rat model, the myoendothelial coupling density was unchanged and the media-to-lumen ratio increased146 (Table 3). In the mesenteric artery from the hypertensive diet-induced obese rat, EDH was due to an increased contribution of IKCa and decreased SKCa and Kir, compared with normotensive control.
In cremaster muscle arterioles (as a typical microvascular resistance vessel) of the hypertensive diet-induced obese rat, SMC layers, media-to-lumen ratio, wall thickness and medial area were increased; whereas vessel diameter was not significantly different from that in the normotensive control53 (Table 3). As above, these observations again reflect the diversity in remodeling between different vessels in the same model of hypertension; and that also occur between models of hypertension.147 Interestingly, in the hypertensive diet-induced obese rat, cremaster muscle arteriole endothelial caveolae density was decreased, and NO synthase (S) activity and NO activity induced;20 all of which contrast to observations from the conduit saphenous artery in this model.141
In the caudal artery of the hypertensive adult SHR rat, SMC layers, medial area and myoendothelial gap junction density is increased, and vessel diameter decreased compared with age-matched normotensive WKY.15 In contrast to the hypertensive diet-induced obese rat model, such changes are consistent with the general view that such increased medial area and reduced lumen diameter may underlie elevated BP. However, the implications of such observations are complicated by the diversity in mechanisms between vascular beds and the genetic diversity between comparative strains of SHR between laboratories (for example, refs 148, 149).
Based on these data, and as above, the ultrastructural characteristics of remodeling of the vessel wall in hypertension are diverse with differences among various vascular beds in the same model of hypertension as well as between models and vascular beds. Reflecting on this, there are diverse changes in the contribution of mechanisms of EDH and NO between beds in the same animal model. For EDH and related myoendothelial gap junctional coupling these involve dramatic changes from absence to presence and other subtle changes such as alterations in the specific contribution of K+ channels.146 However, these data do not support a direct link between the ultrastructural remodeling, EDR and the presence of hypertension.
Elastic lamina, extracellular matrix (ECM) and integrins
The vascular ECM contains several elements, including elastin, collagen and integrins, as well as various other proteins, which collectively have a significant mechanical role in the vascular wall; serving as a mechanism to influence stiffness and elasticity, with clear implications for remodeling in hypertension (for review, see refs 150, 151, 152). Specifically, the ECM modulates several aspects of artery function, which can be altered in hypertension; albeit in a differential manner depending on the vessel location and form of the disease or model. Such ECM activities include interrelated factors, as mechanisms to; (1) sense two-way events where ECM mechanical forces are transferred into SMCs/ECs, and where vasomotor events are transmitted to the ECM, thereby modulating vascular cell adhesion;152 (2) control mechanical forces associated with increased pulse pressure that are transmitted to the progressively smaller arteries; such as that associated with large vessel stiffening in, for example, hypertension and atherosclerosis; (3) influence cell positioning; including reputed changes in SMC-SMC longitudinal interactions that occur with chronically elevated constriction (albeit in culture conditions);153, 154 (4) provide communication pathways between EC and SMC projections, contacts and related coupling sites associated with gap junction and/or microdomain signalling activity;16, 137, 155 and (5) facilitate the control of longitudinal and radial force generation associated with tissue movement.152
PVAT
In most in vitro studies of vascular structure and function, the PVAT layer outside the adventitia was generally removed, partly because it was thought to have little if any vascular function, and also as it is easier to study vessel structure and function in the absence of said layer (for example, refs 138, 144). However, adipose tissue is an important endocrine and metabolic organ. It is known to release many substances that can cause vascular SM relaxation and constriction.153 These include leptin,156 hydrogen sulfite,157 omentin,158 adiponectin159 and palmitic acid methyl ester.160 Notably, PVAT causes dilation of mesenteric resistance arteries;161 and conduit vessels such as human internal thoracic artery,11 as well as in large veins of the rat.12 Importantly, in pressurized mesenteric arteries, lumen diameter is larger in arteries with intact PVAT than in arteries with PVAT removed, in both control and hypertensive rats, indicating that factors released by PVAT are functional under resting conditions, and that it can play an important role in the control of tone.105, 113
PVAT modulates vascular function through two distinct mechanisms: (1) the release of a transferable relaxing factor, which induces EDR through NO release and subsequent KCa channel activation and (2) an endothelium-independent mechanism involving hydrogen peroxide and a subsequent activation of soluble guanylyl cyclase.162 Subsequent studies have shown that this transferable relaxation factor acting through the endothelium is Ang-(1–7), with a mechanism involving Mas receptors and NO release.113, 160 Using Mas-knockout and wild-type mice, it was confirmed that Mas receptors are essential in mediating the EDR induced by PVAT; thereby highlighting the important role of Ang-(1–7) in the control of vascular function through PVAT.160 In contrast, in the vein, the endothelium-independent PVAT-mediated mechanism inhibiting constriction is absent.163
PVAT function is altered under pathological conditions such as obesity,158, 164, 165 and hypertension,105, 156, 163, 166 and is characterized mostly by an attenuation of PVAT effects on agonist-mediated constriction. However, in diabetes, acute and chronic hyperglycemia potentiated PVAT-modulated EDR; with hyperglycemia having no effect on the endothelium-independent relaxation pathway.167 Interestingly, an absence of adipose tissue (lipoatrophy) induces hypertension in these animals; which appears likely owing to the absence of a PVAT enhanced contractile response to agonists, and an upregulation of vascular AT1 receptors.162 PVAT also potentiates vasoconstriction induced by nerve stimulation through superoxide-mediated by Ang II production;13, 168 and potentially via PVAT-mediated activation of SM large conductance KCa,168 and macrophage activity.169
Antihypertensive treatment also affects PVAT function. Treatment of SHR with atorvastatin restored the PVAT-mediated inhibition of noradrenaline-induced constriction.170 Chronic treatment of obese mice with melatonin also restored the anti-contractile effect of PVAT.171 Whether PVAT is involved in the modulation of vascular remodeling in hypertension is largely unknown. However, given that Ang-(1–7) is the PVAT-mediated EDR factor, and that per above, PVAT function is altered in hypertension, the role of Ang-(1–7) on vascular remodeling in hypertension deserves further study. Notably, SHR treatment with an ACEI and an angiotensin receptor antagonist increased plasma levels of Ang-(1–7),172 and Ang-(1–7) inhibits SM cell growth.173 Thus, it is feasible that vascular remodeling associated with ACEI treatment is associated with Ang-(1–7), in at least some models and forms of hypertension.
An emerging area of research relates to the modulation of drug action by PVAT. Propofol, a commonly used anesthetic, causes hypotension owing to vascular SM relaxation, through its direct or indirect vasodilator effects (see ref. 174). PVAT enhances the relaxation effect induced by propofol in rat aorta through both ED and endothelium-independent pathways.175 In the ED pathway, propofol induced NO release via PVAT and activation of endothelial potassium channels to hyperpolarize SM, resulting in relaxation; whereas the endothelium-independent pathway involved the release of hydrogen peroxide by PVAT and subsequent activation of soluble guanylyl cyclase.175 This is in contrast to thiopental, another popular anesthetic until recently, where thiopental-induced relaxation was acting through an endothelium-independent pathway, and the presence of PVAT, endothelium, or both attenuated this relaxation response induced through Ang II-dependent and ED mechanisms, respectively.175 The interaction of PVAT with drugs (and notably, anesthetics with broad and as yet uncharacterized effects), including those in the treatment of hypertension and diabetes, will be an important area for future work.
Of note, PVAT is absent in cerebral arteries and intramuscular arteries of the heart, and therefore local control of vascular function by substances released by PVAT is absent; although such vessels may still react to circulating factors released by adipose tissue. Indeed, further work in this area may provide insight into the disease process leading to the development of stroke and coronary artery disease.
Conclusions
Vascular changes in hypertension are complex, and are closely associated with both the development of hypertension alone, and the response to changes in the circulating levels of hormones and vasoactive compounds. The specific composition of any given artery and the changes that occur therein in disease, differ within and between vascular beds, in development, ageing and between species and sexes. The role of PVAT in the modulation of vascular structure under various disease states such as hypertension, obesity and metabolic syndrome is an emerging area of research, and is likely to contribute to the heterogeneity described above.
Among the in vitro methods used to study vascular structure, it is critical to understand and appreciate the limitations of the methods used. For example, the use of the vascular myograph system is ideal for functional studies (pioneered by Mulvany and Halpern176), but is not suitable for quantitative morphological work; particularly on small diameter vessels (for review/summary, see refs 177, 178). For meaningful morphometric studies, blood vessels must be fixed in situ in a consistent and ideally a maximally relaxed state, so that accurate measurement of lumen size and vessel wall parameters can be carried out on tissue in a uniform state between preparations (for review/summary, see ref. 177).
The use of cell and organ culture to study the pathological processes in hypertension is problematic. Although the use of these systems has yielded significant advances in the understanding of vascular cell and tissue function at the cellular and molecular levels, they are most useful in ‘proof of principle’ experiments, such as demonstrating that Ang II induce SMC, or blood vessel hypertrophy. However, when utilized to study changes between hypertensive and normotensive vessels, or cells derived from these vessels, culture conditions rather than tissue origin significantly influence the results. Other limitations in such methods include phenotypic change owing to the culture methods themselves, primarily as stresses on the isolated cells and organs; as being artificial from the intact vessel state. Unfortunately, the limitations of the use of cell and tissue culture are often underplayed or ignored. Indeed, when comparisons of culture and the intact vessel state are conducted, the results generated therein are usually different, thus supporting critical consideration of the culture state and the data generated therein.
Differences in the etiology of specific forms of hypertension occur, and the characteristics of vessel remodeling reported in hypertension show significant variation in the literature; even for the same vessel under apparently equivalent conditions. Other factors that contribute differences in reported morphology, but that are often overlooked, include differences in age, sex, genotype between laboratories,149 as well as differences in methodology of how remodeling was assessed; including drawing conclusions from disparate data sets (such as isolated cell to intact vessels); without adequate consideration of these issues.
Understanding the fundamental properties of artery function and how these relate to signalling mechanisms in real tissues is key to translating isolated cell and intact tissue data. Indeed, the ultimate aim of developing new treatments to correct vascular dysfunction requires understanding and recognition of the limitations of the methodologies used.
References
Sandow SL, Grayson TH . Limits of isolation and culture: intact vascular endothelium and BKCa . Am J Physiol 2009; 297: H1–H7.
Barton M . Obesity and aging: determinants of endothelial cell dysfunction and atherosclerosis. Pflugers Arch 2010; 460: 825–837.
Sandow SL, Senadheera S, Bertrand PP, Murphy TV, Tare M . Myoendothelial contacts, gap junctions, and microdomains: anatomical links to function? Microcirculation 2012; 19: 403–415.
Sandow SL . Factors, fiction and EDHF. Clin Exp Pharmacol Physiol 2004; 31: 563–570.
Greenwald SE . Ageing of the conduit arteries. J Pathol 2007; 211: 157–172.
Wolinsky H, Glagov S . Nature of species differences in the medial distribution of aortic vasa vasorum in mammals. Circ Res 1967; 20: 409–421.
Tanaka H, Zaima N, Sasaki T, Hayasaka T, Goto-Inoue N, Onoue K, Ikegami K, Morita Y, Yamamoto N, Mano Y, Sano M, Saito T, Sato K, Konno H, Setou M, Unno N . Adventitial vasa vasorum arteriosclerosis in abdominal aortic aneurysm. PLoS ONE 2013; 8: e57398.
Lee RMKW . Structural alterations of blood vessels in hypertensive rats. Can J Physiol Pharmacol 1987; 65: 1528–1535.
Lee RMKW, Forrest JB, Garfield RE, Daniel EE . Ultrastructural changes in mesenteric arteries from SHR. A morphometric study. Blood Vessels 1983; 20: 72–91.
Lee RMKW, Garfield RE, Forrest JB, Daniel EE . Morphometric study of structural changes in the mesenteric blood vessels of SHR. Blood Vessels 1983; 20: 57–71.
Gao YJ, Zeng ZH, Teoh K, Sharma AM, Abouzahr L, Cybulsky I, Lamy A, Semelhago L, Lee RMKW . Perivascular adipose tissue modulates vascular function in the human internal thoracic artery. J Thor Cardiovasc Surg 2005; 130: 1130–1136.
Lu C, Su LY, Gao YJ, Lee RMKW . Modulation of vein function by perivascular adipose tissue. Eur J Pharmacol 2011; 657: 111–116.
Lu C, Su LY, Lee RM, Gao YJ . Mechanisms for perivascular adipose tissue-mediated potentiation of vascular contraction to perivascular neuronal stimulation: the role of adipocyte-derived angiotensin II. Eur J Pharmacol 2010; 634: 107–112.
Sandow SL, Hill CE . Incidence of myoendothelial gap junctions in the proximal and distal mesenteric arteries of the rat is suggestive of a role in endothelium-derived hyperpolarizing factor-mediated responses. Circ Res 2000; 86: 341–346.
Sandow SL, Bramich NJ, Bandi HP, Rummery NM, Hill CE . Structure, function, and endothelium-derived hyperpolarizing factor in the caudal artery of the SHR and WKY rat. Arterioscler Thromb Vascr Biol 2003; 23: 822–828.
Sandow SL, Gzik DJ, Lee RMKW . Arterial internal elastic lamina holes: relationship to function? J Anat 2009; 214: 258–266.
Sandow SL, Senadheera S, Bertrand PP, Murphy TV, Tare M . Myoendothelial contacts, gap junctions and microdomains: anatomical links to function? Microcirc 2012; 19: 403–415.
Sandow SL, Looft-Wilson R, Doran B, Grayson TH, Segal SS, Hill CE . Expression of homocellular and heterocellular gap junctions in hamster arterioles and feed arteries. Cardiovasc Res 2003; 60: 643–653.
Hirst GD, Edwards FR, Gould DJ, Sandow SL, Hill CE . Electrical properties of iridial arterioles of the rat. Am J Physiol 1997; 273: H2465–H2472.
Howitt L, Grayson TH, Morris MJ, Sandow SL, Murphy TV . Dietary obesity increases NO and inhibits BKCa-mediated, endothelium-dependent dilation in rat cremaster muscle artery: association with caveolins and caveolae. Am J Physiol 2012; 302: H2464–H2476.
Faraci FM, Heistad DD . Regulation of large cerebral arteries and cerebral microvascular pressure. Circ Res 1990; 66: 8–17.
Bohlen HG . Localization of vascular resistance changes during hypertension. Hypertension 1986; 8: 181–183.
Bohlen HG, Gore RW, Hutchins PM . Comparison of microvascular pressures in normal and SHR. Microvasc Res 1977; 13: 125–130.
Christensen KL, Mulvany MJ . Mesenteric arcade arteries contribute substantially to vascular resistance in conscious rats. J Vasc Res 1993; 30: 73–79.
Smeda JS, RMKW Lee, Forrest JB . Structural and reactivity alterations in the renal vasculature of SHR prior to and during established hypertension. Circ Res 1988; 63: 518–533.
Miller BG, Conners BA, Bohlen HG, Evan AP . Cell and wall morphology of intestinal arterioles from 4-6 and 17-19 wk old Wistar-Kyoto and SHR. Hypertension 1987; 9: 59–68.
Christensen KL, Mulvany MJ . Location of resistance arteries. J Vasc Res 2001; 38: 1–12.
Lee RMKW, Smeda JS . Primary versus secondary structural changes of the blood vessels in hypertension. Can J Physiol Pharmacol 1985; 63: 392–401.
Owens GK . Differential effects of antihypertensive drug therapy on vascular smooth muscle cell hypertrophy, hyperploidy, and hyperplasia in the SHR. Circ Res 1985; 56: 525–536.
Owens GK, Rabinovitch PS, Schwartz SM . Smooth muscle cell hypertrophy versus hyperplasia in hypertension. Proc Nat Acad Sci USA 1981; 78: 7759–7763.
Owens GK, Schwartz SM . Alterations in vascular smooth muscle mass in the SHR. Role of cellular hypertrophy, hyperploidy, and hyperplasia. Circ Res 1982; 51: 280–289.
Lee RMKW, Tsoporis J, Wang RRJ . Influence of chronic nadolol treatment on blood pressure and vascular changes in SHR. Can J Physiol Pharmacol 1992; 70: 1261–1270.
RMKW Lee, Garfield RE, Forrest JB, Daniel EE . Morphometric study of structural changes in the mesenteric blood vessels of SHR. Blood Vessels 1983; 20: 57–71.
Smeda JS, Lee RMKW . Effect of hydralazine on the mesenteric vasculature of hypertensive rats. Hypertension 1991; 17: 526–533.
Gray SD . Anatomical and physiological aspects of cardiovascular function in Wistar-Kyoto and SHR at birth. Clin Sci 1982; 63: 383 s–385 s.
Nordborg C, Johansson BB . Morphometric study on cerebral vessels in SHR. Stroke 1980; 11: 266–270.
Hansen TR, Abrams GD, Bohr DF . Role of pressure in structural and functional changes in arteries of hypertensive rats. Circ Res 1974; 34/35 ((SI)): 101–107.
Wolinsky H . Response of the rat aortic media to hypertension. Morphological and chemical studies. Circ Res 1970; 26: 507–522.
Berry CL, Henrichs KJ . Morphometric investigation of hypertrophy in the arteries of DOCA-hypertensive rats. J Path 1982; 136: 85–94.
Lee RMKW, Richardson M, McKenzie R . Vascular changes associated with deoxycorticosterone-NaCl- induced hypertension. Blood Vessels 1989; 26: 137–156.
Bevan RD, van Marthens E, Bevan JA . Hyperplasia of vascular smooth muscle in experimental hypertension in the rabbit. Circ Res 1976; 38: II-58.
Wolinsky H . Long-term effects of hypertension on the rat aortic wall and their relation to concurrent aging changes. Circ Res 1972; 30: 301–309.
Olivetti G, Anversa P, Melissari M, Loud AV . Morphometry of medial hypertrophy in the rat thoracic aorta. Lab Invest 1980; 42: 559–565.
Lee RMKW, Triggle CR . Morphometric study of mesenteric arteries from genetically hypertensive Dahl strain rats. Blood Vessels 1986; 23: 199–224.
Yoo SM, Choi SH, Jung MD, Lim SC, Baek SH . Short-term use of telmisartan attenuates oxidation and improves Prdx2 expression more than antioxidant β-blockers in the cardiovascular systems of spontaneously hypertensive rats. Hypertens Res 2015; 38: 106–115.
Owens GK, Schwartz SM . Vascular smooth muscle cell hypertrophy and hyperploidy in the Goldblatt hypertensive rat. Circ Res 1983; 53: 491–501.
Lee RMKW . Vascular changes at the prehypertensive phase in the mesenteric arteries from SHR. Blood Vessels 1985; 22: 105–126.
Smeda JS, RMKW Lee, Forrest JB . Prenatal and postnatal hydralazine treatment does not prevent renal vessel wall thickening in SHR despite the absence of hypertension. Circ Res 1988; 63: 534–542.
Jespersen LT, Nyborg NCB, Pedersen OL, Mikkelsen EO, Mulvany MJ . Cardiac mass and peripheral vascular structure in hydralazine-treated SHR. Hypertension 1985; 7: 734–741.
Lee RMKW, Berecek KH, Tsoporis J, McKenzie R, Triggle CR . Prevention of hypertension and vascular changes by captopril treatment. Hypertension 1991; 17: 141–150.
Dickhout JG . Lee RMKW. Increased medial smooth muscle cell length is responsible for vascular hypertrophy in young hypertensive rats. Am J Physiol 2000; 279: H2085–H2094.
Mangiarua EI, Lee RMKW . Morphometric study of cerebral arteries from SHR and stroke-prone SHR. J Hypertens 1992; 10: 1183–1190.
Howitt L, Sandow SL, Grayson TH, Ellis ZE, Morris MJ, Murphy TV . Differential effects of diet-induced obesity on BKCaβ1-subunit expression and function in rat skeletal muscle arterioles and small cerebral arteries. Am J Physiol 2011; 301: H29–H40.
Greenberg S, Palmer EC, Wilborn WM . Pressure-independent hypertrophy of veins and pulmonary arteries of SHR. Characterization of function, structural and histochemical changes. Clin Sci Mol Med 1978; 55: 31S–36S.
Greenberg S, Gaines K, Sweatt D . Evidence for circulating factors as a cause of venous hypertrophy in SHR. Am J Physiol 1981; 241: H421–H430.
Aalkjaer C, Mulvany MJ . Morphological and mechanical properties of small mesenteric arteries and veins in SHR. Acta Physiol Scand 1979; 107: 309–312.
Mulvany MJ, Ljung B, Stoltze M, Kjellstedt A . Contractile and morphological properties of the portal vein in SHR and Wistar-Kyoto rats. Blood Vessels 1980; 17: 202–215.
Kanbe T, Nara Y, Tagami M, Yamori Y . Studies of hypertension-induced vascular hypertrophy in cultured smooth muscle cells from SHR. Hypertension 1983; 5: 887–892.
Yamori Y, Igawa T, Tagami M, Kanbe T, Nara Y, Nihara M, Horie R . Humoral trophic influence on cardiovascular structural changes in hypertension. Hypertension 1984; 6 (SIII): 27–32.
Hadrava V, Tremblay J, Hamet P . Abnormalities in growth characteristics of aortic smooth muscle cells in SHR. Hypertension 1989; 13: 589–597.
Hadrava V, Tremblay J, Hamet P . Intrinsic factors involved in vascular smooth muscle cell proliferation in hypertension. Clin Invest Med 1991; 14: 535–544.
Saltis J, Bobik A . Vascular smooth muscle growth in genetic hypertension: evidence for multiple abnormalities in growth regulatory pathways. J Hypertens 1992; 10: 635–643.
Rosen EM, Goldberg ID, Shapiro HM, Levenson SE, Halpin PA, Faraggi D . Strain and site dependence of polyploidization of cultured rat smooth muscle. J Cell Physiol 1986; 128: 337–344.
De Mey JGR, Uitendaal MP, Boonen HCM, Schiffers PMH, Fazzi GE . Growth responses in isolated elastic, muscular and resistance-sized arterial segments of the rat. Blood Vessels 1991; 28: 372–385.
Waldbillig DK, Pang SC . Differential proliferation of rat aortic and mesenteric smooth muscle cells in culture. Histol Histopathol 1992; 7: 199–207.
Owens GK . Determinants of angiotensin II-induced hypertrophy versus hyperplasia in vascular smooth muscle. Drug Dev Res 1993; 29: 83–87.
Altara R, Manca M, Brandão RD, Zeidan A, Booz GW, Zouein FA . Emerging importance of chemokine receptor CXCR3 and its ligands in cardiovascular diseases. Clin Sci 2016; 130: 463–478.
Kim HY, Cha HJ, Kim HS . CCL5 upregulates IL-10 expression and partially mediates the antihypertensive effects of IL-10 in the vascular smooth muscle cells of spontaneously hypertensive rats. Hypertens Res 2015; 38: 666–674.
Daemen MJAP, De Mey JGR . Regional heterogeneity of arterial structural changes. Hypertension 1995; 25: 464–473.
De Mey JG, Uitendaal MP, Boonen HC, Vrijdag MJ, Daemen MJ, Struyker-Boudier HA . Acute and long-term effects of tissue culture on contractile reactivity in renal arteries of the rat. Circ Res 1989; 65: 1125–1135.
Warshaw DM, Mulvany MJ, Halpern W . Mechanical and morphological properties of arterial resistance vessels in young and old SHR. Circ Res 1979; 45: 250–259.
Warshaw DM, Root DT, Halpern W . Effects of antihypertensive drug therapy on the morphology and mechanics of resistance arteries from SHR. Blood Vessels 1980; 17: 257–270.
Heagerty AM, Aalkjaer C, Bund SJ, Korsgaard N, Mulvany MJ . Small artery structure in hypertension: dual processes of remodeling and growth. Hypertension 1993; 21: 391–397.
Mulvany MJ, Baandrup U, Gundersen HJG . Evidence for hyperplasia in mesenteric resistance vessels of SHR using a three-dimensional disector. Circ Res 1985; 57: 794–800.
Sterio DC . The unbiased estimator of number and sizes of arbitrary particles using the dissector. J Microsc 1984; 134: 127–136.
Lee RMKW, McKenzie R, Roy M . Ultrastructure and morphometric measurements of mesenteric arteries from newborn SHR. Blood Vessels 1988; 25: 105–114.
Yang H, Morton W, RMKW Lee, Forrest JB . Autoradiographic study of smooth muscle cell proliferation in SHR. Clin Sci 1989; 76: 475–478.
De Mey JGR, MJAP Daemen, Boonen HCM, Bosman FT, Dijkstra EH, Fazzi GE, Janssen GM, Schiffers PM, Struyker-Boudier HA, Vridag MJ . In vivo DNA synthesis is not uniformly increased in arterial smooth muscle of young SHR. J Hypertens 1991; 9: 695–701.
Dickhout JG, Lee RMKW . Apoptosis in the muscular arteries from young SHR. J Hypertens 1999; 17: 1413–1419.
Rizzoni D, Rodella L, Porteri E, Rezzani R, Guelfi D, Piccoli A, Castellano M, Muiesan ML, Bianchi R, Rosei EA . Time course of apoptosis in small resistance arteries of SHR. J Hypertens 2000; 18: 885–891.
Intengan HD, Schiffrin EL . Vascular remodeling in hypertension: roles of apoptosis, inflammation, and fibrosis. Hypertension 2001; 38: 581–587.
Zicha J, Kunes J . Ontogenetic aspects of hypertension development: analysis in the rat. Physiol Rev 1999; 79: 1227–1282.
Dickhout JG, Lee RMKW . Structural and functional analysis of small arteries from young SHR. Hypertension 1997; 29: 781–789.
Arribas SM, Gonzalez C, Graham D, Dominiczak AF, McGrath JC . Cellular changes induced by chronic NO inhibition in intact rat basilar arteries revealed by confocal microscopy. J Hypertens 1997; 15: 1685–1693.
Arribas SM, Gonzalez JM, Briones AM, Somoza B, Daly CJ, Vila E, Gonzalez MC, McGrath JC . Confocal myography for the study of hypertensive vascular remodelling. Clin Hemorheol Microcirc 2007; 37: 205–210.
Arribas SM, Hillier C, Gonzalez C, McGrory S, Dominiczak AF, McGrath JC . Cellular aspects of vascular remodeling in hypertension revealed by confocal microscopy. Hypertension 1997; 30: 1455–1464.
Schulze-Bauer CA, Regitnig P, Holzapfel GA . Mechanics of the human femoral adventitia including the high-pressure response. Am J Physiol 2002; 282: H2427–H2440.
Diep QN, Amiri F, Touyz RM, Cohn JS, Endemann D, Neves MF, Schiffrin EL . PPARà activator effects on Ang II-induced vascular oxidative stress and inflammation. Hypertension 2002; 40: 866–871.
Yang L, Gao YJ, Lee RM . The effects of quinapril and atorvastatin on artery structure and function in adult SHR. Eur J Pharmacol 2005; 518: 145–151.
Julius S, Majahalme S . The changing face of sympathetic overactivity in hypertension. Ann Med 2000; 32: 365–370.
Rizzoni D, Perlini S, Mircoli L, Porteri E, Franzelli C, Castellano M, Rosei EA, Ferrari AU . Enhanced vascular reactivity in the sympathectomized rat: studies in vivo and in small isolated resistance arteries. J Hypertens 2000; 18: 1041–1049.
Dickhout JG, Lee RMKW . Blood pressure and heart rate development in young SHR. Am J Physiol 1998; 274: H794–H800.
Lee RMKW Structural and functional consequence of antihypertensive treatments on blood vessels. In: Lee RMKW (ed.). Blood Vessels Changes in Hypertension: Structure and Function Volume I. CRC Press: Boca Raton, FL, USA. 1989, pp 163–190.
Mangiarua EI, Lee RMKW . Increased sympathetic innervation in the cerebral and mesenteric arteries of hypertensive rats. Can J Physiol Pharmacol 1990; 68: 492–499.
Lee RMKW, Triggle CR, Cheung DW, Coughlin MD . Structural and functional consequence of neonatal sympathectomy on the blood vessels of SHR. Hypertension 1987; 10: 328–338.
Lee RMKW, Borkowski KR, Leenen FHH, Tsoporis J, Coughlin M . Combined effect of neonatal sympathectomy and adrenal demedullation on blood pressure and vascular changes in SHR. Circ Res 1991; 69: 714–721.
Lee RMKW, Gzik DJ . Sympatholytic intervention and vascular remodelling. Basic Res Cardiol 1991; 86: 55–64.
Henning RJ, Sawmiller DR . Vasoactive intestinal peptide: cardiovascular effects. Cardiovasc Res 2001; 49: 27–37.
Jovanovic A, Jovanovic S, Tulic I, Grbovic L . Predominant role for NO in the relaxation induced by VIP in human uterine artery. Mol Hum Reprod 1998; 4: 71–76.
Lee RMKW, Nagahama M, McKenzie R, Daniel EE . Peptide-containing nerves around blood vessels of stroke-prone SHR. Hypertension 1988; 11: I-117.
Scotland RS, Chauhan S, Davis C, De FC, Hunt S, Kabir J, Kotsonis P, Oh U, Ahluwalia A . Vanilloid receptor TRPV1, sensory C-fibers, and vascular autoregulation: a novel mechanism involved in myogenic constriction. Circ Res 2004; 95: 1027–1034.
Scott TM, Pang SC-N . Changes in jijunal arteries in SHR and normotensive rats following neonatal treatment with capsaicin. Acta Stereol 1983; 2/1: 127–133.
Ibrahim J, Schachter M, Hughes AD, Sever PS . Role of polyamines in hypertension induced by angiotensin II. Cardiovasc Res 1995; 29: 50–56.
Li JS, Touyz RM, Schiffrin EL . Effects of AT1 and AT2 angiotensin receptor antagonists in angiotensin II-infused rats. Hypertension 1998; 31: 487–492.
Lee RMKW, Ding L, Lu C, Su LY, Gao YJ . Alteration of perivascular adipose tissue function in AngII-induced hypertension. Can J Physiol Pharmacol 2009; 87: 944–953.
Gillies LK, Lu M, Wang H, Lee RMKW . AT1 receptor antagonist treatment caused persistent arterial functional changes in young SHR. Hypertension 1997; 30: 1471–1478.
Rizzoni D, Castellano M, Porteri E, Bettoni G, Muiesan ML, Cinelli A, Zulli R, Rosei EA . Prolonged effects of short-term fosinopril on blood pressure and vascular morphology and function in rats. Am J Hypertens 1997; 10: 1034–1043.
Rizzoni D, Castellano M, Porteri E, Bettoni G, Muiesan ML, Rosei EA . Delayed development of hypertension after short-term nitrendipine treatment. Hypertension 1994; 24: 131–139.
Gillies LK, Werstiuk ES, Lee RMKW . Cross-over study comparing effects of treatment with an angiotensin converting enzyme inhibitor and an angiotensin II type 1 receptor antagonist on cardiovascular changes in hypertension. J Hypertens 1998; 16: 477–486.
Kohara K, Brosnihan KB, Chappell MC, Khosla MC, Ferrario CM . Angiotensin-(1-7). A member of circulating angiotensin peptides. Hypertension 1991; 17: 131–138.
Yamamoto K, Chappell MC, Brosnihan KB, Ferrario CM . In vivo metabolism of angiotensin I by neutral endopeptidase (EC 3.4.24.11) in SHR. Hypertension 1992; 19: 692–696.
Benter IF, Ferrario CM, Morris M, Diz DI . Antihypertensive actions of angiotensin-(1-7) in SHR. Am J Physiol 1995; 269: H313–H319.
Lee RMKW, Lu C, Su LY, Gao YJ . Endothelium-dependent relaxation factor released by perivascular adipose tissue. J Hypertens 2009; 27: 782–790.
Yu C, Chen J, Guan W, Han Y, Wang WE, Wang X, Wang H, Jose PA, Zeng C . Activation of the D4 dopamine receptor attenuates proliferation and migration of vascular smooth muscle cells through downregulation of AT1a receptor expression. Hypertens Res 2015; 38: 588–596.
DeBlois D, Tea BS, Dam TV, Tremblay J, Hamet P . Smooth muscle apoptosis during vascuar regression in SHR. Hypertension 1997; 29: 340–349.
Yang L, Gao YJ, Lee RMKW . Quinapril effects on resistance artery structure and function in hypertension. Naunyn Schmiedebergs Arch Pharmacol 2004; 370: 444–451.
Dukacz SA, Feng MG, Yang LF, RMKW Lee, Kline RL . Abnormal renal medullary response to angiotensin II in SHR is corrected by long-term enalapril treatment. Am J Physiol 2001; 280: R1076–R1084.
Lee RMKW, Delaney KH, Lu M . Perindopril treatment prolonged the lifespan of SHR. J Hypertens 1995; 13: 471–476.
Wang H, Delaney KH, Kwiecien JM, Smeda JS, Lee RMKW . Prevention of stroke with perindopril treatment in stroke-prone SHR. Clin Invest Med 1997; 20: 327–338.
Wang H, Smeda JS, Lee RMKW . Prevention of stroke and preservation of the functions of cerebral arteries by treatment with perindopril in stroke-prone SHR. Can J Physiol Pharmacol 1998; 76: 26–34.
Smeda JS, VanVliet BN, King SR . Stroke-prone SHRlose their ability to auto-regulate cerebral blood flow prior to stroke. J Hypertens 1999; 17: 1697–1705.
Tohda K, Masuda H, Kawamura K, Shozawa T . Difference in dilatation between endothelium-preserved and -desquamated segments in the flow-loaded rat common carotid artery. Arterioscler Thromb 1992; 12: 519–528.
Tulis DA, Unthank JL, Prewitt RL . Flow-induced arterial remodeling in rat mesenteric vasculature. Am J Physiol 1998; 274: H874–H882.
Buus CL, Pourageaud F, Fazzi GE, Janssen G, Mulvany MJ, De Mey JG . Smooth muscle cell changes during flow-related remodeling of rat mesenteric resistance arteries. Circ Res 2001; 89: 180–186.
Koller A, Huang A . Development of NO and prostaglandin mediation of shear stress-induced arteriolar dilation with aging and hypertension. Hypertension 1999; 34: 1073–1079.
Henrion D, Iglarz M, Levy BI . Chronic endothelin-1 improves NO-dependent flow-induced dilation in resistance arteries from normotensive and hypertensive rats. Arterioscler Thromb Vasc Biol 1999; 19: 2148–2153.
Sun D, Huang A, Koller A, Kaley G . Endothelial KCa channels mediate flow-dependent dilation of arterioles of skeletal muscle and mesentery. Microvasc Res 2001; 61: 179–186.
Iriuchijima J . Regional blood flow in conscious SHR. Jpn J Physiol 1983; 33: 41–50.
Iida N . Different flow regulation mechanisms between celiac and mesenteric vascular beds in conscious rats. Hypertension 1995; 25: 260–265.
Lominadze D, Joshua IG, Schuschke DA . Blood flow shear rates in arterioles of SHR at early and established stages of hypertension. Clin Exp Hypertens 2001; 23: 317–328.
Tsukada K, Minamitani H, Sekizuka E, Oshio C . Image correlation method for measuring blood flow velocity in microcirculation: correlation 'window' simulation and in vivo image analysis. Physiol Meas 2000; 21: 459–471.
Unthank JL, Fath SW, Burkhart HM, Miller SC, Dalsing MC . Wall remodeling during luminal expansion of mesenteric arterial collaterals in the rat. Circ Res 1996; 79: 1015–1023.
Langille BL, O'Donnell F . Reductions in arterial diameter produced by chronic decreases in blood flow are endothelium-dependent. Science 1986; 231: 405–407.
Gairhe S, Bauer NN, Gebb SA, McMurtry IF . Myoendothelial gap junctional signaling induces differentiation of pulmonary arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2011; 301: L527–L535.
Gairhe S, Bauer NN, Gebb SA, McMurtry IF . Serotonin passes through myoendothelial gap junctions to promote pulmonary arterial smooth muscle cell differentiation. Am J Physiol Lung Cell Mol Physiol 2012; 303: L767–L767.
Gao YJ, Yang LF, Stead S, Lee RMKW . Flow-induced vascular remodeling in the mesenteric artery of SHR. Can J Physiol Pharmacol 2008; 86: 737–744.
Ellinsworth DC, Earley S, Murphy TV, Sandow SL . Endothelial control of vasodilation: integration of myoendothelial microdomain signalling and modulation by epoxyeicosatrienoic acids. Pflugers Arch 2014; 466: 389–405.
Giles TD, Sander GE, Nossaman BD, Kadowitz PJ . Impaired vasodilation in the pathogenesis of hypertension: focus on NO, EDHFs, and prostaglandins. J Clin Hypertens 2012; 14: 198–205.
Mulvany MJ, Baumbach GL, Aalkjaer C, Heagerty AM, Korsgaard N, Schiffrin EL, Heistad DD . Vascular remodeling. Letter to the Editor. Hypertension 1996; 28: 505–506.
Chadha PS, Haddock RE, Howitt L, Morris MJ, Murphy TV, Grayson TH, Sandow SL . Obesity up-regulates IKCa and myoendothelial gap junctions to maintain endothelial vasodilator function. J Pharmacol Exp Ther 2010; 335: 284–293.
Grayson TH, Chadha PS, Bertrand PP, Chen H, Morris MJ, Senadheera S, Murphy TV, Sandow SL . Increased caveolae density and caveolin-1 expression accompany impaired NO-mediated vasorelaxation in diet-induced obesity. Histochem Cell Biol 2013; 139: 309–321.
Sandow SL, Goto K, Rummery NM, Hill CE . Developmental changes in myoendothelial gap junction mediated vasodilator activity in the rat saphenous artery. J Physiol 2004; 556: 875–886.
Tran CH, Welsh DG . The differential hypothesis: a provocative rationalization of the conducted vasomotor response. Microcirc 2010; 17: 226–236.
Hill-Eubanks DC, Gonzales AL, Sonkusare SK, Nelson MT . Vascular TRP channels: performing under pressure and going with the flow. Physiology 2014; 29: 343–360.
Tran CH, Kurjiaka DT, Welsh DG . Emerging trend in second messenger communication and myoendothelial feedback. Front Physiol 2014; 5: 243.
Haddock RE, Grayson TH, Morris MJ, Howitt L, Chadha PS, Sandow SL . Diet-induced obesity impairs endothelium-derived hyperpolarization via altered potassium channel signaling mechanisms. PLoS ONE 2011; 6: e16423.
Kantachuvesiri S, Fleming S, Peters J, Peters B, Brooker G, Lammie AG, McGrath I, Kotelevtsev Y, Mullins JJ . Controlled hypertension, a transgenic toggle switch reveals differential mechanisms underlying vascular disease. J Biol Chem 2001; 276: 36727–36733.
Louis WJ, Howes LG . Genealogy of the SHR and Wistar-Kyoto rat strains: implications for studies of inherited hypertension. J Cardiovasc Pharmacol 1990; 16: S1–S5.
Bernatova I, Conde MV, Kopincova J, Gonzalez MC, Puzserova A, Arribas SM . Endothelial dysfunction in SHR: focus on methodological aspects. J Hypertens 2009; 27: S27–S31.
Heerkens EH, Izzard AS, Heagerty AM . Integrins, vascular remodeling, and hypertension. Hypertension 2007; 49: 1–4.
Intengan HD, Schiffrin EL . Structure and mechanical properties of resistance arteries in hypertension: role of adhesion molecules and extracellular matrix determinants. Hypertension 2000; 36: 312–318.
Hill MA, Nourian Z, Ho I-L, Clifford PS, Martinez-Lemus L, Meininger GA . Small artery elastin distribution and architecture-focus on the three dimensional organization. Microcirculation (e-pub ahead of print 30 June 2016; doi: 10.1111/micc.12294).
Martinez-Lemus LA, Hill MA, Meininger GA . The plastic nature of the vascular wall: a continuum of remodeling events contributing to control of arteriolar diameter and structure. Physiology 2009; 24: 45–57.
Heerkens EH, Shaw L, Ryding A, Brooker G, Mullins JJ, Austin C, Ohanian V, Heagerty AM . alphaV integrins are necessary for eutrophic inward remodeling of small arteries in hypertension. Hypertension 2006; 47: 281–287.
Bloksgaard M, Leurgans TM, Nissen I, Jensen PS, Hansen ML, Brewer JR, Bagatolli LA, Marcussen N, Irmukhamedov A, Rasmussen LM, De Mey JG . Elastin organization in pig and cardiovascular disease patients' pericardial resistance arteries. J Vasc Res 2015; 52: 1–11.
Galvez B, de Castro J, Herold D, Dubrovska G, Arribas S, Gonzalez MC, Aranguez I, Luft FC, Ramos MP, Gollasch M, Fernandez Alfonso MS . Perivascular adipose tissue and mesenteric vascular function in spontaneously hypertensive rats. Arterioscler Thromb Vasc Biol 2006; 26: 1297–1302.
Fang L, Zhao J, Chen Y, Ma T, Xu G, Tang C, Liu X, Geng B . Hydrogen sulfide derived from periadventitial adipose tissue is a vasodilator. J Hypertens 2009; 27: 2174–2185.
Yamawaki H, Tsubaki N, Mukohda M, Okada M, Hara Y . Omentin, a novel adipokine, induces vasodilation in rat isolated blood vessels. Biochem Biophys Res Commun 2010; 393: 668–672.
Fesus G, Dubrovska G, Gorzelniak K, Kluge R, Huang Y, Luft FC, Gollasch M . Adiponectin is a novel humoral vasodilator. Cardiovasc Res 2007; 75: 719–727.
Lee RMKW, Bader M, Alenina N, Santos RAS, Gao YJ, Lu C . The role of Mas receptors in modulating relaxation induced by perivascular adipose tissue. Life Sciences 2011; 89: 467–472.
Verlohren S, Dubrovska G, Tsang SY, Essin K, Luft FC, Huang Y, Gollasch M . Visceral periadventitial adipose tissue regulates arterial tone of mesenteric arteries. Hypertension 2004; 44: 271–276.
Takemori K, Gao YJ, Ding L, Lu C, Su LY, An WS, Vinson C, Lee RM . Elevated blood pressure in transgenic lipoatrophic mice and altered vascular function. Hypertension 2007; 49: 365–372.
Lu C, Su LY, Lee RM, Gao YJ . Alterations in perivascular adipose tissue structure and function in hypertension. Eur J Pharmacol 2011; 656: 68–73.
Greenstein AS, Khavandi K, Withers SB, Sonoyama K, Clancy O, Jeziorska M, Laing I, Yates AP, Pemberton PW, Malik RA, Heagerty AM . Local inflammation and hypoxia abolish the protective anticontractile properties of perivascular fat in obese patients. Circulation 2009; 119: 1661–1670.
Aghamohammadzadeh R, Withers S, Lynch F, Greenstein A, Malik R, Heagerty A . Perivascular adipose tissue from human systemic and coronary vessels: the emergence of a new pharmacotherapeutic target. Br J Pharmacol 2012; 165: 670–682.
Gao YJ, Holloway AC, Su LY, Takemori K, Lu C, Lee RMKW . Effects of fetal and neonatal exposure to nicotine on blood pressure and perivascular adipose tissue function in adult life. Eur J Pharmacol 2008; 590: 264–268.
Lee RM, Lu C, Su LY, Werstuck G, Gao YJ . Effects of hyperglycemia on the modulation of vascular function by perivascular adipose tissue. J Hypertens 2009; 27: 118–131.
Gao YJ, Takemori K, Su LY, An WS, Lu C, Sharma AM, Lee RMKW . Perivascular adipose tissue promotes vasoconstriction: the role of superoxide anion. Cardiovasc Res 2006; 71: 363–373.
Withers SB, Agabiti-Rosei C, Livingstone DM, Little MC, Aslam R, Malik RA, Heagerty AM . Macrophage activation is responsible for loss of anticontractile function in inflamed perivascular fat. Arterioscler Thromb Vasc Biol 2011; 31: 908–913.
Zeng ZH, Zhang ZH, Luo BH, He WK, Liang LY, He CC, Su CJ . The functional changes of the perivascular adipose tissue in spontaneously hypertensive rats and the effects of atorvastatin therapy. Clin Exp Hypertens 2009; 31: 355–363.
Agabiti-Rosei C, De CC, Rossini C, Porteri E, Rodella LF, Withers SB, Heagerty AM, Favero G, Agabiti-Rosei E, Rizzoni D, Rezzani R . Anticontractile activity of perivascular fat in obese mice and the effect of long-term treatment with melatonin. J Hypertens 2014; 32: 1264–1274.
Iyer SN, Chappell MC, Averill DB, Diz DI, Ferrario CM . Vasodepressor actions of angiotensin-(1-7) unmasked during combined treatment with lisinopril and losartan. Hypertension 1998; 31: 699–705.
Freeman EJ, Chisolm GM, Ferrario CM, Tallant EA . Angiotensin-(1-7) inhibits vascular smooth muscle cell growth. Hypertension 1996; 28: 104–108.
Kassam SI, Lu C, Buckley N, Lee RM . The mechanisms of propofol-induced vascular relaxation and modulation by perivascular adipose tissue and endothelium. Anesth Analg 2011; 112: 1339–1345.
Kassam SI, Lu C, Buckley N, Gao YJ, Lee RM . Modulation of thiopental-induced vascular relaxation and contraction by perivascular adipose tissue and endothelium. Br J Anaesth 2012; 109: 177–184.
Mulvany MJ, Halpern W . Contractile properties of small arterial resistance vessel in SHR and normotensive rats. Circ Res 1977; 41: 19–26.
Schiffrin EL, Hayoz D . How to assess vascular remodelling in small and medium-sized muscular arteries in humans. J Hypertens 1997; 15: 571–584.
Bund SJ, Lee RMKW . Arterial structural changes in hypertension: a consideration of methodology, terminology and functional consequence. J Vasc Res 2003; 40: 547–557.
Wu W, Zhang Y, Ballew JR, Fink G, Wang DH . Development of hypertension induced by subpressor infusion of Ang II: role of sensory nerves. Hypertension 2000; 36: 549–552.
Frühbeck G . The adipose tissue as a source of vasoactive factors. Curr Med Chem Cardiovasc Hematol Agents 2004; 2: 197–20.
Lynch FM, Withers SB, Yao Z, Werner ME, Edwards G, Weston AH, Heagerty AM . Perivascular adipose tissue-derived adiponectin activates BKCa channels to induce anticontractile responses. Am J Physiol Heart Circ Physiol 2013; 304: H786–H795.
Acknowledgements
Supported by the Brain Foundation, Australia (SLS).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Lee, R., Dickhout, J. & Sandow, S. Vascular structural and functional changes: their association with causality in hypertension: models, remodeling and relevance. Hypertens Res 40, 311–323 (2017). https://doi.org/10.1038/hr.2016.145
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hr.2016.145
Keywords
This article is cited by
-
Inhibition of Notch3/Hey1 ameliorates peribiliary hypoxia by preventing hypertrophic hepatic arteriopathy in biliary atresia progression
Histochemistry and Cell Biology (2024)
-
Neurovascular dysfunctions in hypertensive disorders of pregnancy
Metabolic Brain Disease (2021)
-
Association between epicardial adipose tissue thickness and parameters of target organ damage in patients undergoing coronary angiography
Hypertension Research (2019)
-
Brain arterial dilatation modifies the association between extracranial pulsatile hemodynamics and brain perivascular spaces: the Northern Manhattan Study
Hypertension Research (2019)
-
Role of activating transcription factor 3 in fructose-induced metabolic syndrome in mice
Hypertension Research (2018)
